
Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Virus and Infection
2.2. Mice and Tissue Collection
2.3. Determination of Lung Viral Titers
2.4. Bronchoalveolar Lavage (BAL)
2.5. Flow Cytometry
2.6. PCR and Gene Expression
2.7. Histopathology
2.8. Statistics
3. Results
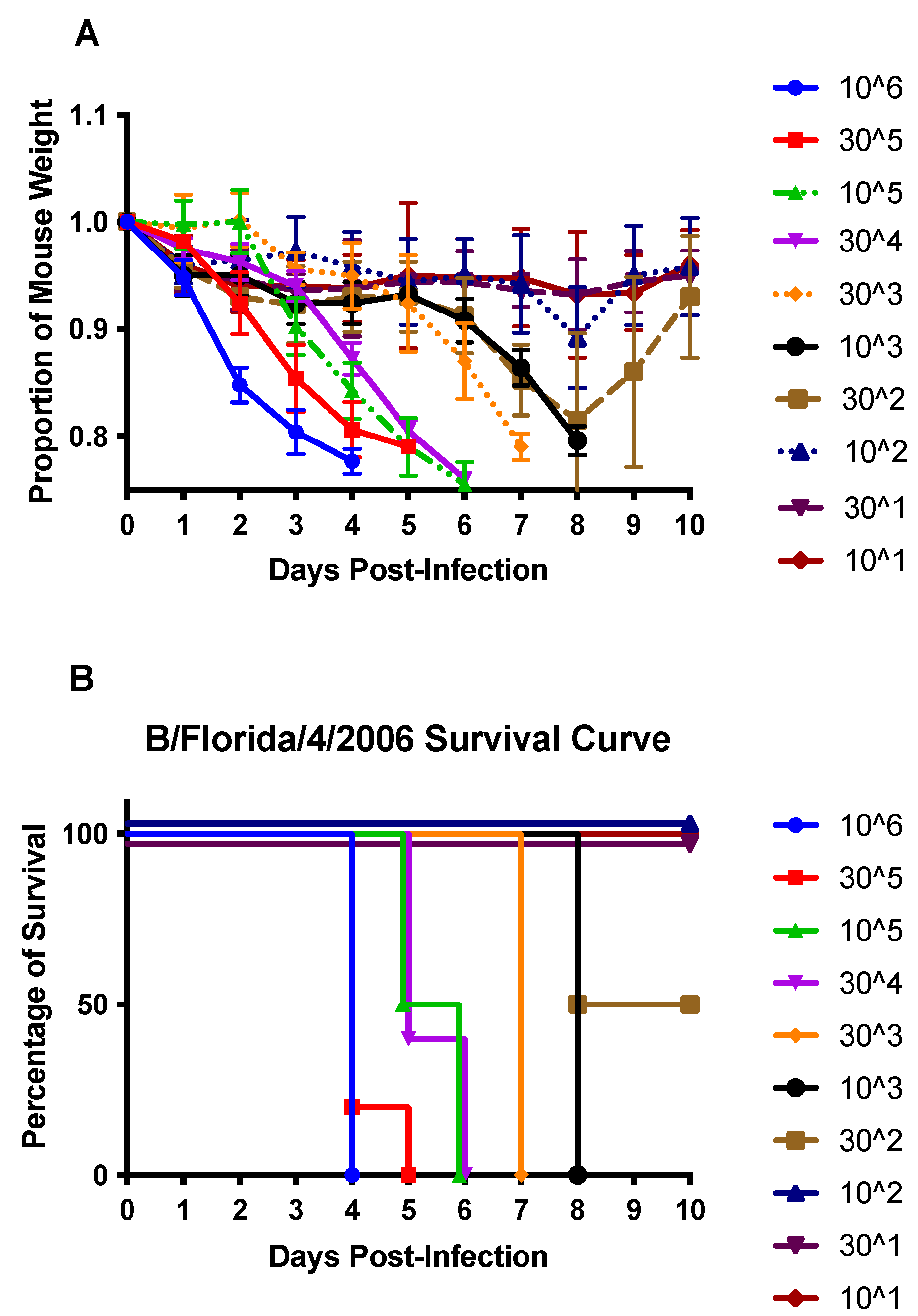
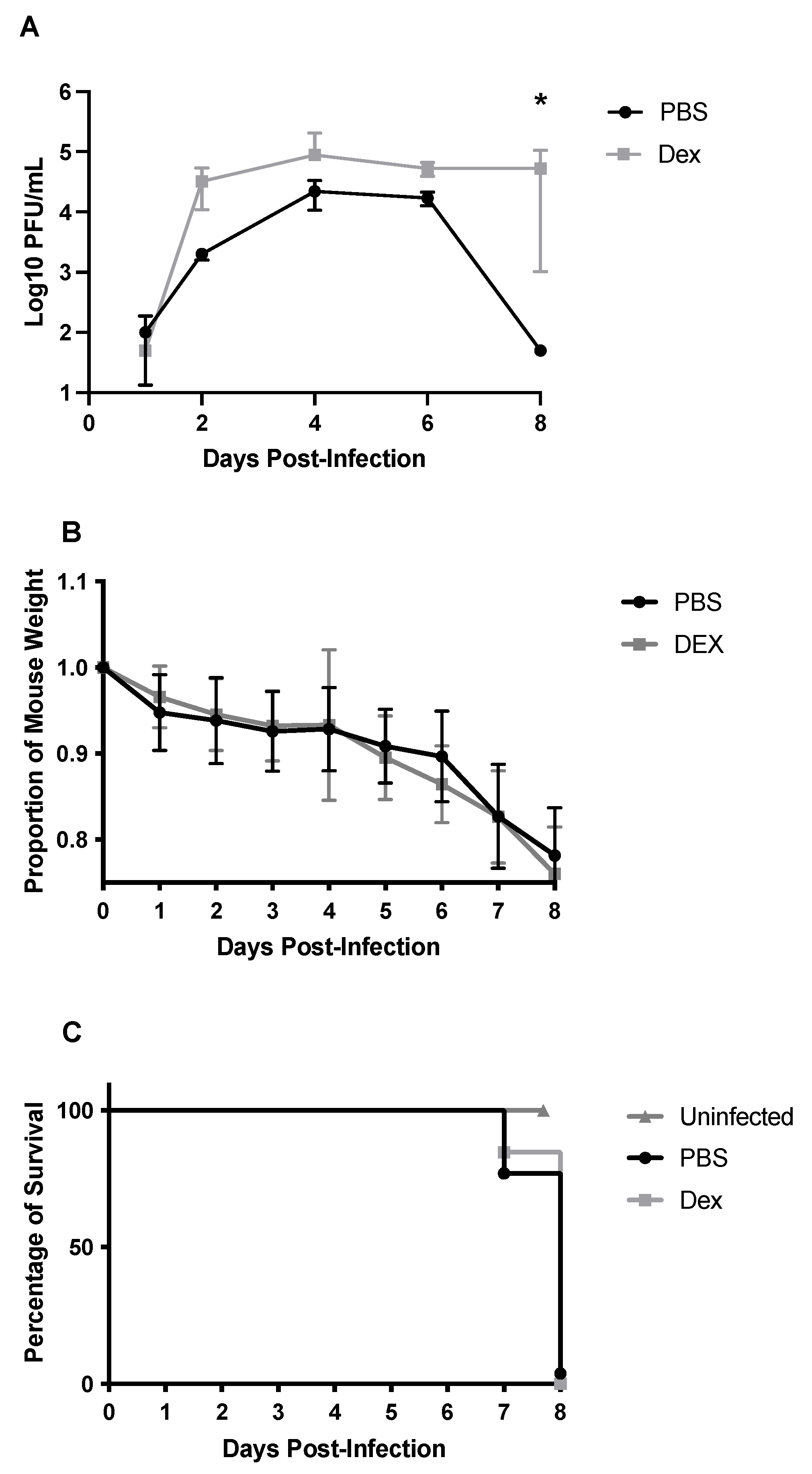
3.1. DEX Treatment Does Not Reduce Mortality
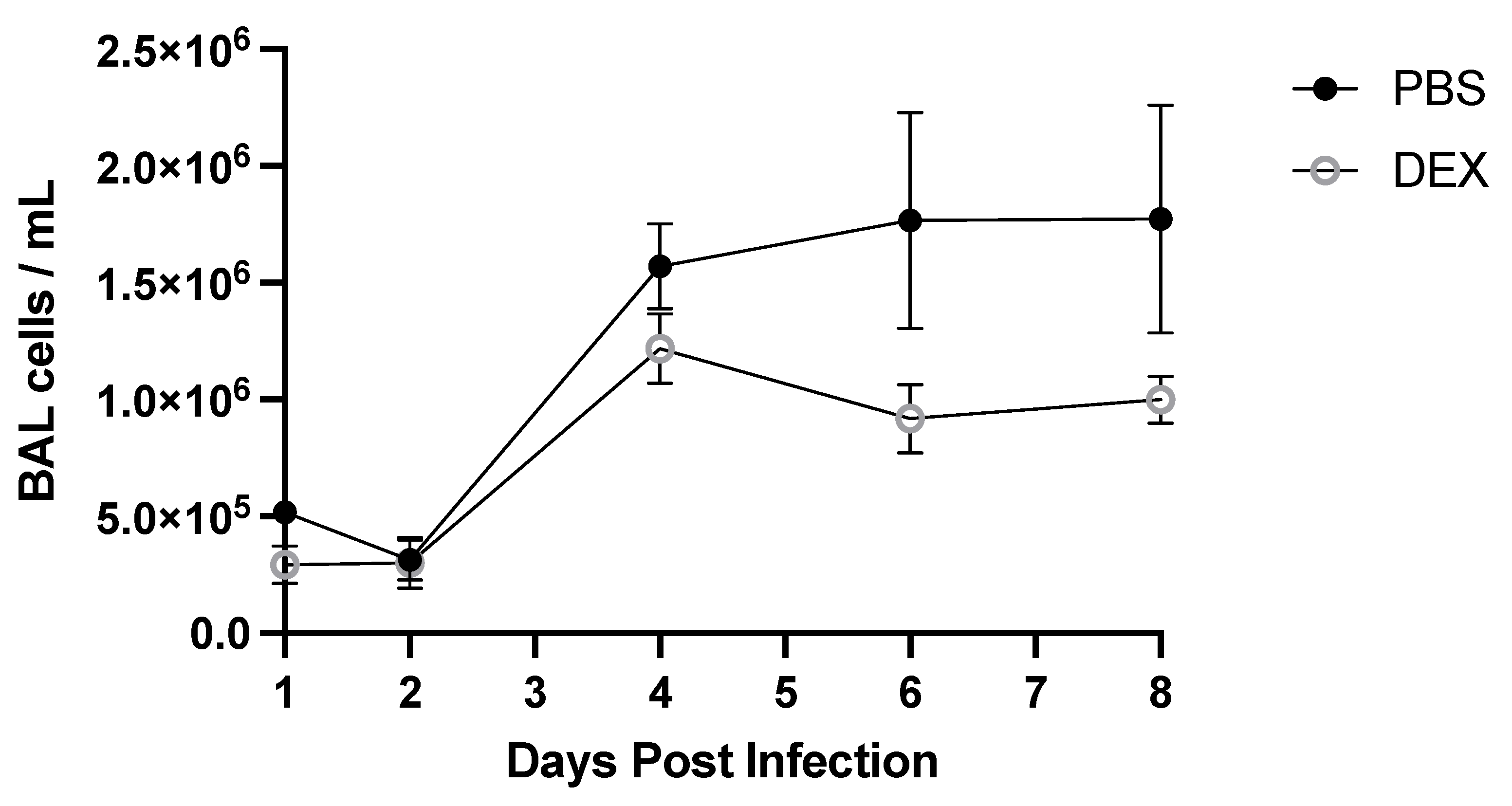
3.2. BAL Cell Response to B/Florida/04/2006 Infection and DEX Treatment
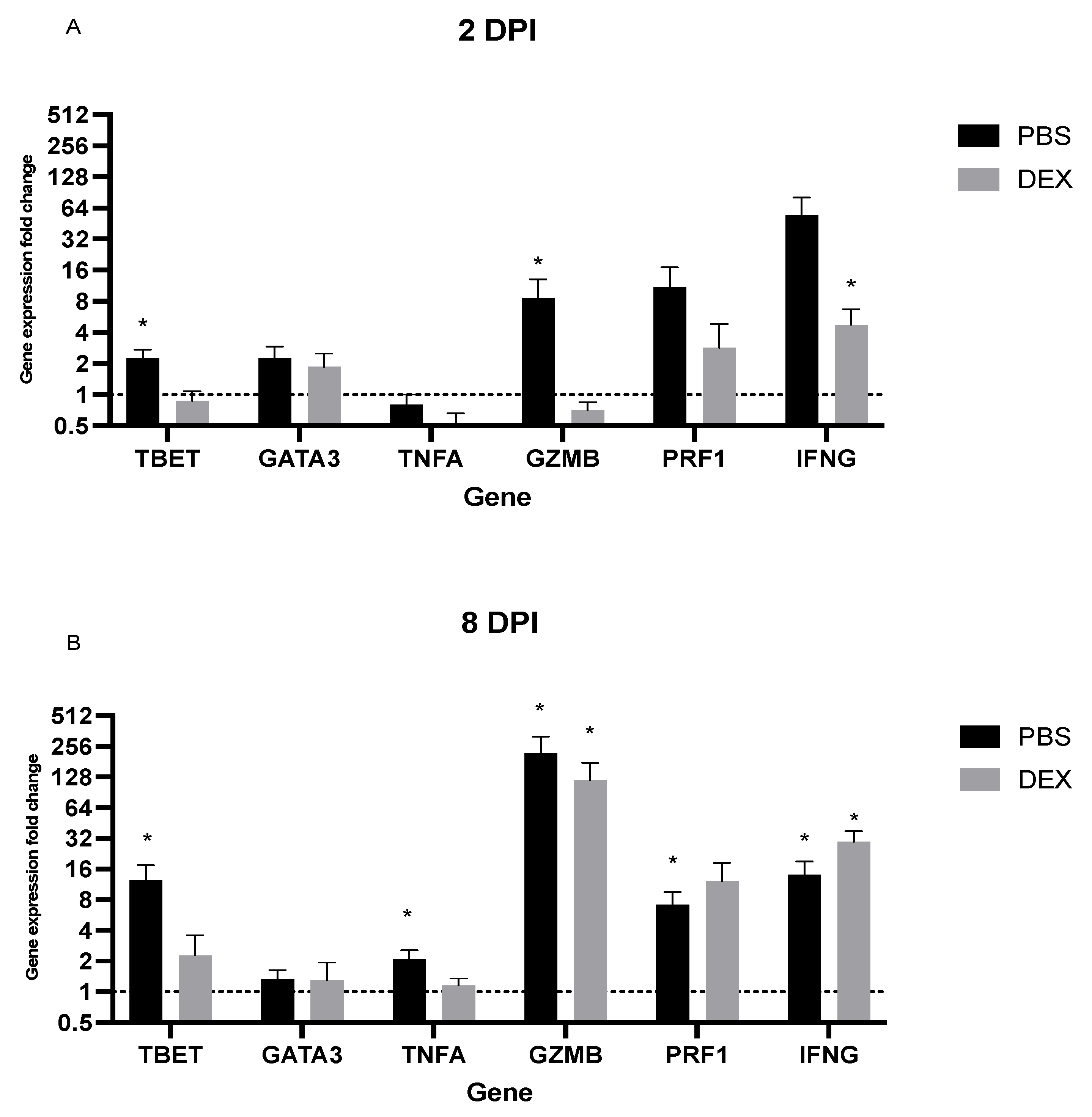
3.3. Lung Gene Expression in Response to IBV Infection
3.4. DEX Treatment Reduces Lung Histopathology Associated with IBV Infection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caini, S.; Kusznierz, G.; Garate, V.V.; Wangchuk, S.; Thapa, B.; Júnior, F.J.D.P.; De Almeida, W.A.F.; Njouom, R.; Fasce, R.A.; Bustos, P.; et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE 2019, 14, e0222381. [Google Scholar] [CrossRef] [PubMed]
- Tafalla, M.; Buijssen, M.; Geets, R.; Noordegraaf-Schouten, M.V. A comprehensive review of the epidemiology and disease burden of Influenza B in 9 European countries. Hum. Vaccines Immunother. 2016, 12, 993–1002. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gutiérrez-Pizarraya, A.; Pérez-Romero, P.; Alvarez, R.; Aydillo, T.; Osorio-Gómez, G.; Milara-Ibáñez, C.; Sánchez, M.; Pachón, J.; Cordero, E. Unexpected severity of cases of influenza B infection in patients that required hospitalization during the first postpandemic wave. J. Infect. 2012, 65, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chaves, S.S.; Perez, A.; D’Mello, T.; Kirley, P.D.; Yousey-Hindes, K.; Farley, M.M.; Harris, M.; Sharangpani, R.; Lynfield, R.; et al. Comparing Clinical Characteristics Between Hospitalized Adults With