Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia

 ,
, 
 , ,
, ,  , and
, and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Models and Treatment
2.2. Cell Culture and Differentiation
2.3. Stably Silenced Murine Myoblast C2C12 Cell Line
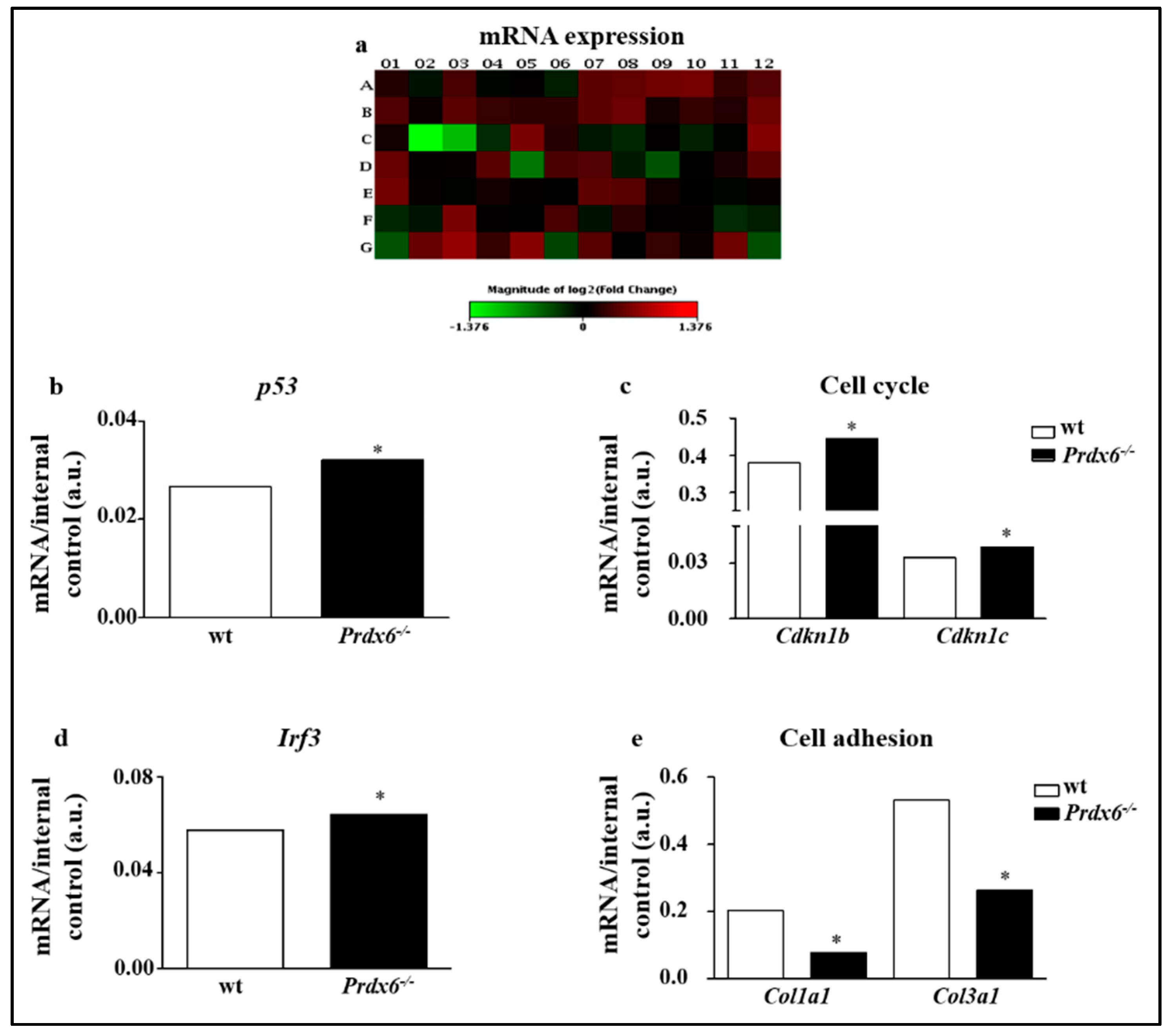
2.4. RT2 Profiler PCR Senescence Array
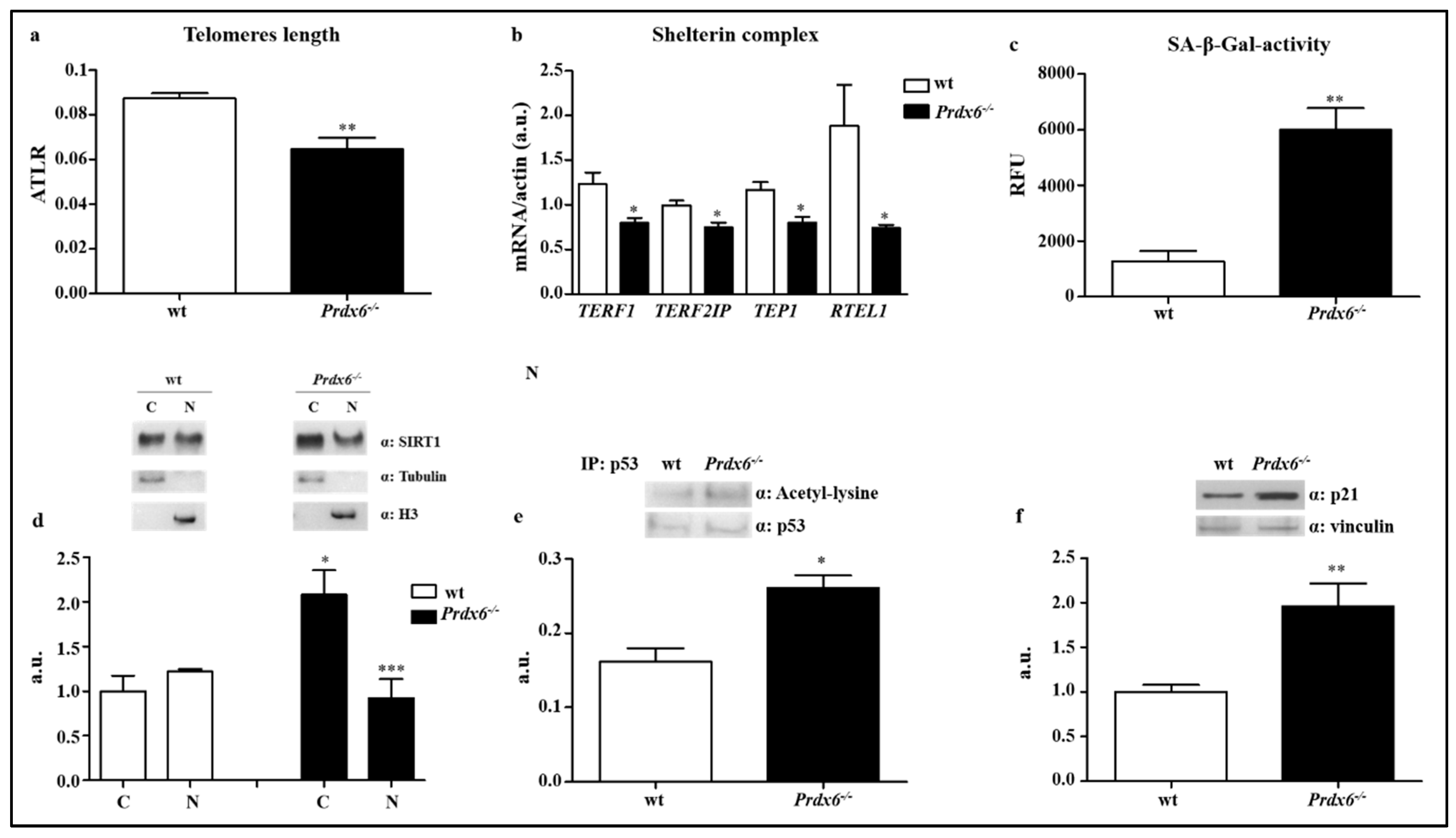
2.5. DNA Isolation and Average Telomere Length Ratio
2.6. Senescence-Associated Beta-Galactosidase Activity
