Vitamin D Deficiency and the Risk of Cerebrovascular Disease

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
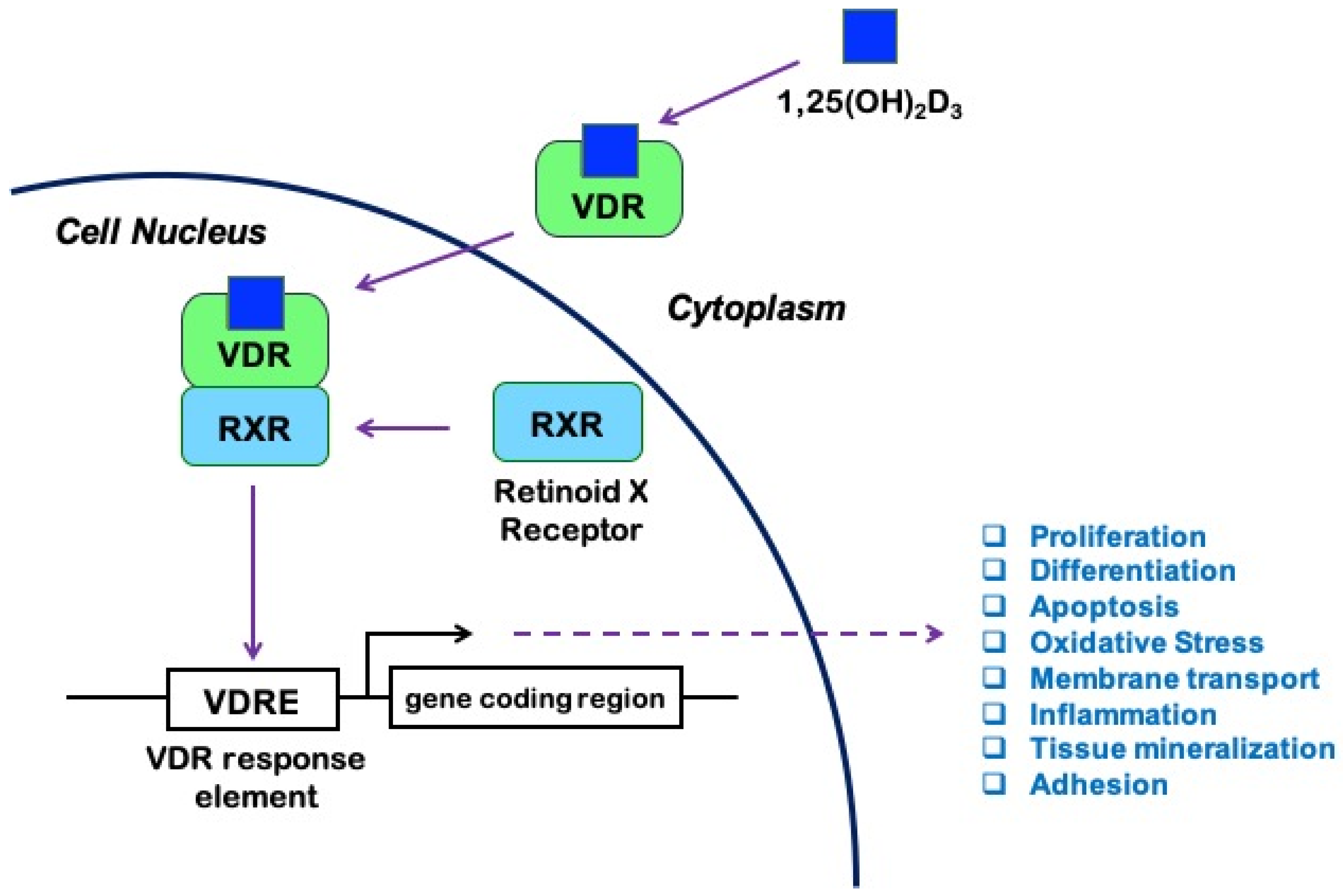
1. Sources, Metabolism, and Pleiotropic Functions of Vitamin D
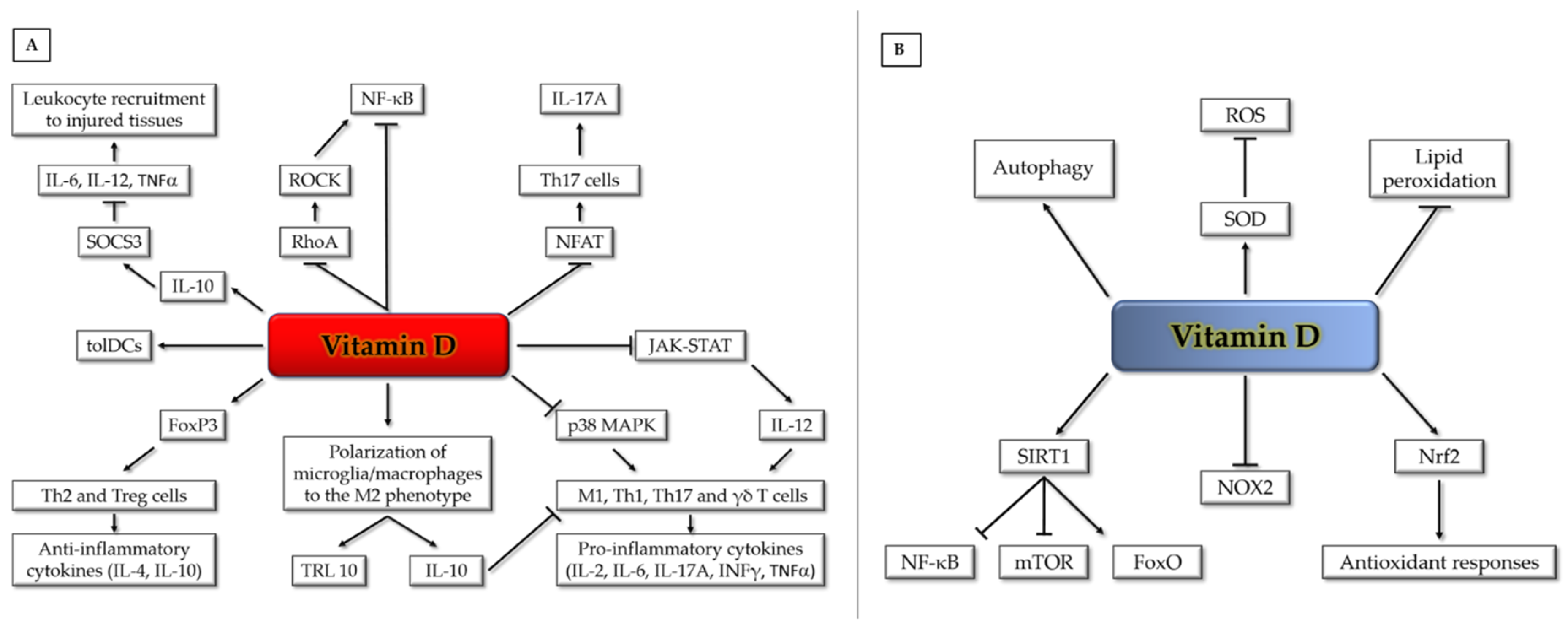
2. Anti-inflammatory Properties of Vitamin D
3. Antioxidant Properties of Vitamin D
4. Determinants of Vitamin D Status and Related Health Outcomes
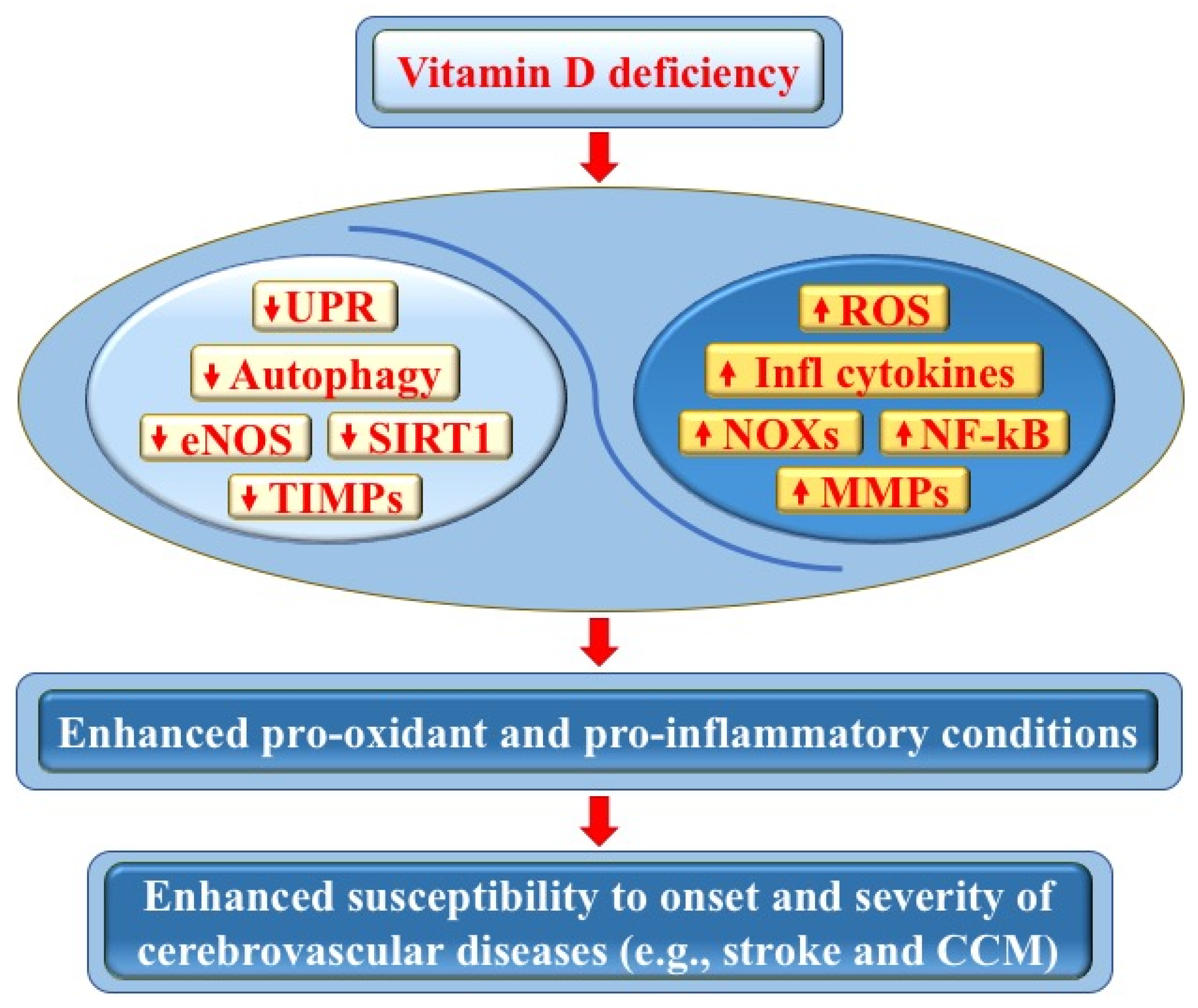
5. Vitamin D Deficiency and Its Impact on Cerebrovascular Diseases
5.1. Vitamin D Deficiency and Stroke
5.2. Vitamin D Deficiency and CCM Disease
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.R.; Kodicek, E. Unique biosynthesis by kidney of a biological active vitamin D metabolite. Nature 1970, 228, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Provvedini, D.M.; Tsoukas, C.D.; Deftos, L.J.; Manolagas, S.C. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science 1983, 221, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Merke, J.; Milde, P.; Lewicka, S.; Hügel, U.; Klaus, G.; Mangelsdorf, D.J.; Haussler, M.R.; Rauterberg, E.W.; Ritz, E. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J. Clin. Investig. 1989, 83, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Brewer, L.D.; Thibault, V.; Chen, K.C.; Langub, M.C.; Landfield, P.W.; Porter, N.M. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J. Neurosci. 2001, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, R.; Williams, R.M.; Xu, M.; Noy, N. Nuclear import of the retinoid X receptor, the vitamin D receptor, and their mutual heterodimer. J. Biol. Chem. 2005, 280, 40152–40160. [Google Scholar] [CrossRef]
- Zhang, J.; Chalmers, M.J.; Stayrook, K.R.; Burris, L.L.; Wang, Y.; Busby, S.A.; Pascal, B.D.; Garcia-Ordonez, R.D.; Bruning, J.B.; Istrate, M.A.; et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 2011, 18, 556–563. [Google Scholar] [CrossRef]
- Lugg, S.T.; Howells, P.A.; Thickett, D.R. Optimal Vitamin D Supplementation Levels for Cardiovascular Disease Protection. Dis. Markers 2015, 2015, 864370. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. Minireview: Vitamin D receptor: New assignments for an already busy receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Lieben, L.; Mathieu, C.; Verstuyf, A.; Carmeliet, G. Vitamin D action: Lessons from VDR and Cyp27b1 null mice. Pediatr. Endocrinol. Rev. 2013, 10 (Suppl. 2), 354–366. [Google Scholar] [PubMed]
- Bikle, D. Nonclassic actions of vitamin D. J. Clin. Endocrinol. Metab. 2009, 94, 26–34. [Google Scholar] [CrossRef]
- Bouillon, R.; Bikle, D. Vitamin D Metabolism Revised: Fall of Dogmas. J. Bone Miner. Res. 2019, 34, 1985–1992. [Google Scholar] [CrossRef]
- Bikle, D. Vitamin D: Production, Metabolism, and Mechanisms of Action. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2017; p. 2000. [Google Scholar]
- Huhtakangas, J.A.; Olivera, C.J.; Bishop, J.E.; Zanello, L.P.; Norman, A.W. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha,25(OH)2-vitamin D3 in vivo and in vitro. Mol. Endocrinol. 2004, 18, 2660–2671. [Google Scholar] [CrossRef]
- Christakos, S.; Li, S.; DeLa Cruz, J.; Verlinden, L.; Carmeliet, G. Vitamin D and Bone. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2019. [Google Scholar] [CrossRef]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)₂vitamin D₃: Genomic and non-genomic mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef]
- Komaba, H.; Kakuta, T.; Fukagawa, M. Management of secondary hyperparathyroidism: How and why? Clin. Exp. Nephrol. 2017, 21, 37–45. [Google Scholar] [CrossRef]
- Daniel, C.; Sartory, N.A.; Zahn, N.; Radeke, H.H.; Stein, J.M. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J. Pharmacol. Exp. Ther. 2008, 324, 23–33. [Google Scholar] [CrossRef]
- Ponsonby, A.L.; McMichael, A.; Van der Mei, I. Ultraviolet radiation and autoimmune disease: Insights from epidemiological research. Toxicology 2002, 181, 71–78. [Google Scholar] [CrossRef]
- Hyppönen, E.; Läärä, E.; Reunanen, A.; Järvelin, M.R.; Virtanen, S.M. Intake of vitamin D and risk of type 1 diabetes: A birth-cohort study. Lancet 2001, 358, 1500–1503. [Google Scholar] [CrossRef]
- Ingraham, B.A.; Bragdon, B.; Nohe, A. Molecular basis of the potential of vitamin D to prevent cancer. Curr. Med. Res. Opin. 2008, 24, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Annweiler, C.; Duval, G.; Karras, S.; Tirabassi, G.; Salvio, G.; Balercia, G.; Kimball, S.; Kotsa, K.; Mascitelli, L.; et al. Vitamin D and cardiovascular disease: From atherosclerosis to myocardial infarction and stroke. Int. J. Cardiol. 2017, 230, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Uberti, F.; Lattuada, D.; Morsanuto, V.; Nava, U.; Bolis, G.; Vacca, G.; Squarzanti, D.F.; Cisari, C.; Molinari, C. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J. Clin. Endocrinol. Metab. 2014, 99, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Nordbrandt, S.P.; Jäättelä, M. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol. Med. 2010, 16, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.P.; Giovannucci, E.; Holmes, M.D.; Bischoff-Ferrari, H.A.; Tworoger, S.S.; Willett, W.C.; Curhan, G.C. Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension 2007, 49, 1063–1069. [Google Scholar] [CrossRef]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Vacek, J.L.; Vanga, S.R.; Good, M.; Lai, S.M.; Lakkireddy, D.; Howard, P.A. Vitamin D deficiency and supplementation and relation to cardiovascular health. Am. J. Cardiol. 2012, 109, 359–363. [Google Scholar] [CrossRef]