Laboratory-Confirmed Influenza A and B Virus Infection. Clin. Infect. Dis. 2014, 59, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Horwood, C.; Hakendorf, P.; Thompson, C. Clinical characteristics and outcomes of influenza A and B virus infection in adult Australian hospitalised patients. BMC Infect. Dis. 2020, 20, 913. [Google Scholar] [CrossRef]
- Jackson, D.; Elderfield, R.A.; Barclay, W.S. Molecular studies of influenza B virus in the reverse genetics era. J. Gen. Virol. 2010, 92, 1–17. [Google Scholar] [CrossRef]
- Österlund, P.; Strengell, M.; Sarin, L.P.; Poranen, M.M.; Fagerlund, R.; Melén, K.; Julkunen, I. Incoming Influenza A Virus Evades Early Host Recognition, while Influenza B Virus Induces Interferon Expression Directly upon Entry. J. Virol. 2012, 86, 11183–11193. [Google Scholar] [CrossRef]
- Lousa, D.; Soares, C.M. Molecular mechanisms of the influenza fusion peptide: Insights from experimental and simulation studies. FEBS Open Bio. 2021, 11, 3253–3261. [Google Scholar] [CrossRef]
- Ma, J.; Li, S.; Li, K.; Wang, X.; Li, S. Effects of the PA-X and PB1-F2 Proteins on the Virulence of the 2009 Pandemic H1N1 Influenza A Virus in Mice. Front. Cell. Infect. Microbiol. 2019, 9, 315. [Google Scholar] [CrossRef]
- Staller, E.; Sheppard, C.M.; Neasham, P.J.; Mistry, B.; Peacock, T.P.; Goldhill, D.H.; Long, J.S.; Barclay, W.S. ANP32 Proteins Are Essential for Influenza Virus Replication in Human Cells. J. Virol. 2019, 93, e00217-19. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Xu, L.; Guo, X.; Wang, W.; Ji, Y.; Lin, C.; Wang, Y.; Wang, X. Selective usage of ANP32 proteins by influenza B virus polymerase: Implications in determination of host range. PLoS Pathog. 2020, 16, e1008989. [Google Scholar] [CrossRef]