2.7. Nuclear and Cytoplasmic Fractionation
2.8. Western Blot
2.9. p53 Immunoprecipitation Assay
2.10. IGF-1 Sera Levels
2.11. Malondialdehyde (MDA) Assay
2.12. Total RNA Extraction and Gene Expression
2.13. Grip Strength Test
2.14. Histology and Immunohistochemical Staining
2.15. Statistical Analysis
3. Results
3.1. Lacking of Prdx6 Induces a Premature Phenotype of Senescence
3.2. Lacking in Prdx6 Impairs SIRT1 Nuclear Translocation Resulting in p53/p21 Pro-Aging Pathway Activation
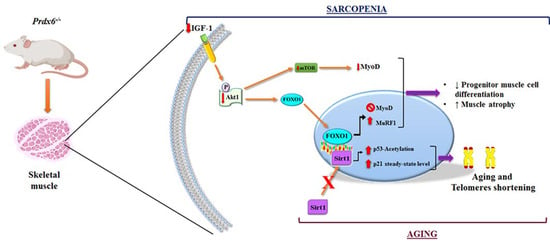
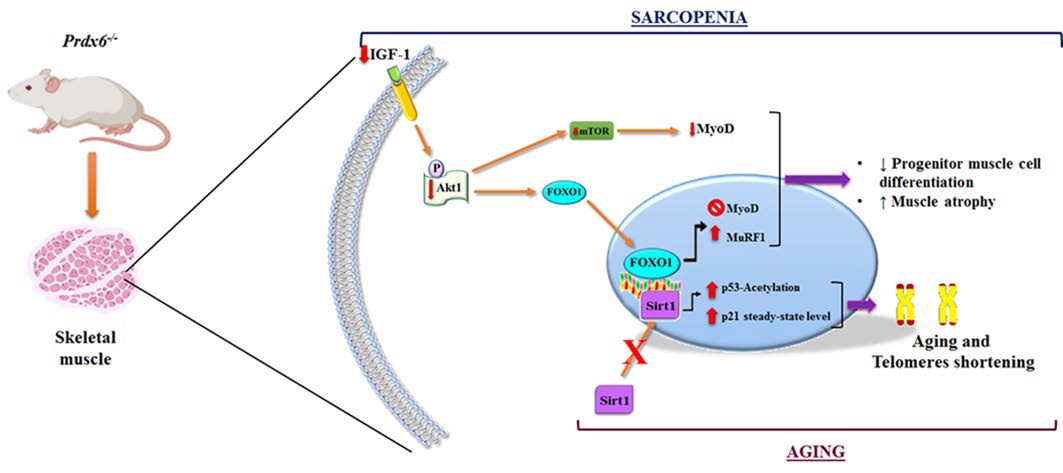
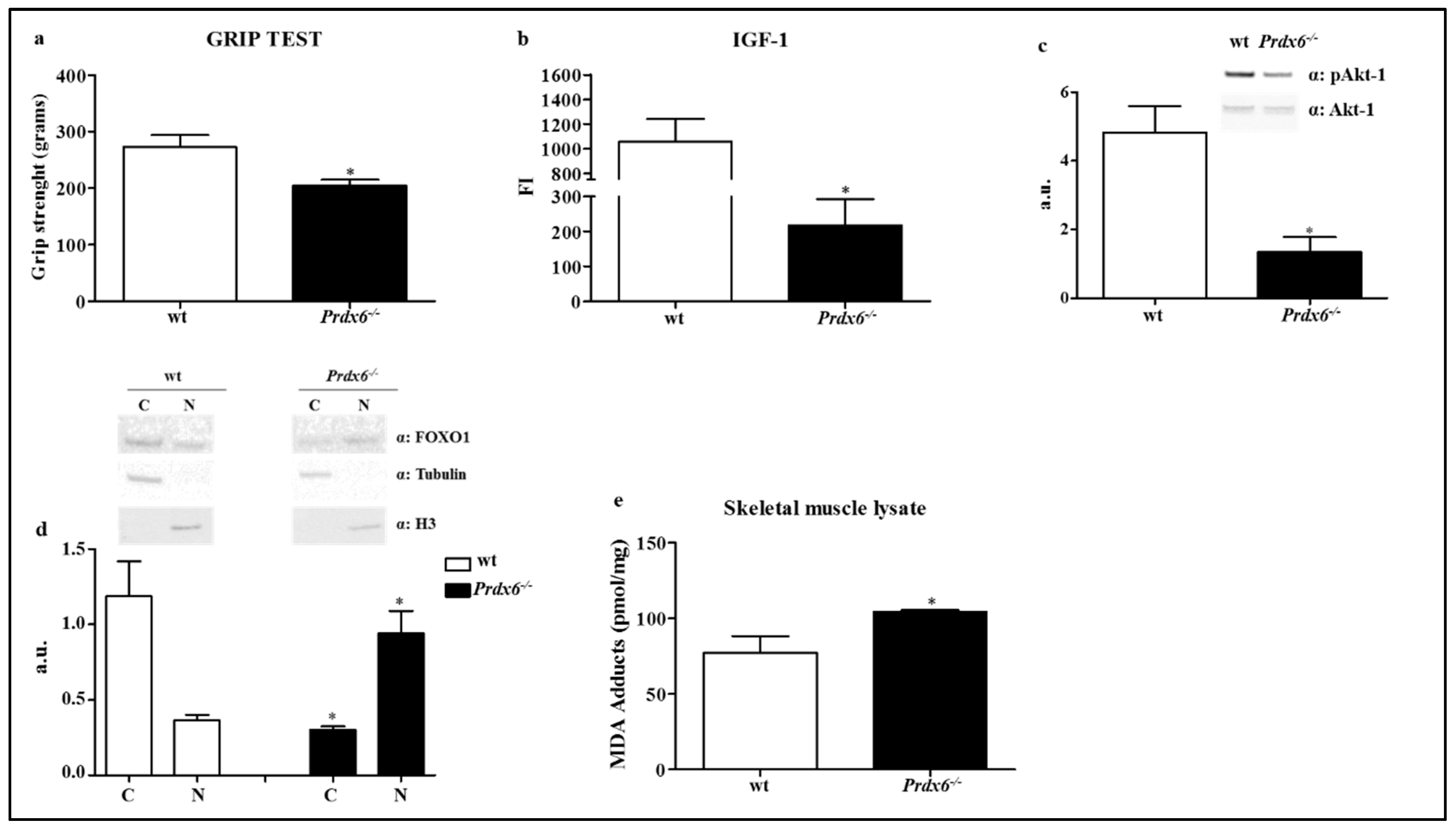
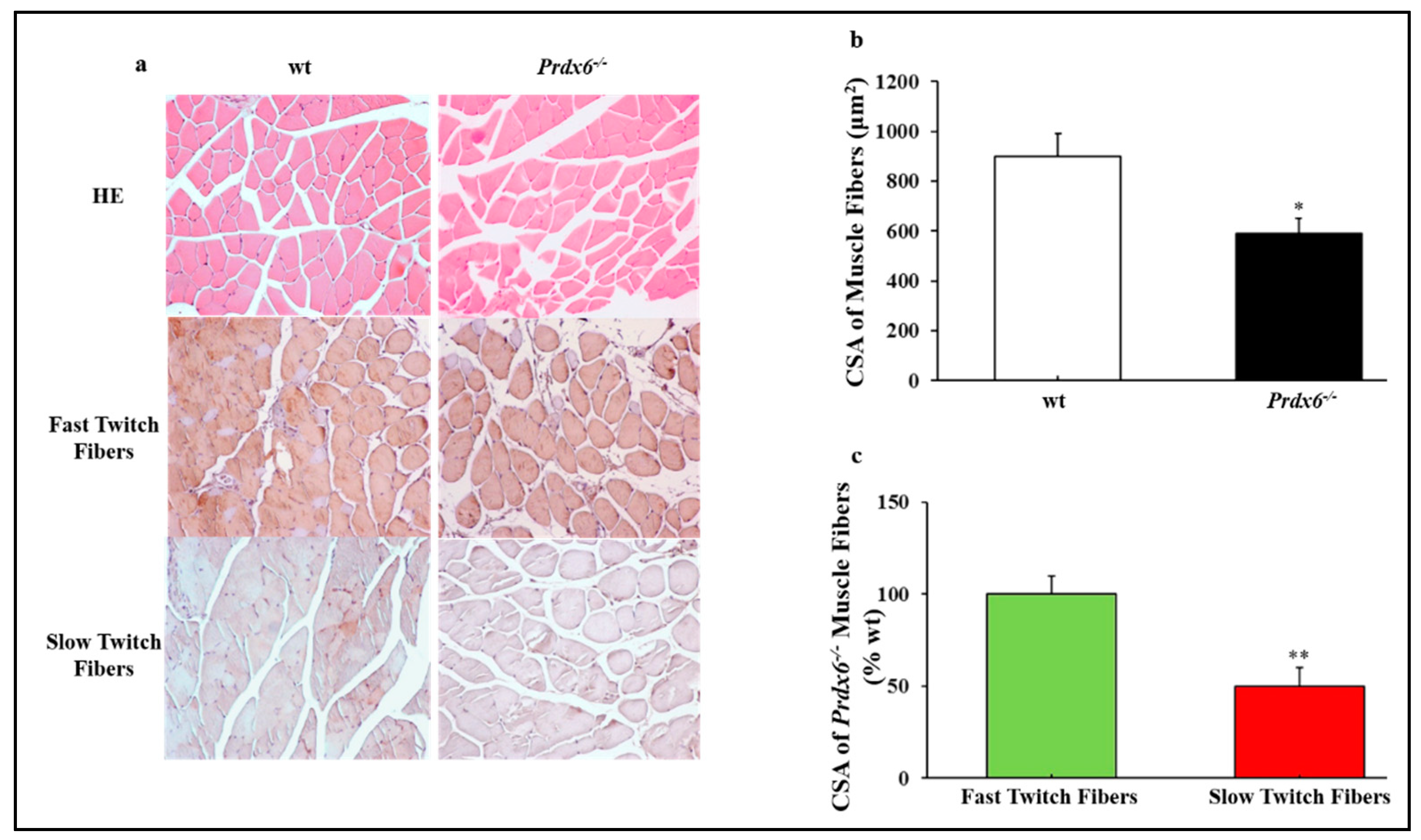
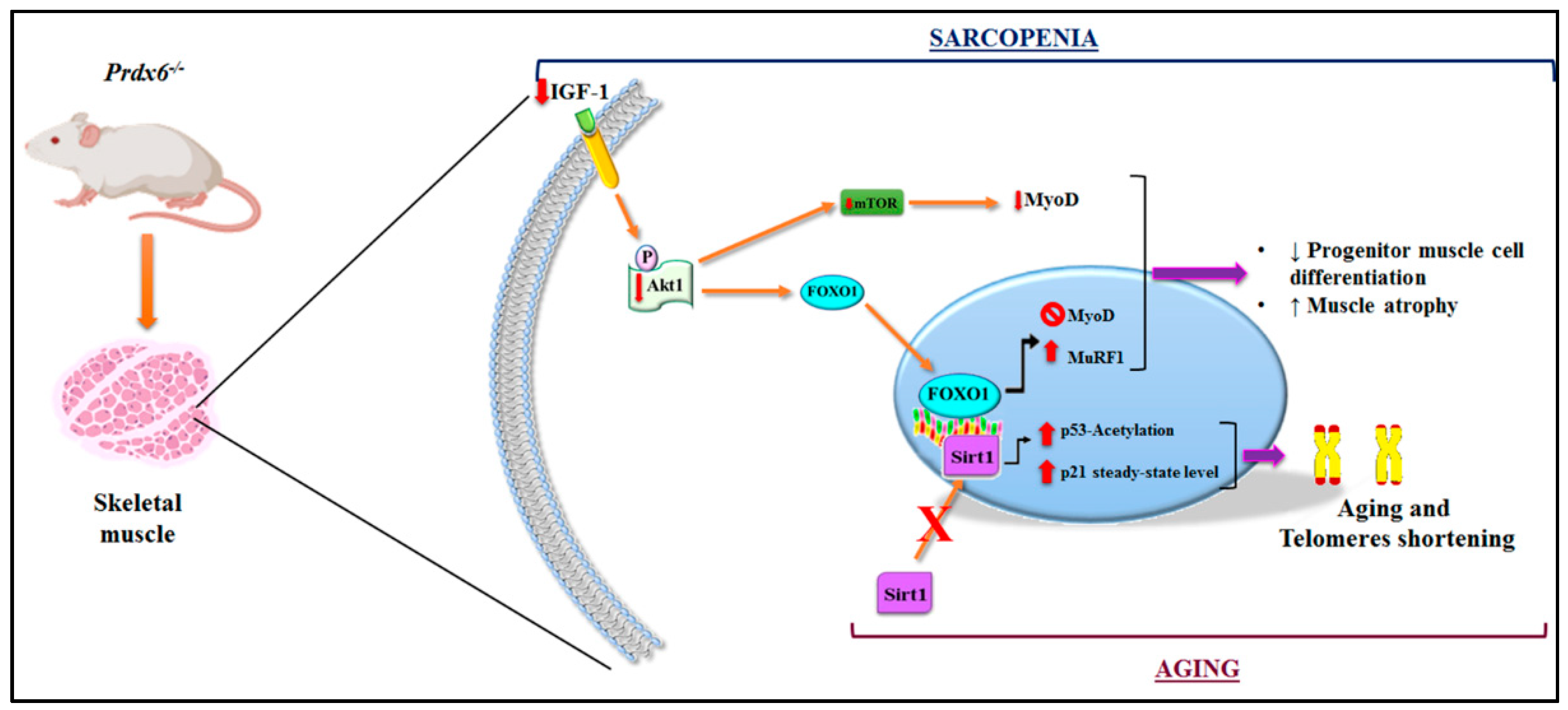