- Song, Y.; Wang, L.; Pittas, A.G.; Del Gobbo, L.C.; Zhang, C.; Manson, J.E.; Hu, F.B. Blood 25-hydroxy vitamin D levels and incident type 2 diabetes: A meta-analysis of prospective studies. Diabetes Care 2013, 36, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, L.C.; Woolley, D.E. The effects of 1 alpha,25-dihydroxyvitamin D(3) on matrix metalloproteinase and prostaglandin E(2) production by cells of the rheumatoid lesion. Arthritis Res. 1999, 1, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wöbke, T.K.; Sorg, B.L.; Steinhilber, D. Vitamin D in inflammatory diseases. Front. Physiol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, W. The Roles of Vitamin D and Its Analogs in Inflammatory Diseases. Curr. Top. Med. Chem. 2016, 16, 1242–1261. [Google Scholar] [CrossRef]
- Garbossa, S.G.; Folli, F. Vitamin D, sub-inflammation and insulin resistance. A window on a potential role for the interaction between bone and glucose metabolism. Rev. Endocr. Metab. Disord. 2017, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, J. Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov. Med. 2011, 11, 325–335. [Google Scholar]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, I.; et al. Vitamin D receptor regulates autophagy in the normal mammary gland and in luminal breast cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186–E2194. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Nakahira, K.; Cloonan, S.M.; Mizumura, K.; Choi, A.M.; Ryter, S.W. Autophagy: A crucial moderator of redox balance, inflammation, and apoptosis in lung disease. Antioxid Redox Signal. 2014, 20, 474–494. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef]
- Retta, S.F.; Glading, A.J. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int. J. Biochem. Cell Biol. 2016, 81, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Eid, N.; Abdelmegeed, M.A.; Sen, A. Interplay of Oxidative Stress, Inflammation, and Autophagy: Their Role in Tissue Injury of the Heart, Liver, and Kidney. Oxid. Med. Cell. Longev. 2018, 2018, 2090813. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, Z.; Liu, Y.; Tu, X.; He, J. Relationship between serum vitamin D levels and inflammatory markers in acute stroke patients. Brain Behav. 2018, 8, e00885. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, D.F.; Lehmann, M.F.; Oliveira, S.R.; Flauzino, T.; Delongui, F.; De Araújo, M.C.; Dichi, I.; Delfino, V.D.; Mezzaroba, L.; Simão, A.N.; et al. Vitamin D deficiency is associated with acute ischemic stroke, C-reactive protein, and short-term outcome. Metab. Brain Dis. 2017, 32, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Giarratana, N.; Smiroldo, S.; Uskokovic, M.; Adorini, L. A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes 2002, 51, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Pantalena, L.C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol. Cell. Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef]
- Korf, H.; Wenes, M.; Stijlemans, B.; Takiishi, T.; Robert, S.; Miani, M.; Eizirik, D.L.; Gysemans, C.; Mathieu, C. 1,25-Dihydroxyvitamin D3 curtails the inflammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology 2012, 217, 1292–1300. [Google Scholar] [CrossRef]
- Evans, M.A.; Kim, H.A.; Ling, Y.H.; Uong, S.; Vinh, A.; De Silva, T.M.; Arumugam, T.V.; Clarkson, A.N.; Zosky, G.R.; Drummond, G.R.; et al. Vitamin D3 supplementation reduces subsequent brain injury and inflammation associated with ischemic stroke. Neuromol. Med. 2018, 20, 147–159. [Google Scholar] [CrossRef]
- Pedersen, L.B.; Nashold, F.E.; Spach, K.M.; Hayes, C.E. 1,25-dihydroxyvitamin D3 reverses experimental autoimmune encephalomyelitis by inhibiting chemokine synthesis and monocyte trafficking. J. Neurosci. Res. 2007, 85, 2480–2490. [Google Scholar] [CrossRef]
- Grishkan, I.V.; Fairchild, A.N.; Calabresi, P.A.; Gocke, A.R. 1,25-Dihydroxyvitamin D3 selectively and reversibly impairs T helper-cell CNS localization. Proc. Natl. Acad. Sci. USA 2013, 110, 21101–21106. [Google Scholar] [CrossRef]
- Yu, X.P.; Bellido, T.; Manolagas, S.C. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1995, 92, 10990–10994. [Google Scholar] [CrossRef] [PubMed]
- Harant, H.; Wolff, B.; Lindley, I.J. 1Alpha,25-dihydroxyvitamin D3 decreases DNA binding of nuclear factor-kappaB in human fibroblasts. FEBS Lett. 1998, 436, 329–334. [Google Scholar] [CrossRef]
- Qian, X.; Zhu, M.; Qian, W.; Song, J. Vitamin D attenuates myocardial ischemia-reperfusion injury by inhibiting inflammation via suppressing the RhoA/ROCK/NF-ĸB pathway. Biotechnol. Appl. Biochem. 2019, 66, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Reddy, G.S.; Kobayashi, T.; Okano, T.; Park, J.; Sharma, S. Nuclear factor of activated T cells (NFAT) as a molecular target for 1alpha,25-dihydroxyvitamin D3-mediated effects. J. Immunol. 1998, 160, 209–218. [Google Scholar]