- Virk, R.K.; Jayakumar, J.; Mendenhall, I.H.; Moorthy, M.; Lam, P.; Linster, M.; Lim, J.; Lin, C.; Oon, L.L.E.; Lee, H.K.; et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 2020, 117, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Okoli, G.N.; Racovitan, F.; Abdulwahid, T.; Hyder, S.K.; Lansbury, L.; Righolt, C.H.; Mahmud, S.M.; Nguyen-Van-Tam, J.S. Decline in Seasonal Influenza Vaccine Effectiveness With Vaccination Program Maturation: A Systematic Review and Meta-analysis. Open Forum Infect. Dis. 2021, 8, ofab069. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-H.; Park, S.-J.; Kwon, H.-I.; Kim, S.M.; Kim, Y.-I.; Song, M.-S.; Choi, E.-J.; Pascua, P.N.Q.; Choi, Y.-K. Mouse adaptation of influenza B virus increases replication in the upper respiratory tract and results in droplet transmissibility in ferrets. Sci. Rep. 2015, 5, 15940. [Google Scholar] [CrossRef]
- de Benedictis, F.M.; Bush, A. Corticosteroids in Respiratory Diseases in Children. Am. J. Respir. Crit. Care Med. 2012, 185, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Aghai, Z.H.; Kumar, S.; Farhath, S.; Kumar, M.A.; Saslow, J.; Nakhla, T.; Eydelman, R.; Strande, L.; Stahl, G.; Hewitt, C.; et al. Dexamethasone Suppresses Expression of Nuclear Factor-kappaB in the Cells of Tracheobronchial Lavage Fluid in Premature Neonates with Respiratory Distress. Pediatr. Res. 2006, 59, 811–815. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.-K.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Li, C.; Yang, P.; Zhang, Y.; Sun, Y.; Wang, W.; Zou, Z.; Xing, L.; Chen, Z.; Tang, C.; Guo, F.; et al. Corticosteroid Treatment Ameliorates Acute Lung Injury Induced by 2009 Swine Origin Influenza A (H1N1) Virus in Mice. PLoS ONE 2012, 7, e44110. [Google Scholar] [CrossRef]