- Mattner, F.; Smiroldo, S.; Galbiati, F.; Muller, M.; Di Lucia, P.; Poliani, P.L.; Martino, G.; Panina-Bordignon, P.; Adorini, L. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D(3). Eur. J. Immunol. 2000, 30, 498–508. [Google Scholar] [CrossRef]
- Muthian, G.; Raikwar, H.P.; Rajasingh, J.; Bright, J.J. 1,25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNgamma axis leading to Th1 response in experimental allergic encephalomyelitis. J. Neurosci. Res. 2006, 83, 1299–1309. [Google Scholar] [CrossRef]
- Zeitelhofer, M.; Adzemovic, M.Z.; Gomez-Cabrero, D.; Bergman, P.; Hochmeister, S.; N’diaye, M.; Paulson, A.; Ruhrmann, S.; Almgren, M.; Tegnér, J.N.; et al. Functional genomics analysis of vitamin D effects on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2017, 114, E1678–E1687. [Google Scholar] [CrossRef]
- Chen, L.; Cencioni, M.T.; Angelini, D.F.; Borsellino, G.; Battistini, L.; Brosnan, C.F. Transcriptional profiling of gamma delta T cells identifies a role for vitamin D in the immunoregulation of the V gamma 9V delta 2 response to phosphate-containing ligands. J. Immunol. 2005, 174, 6144–6152. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chang, T.S.; Jeong, W.; Kang, D. Methods for detection and measurement of hydrogen peroxide inside and outside of cells. Mol. Cells 2010, 29, 539–549. [Google Scholar] [CrossRef]
- Sloka, S.; Silva, C.; Wang, J.; Yong, V.W. Predominance of Th2 polarization by vitamin D through a STAT6-dependent mechanism. J. Neuroinflamm. 2011, 8, 56. [Google Scholar] [CrossRef]
- Nashold, F.E.; Nelson, C.D.; Brown, L.M.; Hayes, C.E. One calcitriol dose transiently increases Helios+ FoxP3+ T cells and ameliorates autoimmune demyelinating disease. J. Neuroimmunol. 2013, 263, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Yamashita, T.; Sasaki, N.; Nakajima, K.; Kita, T.; Shinohara, M.; Ishida, T.; Hirata, K. Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Gorman, S.; Judge, M.A.; Hart, P.H. Topical 1,25-dihydroxyvitamin D3 subverts the priming ability of draining lymph node dendritic cells. Immunology 2010, 131, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 2012, 188, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Kim, J.Y. 1,25-Dihydroxyvitamin D3 Facilitates M2 Polarization and Upregulates TLR10 Expression on Human Microglial Cells. Neuroimmunomodulation 2016, 23, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Boontanrart, M.; Hall, S.D.; Spanier, J.A.; Hayes, C.E.; Olson, J.K. Vitamin D3 alters microglia immune activation by an IL-10 dependent SOCS3 mechanism. J. Neuroimmunol. 2016, 292, 126–136. [Google Scholar] [CrossRef]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Wong, M.S.; Leisegang, M.S.; Kruse, C.; Vogel, J.; Schürmann, C.; Dehne, N.; Weigert, A.; Herrmann, E.; Brüne, B.; Shah, A.M.; et al. Vitamin D promotes vascular regeneration. Circulation 2014, 130, 976–986. [Google Scholar] [CrossRef]
- Sotirchos, E.S.; Bhargava, P.; Eckstein, C.; Van Haren, K.; Baynes, M.; Ntranos, A.; Gocke, A.; Steinman, L.; Mowry, E.M.; Calabresi, P.A. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology 2016, 86, 382–390. [Google Scholar] [CrossRef]
- Wiseman, H. Vitamin D is a membrane antioxidant. Ability to inhibit iron-dependent lipid peroxidation in liposomes compared to cholesterol, ergosterol and tamoxifen and relevance to anticancer action. FEBS Lett. 1993, 326, 285–288. [Google Scholar] [CrossRef]
- Sardar, S.; Chakraborty, A.; Chatterjee, M. Comparative effectiveness of vitamin D3 and dietary vitamin E on peroxidation of lipids and enzymes of the hepatic antioxidant system in Sprague—Dawley rats. Int. J. Vitam. Nutr. Res. 1996, 66, 39–45. [Google Scholar] [PubMed]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Chang, E. 1,25-Dihydroxyvitamin D Decreases Tertiary Butyl-Hydrogen Peroxide-Induced Oxidative Stress and Increases AMPK/SIRT1 Activation in C2C12 Muscle Cells. Molecules 2019, 24, 3903. [Google Scholar] [CrossRef]
- Cui, C.; Song, S.; Cui, J.; Feng, Y.; Gao, J.; Jiang, P. Vitamin D Receptor Activation Influences NADPH Oxidase (NOX). Oxid. Med. Cell. Longev. 2017, 2017, 9245702. [Google Scholar]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef]
- Teixeira, T.M.; Da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef]
- Kallay, E.; Pietschmann, P.; Toyokuni, S.; Bajna, E.; Hahn, P.; Mazzucco, K.; Bieglmayer, C.; Kato, S.; Cross, H.S. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis 2001, 22, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, S.; Porri, D.; De Giuseppe, R.; Manuelli, M.; Alessio, F.; Cena, H. The controversial role of vitamin D as an antioxidant: Results from randomised controlled trials. Nutr. Res. Rev. 2019, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Khan, S.; Naseem, I. Antioxidant Role of Vitamin D in Mice With Alloxan-Induced Diabetes. Can. J. Diabetes 2018, 42, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.C.; Olabisi, O.; Barnett, M.E.; Meng, Y.X.; Martins, D.; Obialo, C.; Lee, J.E.; Nicholas, S.B. The Role of Vitamin D and Oxidative Stress in Chronic Kidney Disease. Int. J. Environ. Res. Public Health 2018, 15, 2701. [Google Scholar] [CrossRef]
- Ambrożewicz, E.; Muszyńska, M.; Tokajuk, G.; Grynkiewicz, G.; Žarković, N.; Skrzydlewska, E. Beneficial Effects of Vitamins K and D3 on Redox Balance of Human Osteoblasts Cultured with Hydroxyapatite-Based Biomaterials. Cells 2019, 8, 325. [Google Scholar] [CrossRef]