- Xu, T.; Qiao, J.; Zhao, L.; He, G.; Li, K.; Wang, J.; Tian, Y.; Wang, H. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur. Respir. J. 2009, 33, 852–860. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Giulietti, A.; Overbergh, L.; Valckx, D.; Decallonne, B.; Bouillon, R.; Mathieu, C. An Overview of Real-Time Quantitative PCR: Applications to Quantify Cytokine Gene Expression. Methods 2001, 25, 386–401. [Google Scholar] [CrossRef]
- Liu, R.; An, L.; Liu, G.; Li, X.; Tang, W.; Chen, X. Mouse lung slices: An ex vivo model for the evaluation of antiviral and anti-inflammatory agents against influenza viruses. Antivir. Res. 2015, 120, 101–111. [Google Scholar] [CrossRef]
- Li, W.; Moltedo, B.; Moran, T.M. Type I Interferon Induction during Influenza Virus Infection Increases Susceptibility to Secondary Streptococcus pneumoniae Infection by Negative Regulation of γδ T Cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef]
- Aoyagi, T.; Newstead, M.W.; Zeng, X.; Kunkel, S.L.; Kaku, M.; Standiford, T.J. IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol. 2017, 10, 1043–1055. [Google Scholar] [CrossRef]
- Chan, R.W.Y.; Leung, C.Y.H.; Nicholls, J.M.; Peiris, J.S.M.; Chan, M.C.W. Proinflammatory Cytokine Response and Viral Replication in Mouse Bone Marrow Derived Macrophages Infected with Influenza H1N1 and H5N1 Viruses. PLoS ONE 2012, 7, e51057. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Sarr, D.; Gingerich, A.D.; Asthiwi, N.M.; Almutairi, F.; Sautto, G.A.; Ecker, J.; Nagy, T.; Kilgore, M.B.; Chandler, J.D.; Ross, T.M.; et al. Dual oxidase 1 promotes antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2017130118. [Google Scholar] [CrossRef]
- Ashtiwi, N.M.; Sarr, D.; Nagy, T.; Reneer, Z.B.; Tripp, R.A.; Rada, B. The Hypothiocyanite and Amantadine Combination Treatment Prevents Lethal Influenza A Virus Infection in Mice. Front. Immunol. 2022, 13, 859033. [Google Scholar] [CrossRef]