- Lee, T.W.; Kao, Y.H.; Chen, Y.J.; Chao, T.F.; Lee, T.I. Therapeutic potential of vitamin D in AGE/RAGE-related cardiovascular diseases. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Ursem, S.; Francic, V.; Keppel, M.; Schwetz, V.; Trummer, C.; Pandis, M.; Aberer, F.; Grübler, M.R.; Verheyen, N.D.; März, W.; et al. The effect of vitamin D supplementation on plasma non-oxidised PTH in a randomised clinical trial. Endocr. Connect. 2019, 8, 518–527. [Google Scholar] [CrossRef]
- Sepidarkish, M.; Farsi, F.; Akbari-Fakhrabadi, M.; Namazi, N.; Almasi-Hashiani, A.; Maleki Hagiagha, A.; Heshmati, J. The effect of vitamin D supplementation on oxidative stress parameters: A systematic review and meta-analysis of clinical trials. Pharmacol. Res. 2019, 139, 141–152. [Google Scholar] [CrossRef]
- Norman, P.E.; Powell, J.T. Vitamin D and cardiovascular disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.J.; Lee, Y.F. Vitamin D and oxidative stress. In Vitamin D: Oxidative Stress, Immunity, and Aging; Gombart, A.F., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 131–150. [Google Scholar]
- Gibson, C.C.; Zhu, W.; Davis, C.T.; Bowman-Kirigin, J.A.; Chan, A.C.; Ling, J.; Walker, A.E.; Goitre, L.; Delle Monache, S.; Retta, S.F.; et al. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 2015, 131, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Zhou, Y.; Bu, F.; Travers-Gustafson, D.; Ye, A.; Xu, X.; Hamm, L.; Gorsage, D.M.; Fang, X.; Deng, H.W.; et al. Factors predicting vitamin D response variation in non-Hispanic white postmenopausal women. J. Clin. Endocrinol. Metab. 2012, 97, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Mazahery, H.; Von Hurst, P.R. Factors Affecting 25-Hydroxyvitamin D Concentration in Response to Vitamin D Supplementation. Nutrients 2015, 7, 5111–5142. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Tao, S.; Liu, H.; Pan, G.; Li, B.; Zhang, Z. The Association between Polymorphisms of Vitamin D Metabolic-Related Genes and Vitamin D. J. Diabetes Res. 2019, 2019, 8289741. [Google Scholar] [CrossRef] [PubMed]
- Balvers, M.G.; Brouwer-Brolsma, E.M.; Endenburg, S.; De Groot, L.C.; Kok, F.J.; Gunnewiek, J.K. Recommended intakes of vitamin D to optimise health, associated circulating 25-hydroxyvitamin D concentrations, and dosing regimens to treat deficiency: Workshop report and overview of current literature. J. Nutr. Sci. 2015, 4, e23. [Google Scholar] [CrossRef]
- Pilz, S.; Zittermann, A.; Trummer, C.; Theiler-Schwetz, V.; Lerchbaum, E.; Keppel, M.H.; Grübler, M.R.; März, W.; Pandis, M. Vitamin D testing and treatment: A narrative review of current evidence. Endocr. Connect. 2019, 8, R27–R43. [Google Scholar] [CrossRef]
- Holick, M.F.; Chen, T.C.; Lu, Z.; Sauter, E. Vitamin D and skin physiology: A D-lightful story. J. Bone Miner. Res. 2007, 22 (Suppl. 2), V28–V33. [Google Scholar] [CrossRef]
- Wacker, M.; Holick, M.F. Sunlight and Vitamin D: A global perspective for health. Dermatoendocrinol 2013, 5, 51–108. [Google Scholar] [CrossRef]
- Jääskeläinen, T.; Itkonen, S.T.; Lundqvist, A.; Erkkola, M.; Koskela, T.; Lakkala, K.; Dowling, K.G.; Hull, G.L.; Kröger, H.; Karppinen, J.; et al. The positive impact of general vitamin D food fortification policy on vitamin D status in a representative adult Finnish population: Evidence from an 11-y follow-up based on standardized 25-hydroxyvitamin D data. Am. J. Clin. Nutr. 2017, 105, 1512–1520. [Google Scholar] [CrossRef]
- Öhlund, I.; Lind, T.; Hernell, O.; Silfverdal, S.A.; Karlsland Åkeson, P. Increased vitamin D intake differentiated according to skin color is needed to meet requirements in young Swedish children during winter: A double-blind randomized clinical trial. Am. J. Clin. Nutr. 2017, 106, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Jayaratne, N.; Hughes, M.C.; Ibiebele, T.I.; Van den Akker, S.; Van der Pols, J.C. Vitamin D intake in Australian adults and the modeled effects of milk and breakfast cereal fortification. Nutrition 2013, 29, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; März, W.; Cashman, K.D.; Kiely, M.E.; Whiting, S.J.; Holick, M.F.; Grant, W.B.; Pludowski, P.; Hiligsmann, M.; Trummer, C.; et al. Rationale and Plan for Vitamin D Food Fortification: A Review and Guidance Paper. Front. Endocrinol. 2018, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, H.M.; Mavroeidi, A.; Aucott, L.A.; Diffey, B.L.; Fraser, W.D.; Ormerod, A.D.; Reid, D.M. Skin color change in Caucasian postmenopausal women predicts summer-winter change in 25-hydroxyvitamin D: Findings from the ANSAViD cohort study. J. Clin. Endocrinol. Metab. 2011, 96, 1677–1686. [Google Scholar] [CrossRef][Green Version]
- Sawicki, C.M.; Van Rompay, M.I.; Au, L.E.; Gordon, C.M.; Sacheck, J.M. Sun-Exposed Skin Color Is Associated with Changes in Serum 25-Hydroxyvitamin D in Racially/Ethnically Diverse Children. J. Nutr. 2016, 146, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.L.; Rees, J.R.; Peacock, J.L.; Mott, L.A.; Amos, C.I.; Bostick, R.M.; Figueiredo, J.C.; Ahnen, D.J.; Bresalier, R.S.; Burke, C.A.; et al. Genetic variants in CYP2R1, CYP24A1, and VDR modify the efficacy of vitamin D3 supplementation for increasing serum 25-hydroxyvitamin D levels in a randomized controlled trial. J. Clin. Endocrinol. Metab. 2014, 99, E2133–E2137. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, D.A.; Walton, R.T.; Griffiths, C.J.; Martineau, A.R. Single nucleotide polymorphisms in the vitamin D pathway associating with circulating concentrations of vitamin D metabolites and non-skeletal health outcomes: Review of genetic association studies. J. Steroid Biochem. Mol. Biol. 2016, 164, 18–29. [Google Scholar] [CrossRef]
- Jiang, T.; Li, L.; Wang, Y.; Zhao, C.; Yang, J.; Ma, D.; Guan, Y.; Zhao, D.; Bao, Y. The Association Between Genetic Polymorphism rs703842 in CYP27B1 and Multiple Sclerosis: A Meta-Analysis. Medicine 2016, 95, e3612. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Ma, T. Associations of Genetic Polymorphisms Relevant to Metabolic Pathway of Vitamin D3 with Development and Prognosis of Childhood Bronchial Asthma. DNA Cell Biol. 2017, 36, 682–692. [Google Scholar] [CrossRef]
- Stjepanovic, M.I.; Mihailovic-Vucinic, V.; Spasovski, V.; Milin-Lazovic, J.; Skodric-Trifunovic, V.; Stankovic, S.; Andjelkovic, M.; Komazec, J.; Momcilovic, A.; Santric-Milicevic, M.; et al. Genes and metabolic pathway of sarcoidosis: Identification of key players and risk modifiers. Arch. Med. Sci. 2019, 15, 1138–1146. [Google Scholar] [CrossRef]
- Malik, S.; Fu, L.; Juras, D.J.; Karmali, M.; Wong, B.Y.; Gozdzik, A.; Cole, D.E. Common variants of the vitamin D binding protein gene and adverse health outcomes. Crit. Rev. Clin. Lab. Sci. 2013, 50, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.; Vogel, U.; Ravn-Haren, G.; Andersen, E.W.; Madsen, K.H.; Nexø, B.A.; Andersen, R.; Mejborn, H.; Bjerrum, P.J.; Rasmussen, L.B.; et al. Common variants in CYP2R1 and GC genes are both determinants of serum 25-hydroxyvitamin D concentrations after UVB irradiation and after consumption of vitamin D₃-fortified bread and milk during winter in Denmark. Am. J. Clin. Nutr. 2015, 101, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Munger, K.L.; Kraft, P.; Hunter, D.J.; De Jager, P.L.; Ascherio, A. Genetic predictors of 25-hydroxyvitamin D levels and risk of multiple sclerosis. J. Neurol. 2011, 258, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Flemming, K.D.; Brown, R.D.; Link, M.J. Seasonal variation in hemorrhage and focal neurologic deficit due to intracerebral cavernous malformations. J. Clin. Neurosci. 2015, 22, 969–971. [Google Scholar] [CrossRef]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef]
- Garland, C.F.; Kim, J.J.; Mohr, S.B.; Gorham, E.D.; Grant, W.B.; Giovannucci, E.L.; Baggerly, L.; Hofflich, H.; Ramsdell, J.W.; Zeng, K.; et al. Meta-analysis of all-cause mortality according to serum 25-hydroxyvitamin D. Am. J. Public Health 2014, 104, e43–e50. [Google Scholar] [CrossRef]
- Pilz, S.; Verheyen, N.; Grübler, M.R.; Tomaschitz, A.; März, W. Vitamin D and cardiovascular disease prevention. Nat. Rev. Cardiol. 2016, 13, 404–417. [Google Scholar] [CrossRef]
- Zittermann, A.; Gummert, J.F. Sun, vitamin D, and cardiovascular disease. J. Photochem. Photobiol. B 2010, 101, 124–129. [Google Scholar] [CrossRef]
- Daubail, B.; Jacquin, A.; Guilland, J.C.; Khoumri, C.; Aboa-Eboulé, C.; Giroud, M.; Béjot, Y. Association between serum concentration of vitamin D and 1-year mortality in stroke patients. Cerebrovasc. Dis. 2014, 37, 364–367. [Google Scholar] [CrossRef]
- Kheiri, B.; Abdalla, A.; Osman, M.; Ahmed, S.; Hassan, M.; Bachuwa, G. Vitamin D deficiency and risk of cardiovascular diseases: A narrative review. Clin. Hypertens. 2018, 24, 9. [Google Scholar] [CrossRef]
- Wajda, J.; Świat, M.; Owczarek, A.J.; Brzozowska, A.; Olszanecka-Glinianowicz, M.; Chudek, J. Severity of Vitamin D Deficiency Predicts Mortality in Ischemic Stroke Patients. Dis. Markers 2019, 2019, 3652894. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, H.M.; Mavroeidi, A.; Fraser, W.D.; Darling, A.L.; Black, A.J.; Aucott, L.; O’Neill, F.; Hart, K.; Berry, J.L.; Lanham-New, S.A.; et al. Sunlight and dietary contributions to the seasonal vitamin D status of cohorts of healthy postmenopausal women living at northerly latitudes: A major cause for concern? Osteoporos Int. 2011, 22, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Harvey, D.J.; Beckett, L.A.; Green, R.; Farias, S.T.; Reed, B.R.; Olichney, J.M.; Mungas, D.M.; DeCarli, C. Vitamin D Status and Rates of Cognitive Decline in a Multiethnic Cohort of Older Adults. JAMA Neurol. 2015, 72, 1295–1303. [Google Scholar] [CrossRef]
- Gibson, C.C.; Davis, C.T.; Zhu, W.; Bowman-Kirigin, J.A.; Walker, A.E.; Tai, Z.; Thomas, K.R.; Donato, A.J.; Lesniewski, L.A.; Li, D.Y. Dietary Vitamin D and Its Metabolites Non-Genomically Stabilize the Endothelium. PLoS ONE 2015, 10, e0140370. [Google Scholar] [CrossRef] [PubMed]
- Mithal, A.; Wahl, D.A.; Bonjour, J.P.; Burckhardt, P.; Dawson-Hughes, B.; Eisman, J.A.; El-Hajj Fuleihan, G.; Josse, R.G.; Lips, P.; Morales-Torres, J.; et al. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos. Int. 2009, 20, 1807–1820. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.W.; Burt, A.; Edwards, T.L.; Zuchner, S.; Scott, W.K.; Martin, E.R.; Vance, J.M.; Wang, L. Vitamin D receptor gene as a candidate gene for Parkinson disease. Ann. Hum. Genet. 2011, 75, 201–210. [Google Scholar] [CrossRef]