- Yim, K.C.; Cragin, R.P.; Boukhvalova, M.S.; Blanco, J.C.; Hamlin, M.È.; Boivin, G.; Porter, D.D.; Prince, G.A. Human metapneumovirus: Enhanced pulmonary disease in cotton rats immunized with formalin-inactivated virus vaccine and challenged. Vaccine 2007, 25, 5034–5040. [Google Scholar] [CrossRef]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85–86. [Google Scholar] [CrossRef]
- Bianchi, M.; Meng, C.; Ivashkiv, L.B. Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proc. Natl. Acad. Sci. USA 2000, 97, 9573–9578. [Google Scholar] [CrossRef]
- Stern, A.; Skalsky, K.; Avni, T.; Carrara, E.; Leibovici, L.; Paul, M. Corticosteroids for pneumonia. Cochrane Database Syst. Rev. 2017, 2017, CD007720. [Google Scholar] [CrossRef]
- Perrone, L.A.; Plowden, J.K.; García-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 Pandemic Influenza Virus Infection Results in Early and Excessive Infiltration of Macrophages and Neutrophils in the Lungs of Mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef]
- Kobasa, D.; Takada, A.; Shinya, K.; Hatta, M.; Halfmann, P.; Theriault, S.; Suzuki, H.; Nishimura, H.; Mitamura, K.; Sugaya, N.; et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 2004, 431, 703–707. [Google Scholar] [CrossRef]
- Vlahos, R.; Stambas, J.; Selemidis, S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 2012, 33, 3–8. [Google Scholar] [CrossRef]
- Baumgarth, N.; Kelso, A. Functionally distinct T cells in three compartments of the respiratory tract after influenza virus infection. Eur. J. Immunol. 1996, 26, 2189–2197. [Google Scholar] [CrossRef]
- Migliorati, G.; Nicoletti, I.; D’Adamio, F.; Spreca, A.; Pagliacci, C.; Riccardi, C. Dexamethasone induces apoptosis in mouse natural killer cells and cytotoxic T lymphocytes. Immunology 1994, 81, 21–26. [Google Scholar]