- Ito, S.; Ohtsuki, S.; Nezu, Y.; Koitabashi, Y.; Murata, S.; Terasaki, T. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1-40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS 2011, 8, 20. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.W.; Tang, Y.L.; Liu, X.; Jiang, J.H.; Li, Q.G.; Yuan, J.Y. Vitamin D: Preventive and therapeutic potential in Parkinson’s disease. Curr. Drug Metab. 2013, 14, 989–993. [Google Scholar] [CrossRef]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef]
- Yiu, Y.F.; Chan, Y.H.; Yiu, K.H.; Siu, C.W.; Li, S.W.; Wong, L.Y.; Lee, S.W.; Tam, S.; Wong, E.W.; Cheung, B.M.; et al. Vitamin D deficiency is associated with depletion of circulating endothelial progenitor cells and endothelial dysfunction in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, E830–E835. [Google Scholar] [CrossRef] [PubMed]
- Talmor, Y.; Golan, E.; Benchetrit, S.; Bernheim, J.; Klein, O.; Green, J.; Rashid, G. Calcitriol blunts the deleterious impact of advanced glycation end products on endothelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F1059–F1064. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef]
- Martorell, S.; Hueso, L.; Gonzalez-Navarro, H.; Collado, A.; Sanz, M.J.; Piqueras, L. Vitamin D Receptor Activation Reduces Angiotensin-II-Induced Dissecting Abdominal Aortic Aneurysm in Apolipoprotein E-Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1587–1597. [Google Scholar] [CrossRef]
- Schedel, M.; Jia, Y.; Michel, S.; Takeda, K.; Domenico, J.; Joetham, A.; Ning, F.; Strand, M.; Han, J.; Wang, M.; et al. 1,25D3 prevents CD8(+)Tc2 skewing and asthma development through VDR binding changes to the Cyp11a1 promoter. Nat. Commun. 2016, 7, 10213. [Google Scholar] [CrossRef]
- Mark, K.A.; Dumas, K.J.; Bhaumik, D.; Schilling, B.; Davis, S.; Oron, T.R.; Sorensen, D.J.; Lucanic, M.; Brem, R.B.; Melov, S.; et al. Vitamin D Promotes Protein Homeostasis and Longevity via the Stress Response Pathway Genes skn-1, ire-1, and xbp-1. Cell Rep. 2016, 17, 1227–1237. [Google Scholar] [CrossRef]
- Zhu, J.; Bing, C.; Wilding, J.P.H. Vitamin D receptor ligands attenuate the inflammatory profile of IL-1β-stimulated human white preadipocytes via modulating the NF-κB and unfolded protein response pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1049–1056. [Google Scholar] [CrossRef]
- López-López, N.; González-Curiel, I.; Treviño-Santa Cruz, M.B.; Rivas-Santiago, B.; Trujillo-Paez, V.; Enciso-Moreno, J.A.; Serrano, C.J. Expression and vitamin D-mediated regulation of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in healthy skin and in diabetic foot ulcers. Arch. Dermatol. Res. 2014, 306, 809–821. [Google Scholar] [CrossRef]
- Freestone, T.; Turner, R.J.; Coady, A.; Higman, D.J.; Greenhalgh, R.M.; Powell, J.T. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1145–1151. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C252–C263. [Google Scholar] [CrossRef] [PubMed]
- Brøndum-Jacobsen, P.; Nordestgaard, B.G.; Schnohr, P.; Benn, M. 25-hydroxyvitamin D and symptomatic ischemic stroke: An original study and meta-analysis. Ann. Neurol. 2013, 73, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Chung, P.W.; Kim, Y.B.; Moon, H.S.; Suh, B.C.; Won, Y.S.; Kim, J.M.; Youn, Y.C.; Kwon, O.S. Serum Vitamin D Status as a Predictor of Prognosis in Patients with Acute Ischemic Stroke. Cerebrovasc. Dis. 2015, 40, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yalbuzdag, S.A.; Sarifakioglu, B.; Afsar, S.I.; Celik, C.; Can, A.; Yegin, T.; Senturk, B.; Guzelant, A.Y. Is 25(OH)D Associated with Cognitive Impairment and Functional Improvement in Stroke? A Retrospective Clinical Study. J. Stroke Cerebrovasc. Dis. 2015, 24, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Daumas, A.; Daubail, B.; Legris, N.; Jacquin-Piques, A.; Sensenbrenner, B.; Denimal, D.; Lemaire-Ewing, S.; Duvillard, L.; Giroud, M.; Béjot, Y. Association between Admission Serum 25-Hydroxyvitamin D Levels and Functional Outcome of Thrombolyzed Stroke Patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zheng, T.; Wang, S.; Wei, L.; Wang, Q.; Sun, Z. Serum 25-hydroxyvitamin D predicts early recurrent stroke in ischemic stroke patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, Y.; Huang, G.; Zhu, J.; Feng, W.; He, J. Association between vitamin D status and cognitive impairment in acute ischemic stroke patients: A prospective cohort study. Clin. Interv. Aging 2018, 13, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Girard, R.; Khanna, O.; Shenkar, R.; Zhang, L.; Wu, M.; Jesselson, M.; Zeineddine, H.A.; Gangal, A.; Fam, M.D.; Gibson, C.C.; et al. Peripheral plasma vitamin D and non-HDL cholesterol reflect the severity of cerebral cavernous malformation disease. Biomark. Med. 2016, 10, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Flemming, K.D. Clinical Management of Cavernous Malformations. Curr. Cardiol. Rep. 2017, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics--2013 update: A report from the American Heart Association. Circulation 2013, 127, e6–e245. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Miller, A.A.; Drummond, G.R.; Kemp-Harper, B.K.; Sobey, C.G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Makariou, S.E.; Michel, P.; Tzoufi, M.S.; Challa, A.; Milionis, H.J. Vitamin D and stroke: Promise for prevention and better outcome. Curr. Vasc. Pharmacol. 2014, 12, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Codelia, V.A.; Sun, G.; Irvine, K.D. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr. Biol. 2014, 24, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Judd, S.E.; Morgan, C.J.; Panwar, B.; Howard, V.J.; Wadley, V.G.; Jenny, N.S.; Kissela, B.M.; Gutiérrez, O.M. Vitamin D deficiency and incident stroke risk in community-living black and white adults. Int. J. Stroke 2016, 11, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wang, M.; Huang, H.; Li, W.; Hu, Y.; Wu, T. Lower Vitamin D Status Is Associated with an Increased Risk of Ischemic Stroke: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Sheerah, H.A.; Eshak, E.S.; Cui, R.; Imano, H.; Iso, H.; Tamakoshi, A.; Group, J.C.C.S. Relationship Between Dietary Vitamin D and Deaths From Stroke and Coronary Heart Disease: The Japan Collaborative Cohort Study. Stroke 2018, 49, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Daubail, B.; Jacquin, A.; Guilland, J.C.; Hervieu, M.; Osseby, G.V.; Rouaud, O.; Giroud, M.; Béjot, Y. Serum 25-hydroxyvitamin D predicts severity and prognosis in stroke patients. Eur. J. Neurol. 2013, 20, 57–61. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, H.; Tong, Y.; Zhang, Z.B. Prognostic value of serum 25-hydroxyvitamin D in patients with stroke. Neurochem. Res. 2014, 39, 1332–1337. [Google Scholar] [CrossRef]
- Turetsky, A.; Goddeau, R.P.; Henninger, N. Low Serum Vitamin D Is Independently Associated with Larger Lesion Volumes after Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2015, 24, 1555–1563. [Google Scholar] [CrossRef]
- Qiu, H.; Wang, M.; Mi, D.; Zhao, J.; Tu, W.; Liu, Q. Vitamin D Status and the Risk of Recurrent Stroke and Mortality in Ischemic Stroke Patients: Data from a 24-Month Follow-Up Study in China. J. Nutr. Health Aging 2017, 21, 766–771. [Google Scholar] [CrossRef]
- Sun, Q.; Pan, A.; Hu, F.B.; Manson, J.E.; Rexrode, K.M. 25-Hydroxyvitamin D levels and the risk of stroke: A prospective study and meta-analysis. Stroke 2012, 43, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Berghout, B.P.; Fani, L.; Heshmatollah, A.; Koudstaal, P.J.; Ikram, M.A.; Zillikens, M.C.; Ikram, M.K. Vitamin D Status and Risk of Stroke: The Rotterdam Study. Stroke 2019, 50, 2293–2298. [Google Scholar] [CrossRef] [PubMed]
- Garcion, E.; Nataf, S.; Berod, A.; Darcy, F.; Brachet, P. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res. Mol. Brain Res. 1997, 45, 255–267. [Google Scholar] [CrossRef]
- Nissou, M.F.; Guttin, A.; Zenga, C.; Berger, F.; Issartel, J.P.; Wion, D. Additional clues for a protective role of vitamin D in neurodegenerative diseases: 1,25-dihydroxyvitamin D3 triggers an anti-inflammatory response in brain pericytes. J. Alzheimers Dis. 2014, 42, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Balasubramanian, P. Role of Vitamin D in the Outcome of Ischemic Stroke- A Randomized Controlled Trial. J. Clin. Diagn. Res. 2017, 11, CC06–CC10. [Google Scholar] [CrossRef]
- Barbarawi, M.; Kheiri, B.; Zayed, Y.; Barbarawi, O.; Dhillon, H.; Swaid, B.; Yelangi, A.; Sundus, S.; Bachuwa, G.; Alkotob, M.L.; et al. Vitamin D Supplementation and Cardiovascular Disease Risks in More Than 83 000 Individuals in 21 Randomized Clinical Trials: A Meta-analysis. JAMA Cardiol. 2019, 4, 765–775. [Google Scholar] [CrossRef]
- Batra, S.; Lin, D.; Recinos, P.F.; Zhang, J.; Rigamonti, D. Cavernous malformations: Natural history, diagnosis and treatment. Nat. Rev. Neurol. 2009, 5, 659–670. [Google Scholar] [CrossRef]
- Goitre, L.; Balzac, F.; Degani, S.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Delle Monache, S.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef]
- Marchi, S.; Retta, S.F.; Pinton, P. Cellular processes underlying cerebral cavernous malformations: Autophagy as another point of view. Autophagy 2016, 12, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Trapani, E.; Corricelli, M.; Goitre, L.; Pinton, P.; Retta, S.F. Beyond multiple mechanisms and a unique drug: Defective autophagy as pivotal player in cerebral cavernous malformation pathogenesis and implications for targeted therapies. Rare Dis. 2016, 4, e1142640. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief. 2018, 16, 929–938. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, N.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef]
- Antognelli, C.; Perrelli, A.; Armeni, T.; Nicola Talesa, V.; Retta, S.F. Dicarbonyl Stress and S-Glutathionylation in Cerebrovascular Diseases: A Focus on Cerebral Cavernous Malformations. Antioxidants 2020, 9, 124. [Google Scholar] [CrossRef]
- Trapani, E.; Retta, S.F. Cerebral cavernous malformation (CCM) disease: From monogenic forms to genetic susceptibility factors. J. Neurosurg. Sci. 2015, 59, 201–209. [Google Scholar]
- Michos, E.D.; Melamed, M.L. Vitamin D and cardiovascular disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 7–12. [Google Scholar] [CrossRef]