- Wargnier, A.; Lafaurie, C.; Legros-Maida, S.; Bourge, J.F.; Sigaux, F.; Sasportes, M.; Paul, P. Down-regulation of human granzyme B expression by glucocorticoids. Dexamethasone inhibits binding to the Ikaros and AP-1 regulatory elements of the granzyme B promoter. J. Biol. Chem. 1998, 273, 35326–35331. [Google Scholar] [CrossRef]
- McAllister, C.S.; Ansaldi, D.; Growcott, E.J.; Zhong, Y.; Quackenbush, D.; Wolff, K.C.; Chen, Z.; Tanaseichuk, O.; Lelais, G.; Barnes, S.W.; et al. Dexamethasone inhibits respiratory syncytial virus-driven mucus production while increasing viral replication without altering antiviral interferon signaling. Virology 2020, 540, 195–206. [Google Scholar] [CrossRef]
- Ullrich, K.A.; Schulze, L.L.; Paap, E.M.; Muller, T.M.; Neurath, M.F.; Zundler, S. Immunology of IL-12: An update on functional activities and implications for disease. EXCLI J. 2020, 19, 1563–1589. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Yoo, J.-K.; Kim, T.S.; Hufford, M.M.; Braciale, T.J. Viral infection of the lung: Host response and sequelae. J. Allergy Clin. Immunol. 2013, 132, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Messmer, U.K.; Pereda-Fernandez, C.; Manderscheid, M.; Pfeilschifter, J. Dexamethasone inhibits TNF-alpha-induced apoptosis and IAP protein downregulation in MCF-7 cells. Br. J. Pharmacol. 2001, 133, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Tripp, R.A.; Tompkins, S.M. Virus-vectored influenza virus vaccines. Viruses 2014, 6, 3055–3079. [Google Scholar] [CrossRef]
- Cardenas-Garcia, S.; Caceres, C.J.; Rajao, D.; Perez, D.R. Reverse genetics for influenza B viruses and recent advances in vaccine development. Curr. Opin. Virol. 2020, 44, 191–202. [Google Scholar] [CrossRef]