- Anagnostis, P.; Athyros, V.G.; Adamidou, F.; Florentin, M.; Karagiannis, A. Vitamin D and cardiovascular disease: A novel agent for reducing cardiovascular risk? Curr. Vasc. Pharmacol. 2010, 8, 720–730. [Google Scholar] [CrossRef]
- Wang, T.J. Vitamin D and Cardiovascular Disease. Annu. Rev. Med. 2016, 67, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Skaaby, T.; Thuesen, B.H.; Linneberg, A. Vitamin D, Cardiovascular Disease and Risk Factors. Adv. Exp. Med. Biol. 2017, 996, 221–230. [Google Scholar] [PubMed]
- Grübler, M.R.; März, W.; Pilz, S.; Grammer, T.B.; Trummer, C.; Müllner, C.; Schwetz, V.; Pandis, M.; Verheyen, N.; Tomaschitz, A.; et al. Vitamin-D concentrations, cardiovascular risk and events—A review of epidemiological evidence. Rev. Endocr. Metab. Disord. 2017, 18, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, T.; Kagawa, N.; Yamamoto, K.; Inouye, K. Metabolism of vitamin D3 by cytochromes P450. Front. Biosci. 2005, 10, 119–134. [Google Scholar] [PubMed]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos. Trans. R Soc. Lond B Biol. Sci. 2016, 371, 20150434. [Google Scholar] [CrossRef] [PubMed]
- Schröder-Heurich, B.; Von Hardenberg, S.; Brodowski, L.; Kipke, B.; Meyer, N.; Borns, K.; Von Kaisenberg, C.S.; Brinkmann, H.; Claus, P.; Von Versen-Höynck, F. Vitamin D improves endothelial barrier integrity and counteracts inflammatory effects on endothelial progenitor cells. FASEB J. 2019, 33, 9142–9153. [Google Scholar] [CrossRef]
- De Luca, E.; Pedone, D.; Moglianetti, M.; Pulcini, D.; Perrelli, A.; Retta, S.F.; Pompa, P.P. Multifunctional Platinum@BSA-Rapamycin Nanocarriers for the Combinatorial Therapy of Cerebral Cavernous Malformation. ACS Omega 2018, 3, 15389–15398. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. [Google Scholar] [CrossRef]
- Abu-el-Maaty, M.A.; Hassanein, S.I.; Gad, M.Z. Polymorphisms in the Vitamin D Pathway in Relation to 25-Hydroxyvitamin D Status and Cardiovascular Disease Incidence: Application to Biomarkers. In Biomarkers in Cardiovascular Disease. Biomarkers in Disease: Methods, Discoveries and Applications; Patel, V., Preedy, V., Eds.; Springer: Dordrecht, The Netherlands, 2016. [Google Scholar]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; Van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Scragg, R.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Sluyter, J.; Murphy, J.; Khaw, K.T.; Camargo, C.A. Effect of Monthly High-Dose Vitamin D Supplementation on Cardiovascular Disease in the Vitamin D Assessment Study: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 608–616. [Google Scholar] [CrossRef]
- Pilz, S.; Tomaschitz, A.; Drechsler, C.; Zittermann, A.; Dekker, J.M.; März, W. Vitamin D supplementation: A promising approach for the prevention and treatment of strokes. Curr. Drug Targets 2011, 12, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Prabhakar, S.; Modi, M.; Bhadada, S.K.; Kalaivani, M.; Lal, V.; Khurana, D. Effect of Vitamin D and calcium supplementation on ischaemic stroke outcome: A randomised controlled open-label trial. Int. J. Clin. Pract. 2016, 70, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Seibert, E.; Lehmann, U.; Riedel, A.; Ulrich, C.; Hirche, F.; Brandsch, C.; Dierkes, J.; Girndt, M.; Stangl, G.I. Vitamin D. Eur. J. Nutr. 2017, 56, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Pilz, S. Vitamin D and Cardiovascular Disease: An Update. Anticancer Res. 2019, 39, 4627–4635. [Google Scholar] [CrossRef]
- Bast, A.; Haenen, G.R. Ten misconceptions about antioxidants. Trends Pharmacol. Sci. 2013, 34, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.; Lim, K.; Thadhani, R.; Manson, J.E. Vitamin D and Atherosclerotic Cardiovascular Disease. J. Clin. Endocrinol. Metab. 2019, 104, 4033–4050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Sutton, A.J.; Riley, R.D. Assessment of publication bias, selection bias, and unavailable data in meta-analyses using individual participant data: A database survey. BMJ 2012, 344, d7762. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Kirkham, J.; Dwan, K.; Kramer, S.; Green, S.; Forbes, A. Bias due to selective inclusion and reporting of outcomes and analyses in systematic reviews of randomised trials of healthcare interventions. Cochrane Database Syst. Rev. 2014, 10, MR000035. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.A.; Perrelli, A.; Ragni, A.; Retta, F.; De Silva, T.M.; Sobey, C.G.; Retta, S.F. Vitamin D Deficiency and the Risk of Cerebrovascular Disease. Antioxidants 2020, 9, 327. https://doi.org/10.3390/antiox9040327
Kim HA, Perrelli A, Ragni A, Retta F, De Silva TM, Sobey CG, Retta SF. Vitamin D Deficiency and the Risk of Cerebrovascular Disease. Antioxidants. 2020; 9(4):327. https://doi.org/10.3390/antiox9040327
Chicago/Turabian StyleKim, Hyun Ah, Andrea Perrelli, Alberto Ragni, Francesca Retta, T. Michael De Silva, Christopher G. Sobey, and Saverio Francesco Retta. 2020. "Vitamin D Deficiency and the Risk of Cerebrovascular Disease" Antioxidants 9, no. 4: 327. https://doi.org/10.3390/antiox9040327
APA StyleKim, H. A., Perrelli, A., Ragni, A., Retta, F., De Silva, T. M., Sobey, C. G., & Retta, S. F. (2020). Vitamin D Deficiency and the Risk of Cerebrovascular Disease. Antioxidants, 9(4), 327. https://doi.org/10.3390/antiox9040327