- Rizzo, G.; Fiorucci, S. PPARs and other nuclear receptors in inflammation. Curr. Opin. Pharmacol. 2006, 6, 421–427. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Kedzierska, K.; Stambas, J.; Doherty, P.C. A question of self-preservation: Immunopathology in influenza virus infection. Immunol. Cell Biol. 2007, 85, 85–92. [Google Scholar] [CrossRef]
- Schwarz, K.B. Oxidative stress during viral infection: A review. Free Radic. Biol. Med. 1996, 21, 641–649. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | FWD | REV |
|---|---|---|
| TBX21 | 5′-TCC GGG AGA ACT TTG AGT CC-′ | 5′-TGG AAG GTC GGG GTA GAA AC-3′ |
| GATA3 | 5′-GTG GTC ACA CTC GGA TTC CT-3′ | 5′-GCA AAA AGG AGG GTT TAG GG-3′ |
| TNFA * | ||
| GZMB * | ||
| INFG | 5′-ATG AAC GCT ACA CAC TGC ATC-3′ | 5′-CCA TCC TTT TGC CAG TTC CTC-3′ |
| PFRN * | ||
| ACTB | 5′-AAG TGT GAC GTT GAC ATC CG-3′ | 5′-GAT CCA CAT CTG GAA GG-3′ |
| IBV (NP) | 5′-GGT TGG ACT TGA CCC TTC ATT A-3′ | 5′-CCA CTA AAG TTC CAC CTC CTT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergeron, H.C.; Reneer, Z.B.; Arora, A.; Reynolds, S.; Nagy, T.; Tripp, R.A. Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice. Vaccines 2022, 10, 1440. https://doi.org/10.3390/vaccines10091440
Bergeron HC, Reneer ZB, Arora A, Reynolds S, Nagy T, Tripp RA. Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice. Vaccines. 2022; 10(9):1440. https://doi.org/10.3390/vaccines10091440
Chicago/Turabian StyleBergeron, Harrison C., Zachary Beau Reneer, Aakash Arora, Stephen Reynolds, Tamas Nagy, and Ralph A. Tripp. 2022. "Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice" Vaccines 10, no. 9: 1440. https://doi.org/10.3390/vaccines10091440
APA StyleBergeron, H. C., Reneer, Z. B., Arora, A., Reynolds, S., Nagy, T., & Tripp, R. A. (2022). Influenza B Virus (IBV) Immune-Mediated Disease in C57BL/6 Mice. Vaccines, 10(9), 1440. https://doi.org/10.3390/vaccines10091440