3.3. Prdx6 Deletion Induces Phenotype of Sarcopenia via IGF-1/Akt-1/FOXO1 Pathway
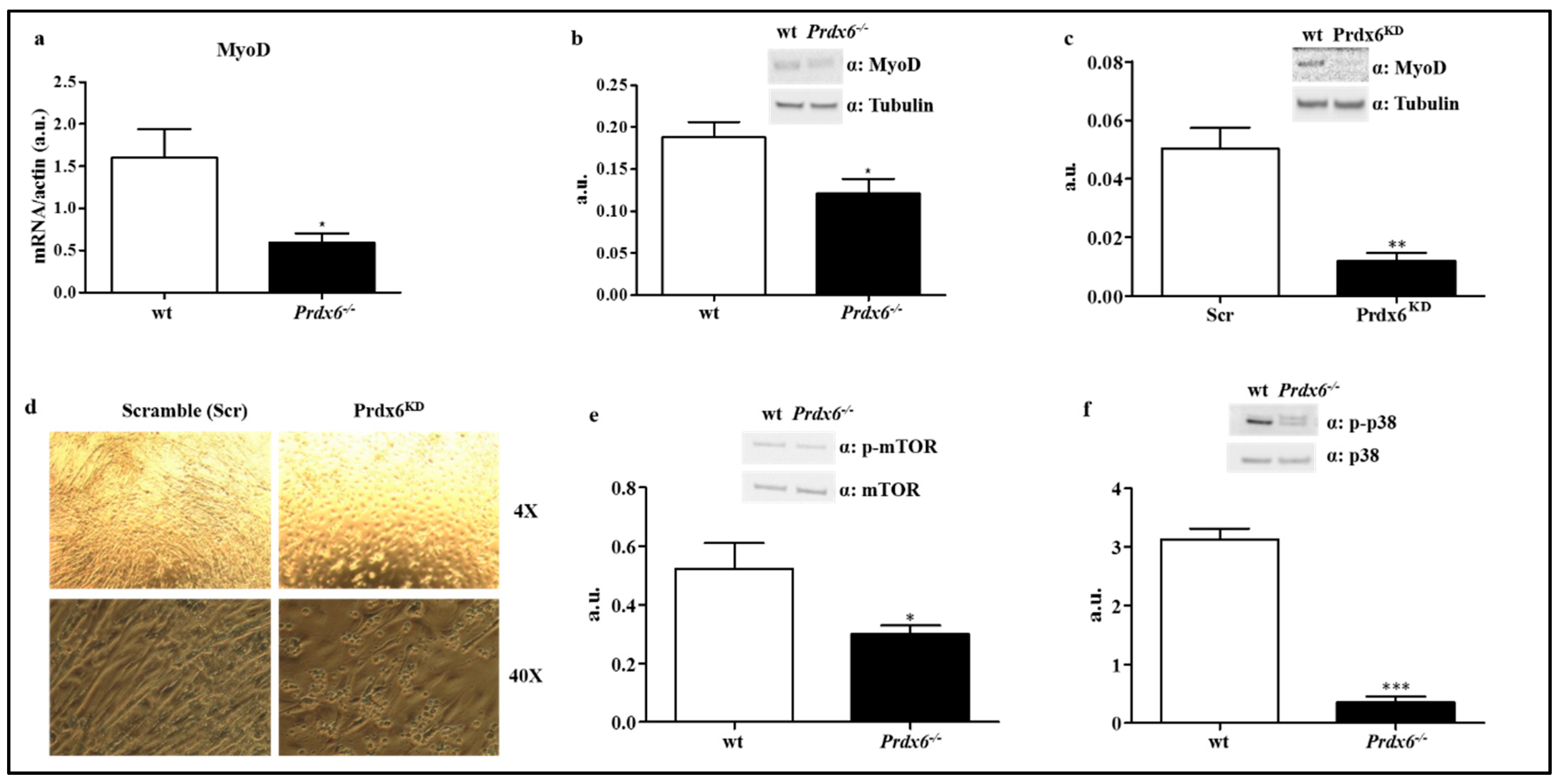
3.4. Loss of Prdx6 Blunts Muscle Differentiation and Proteins Synthesis Leading to Sarcopenia
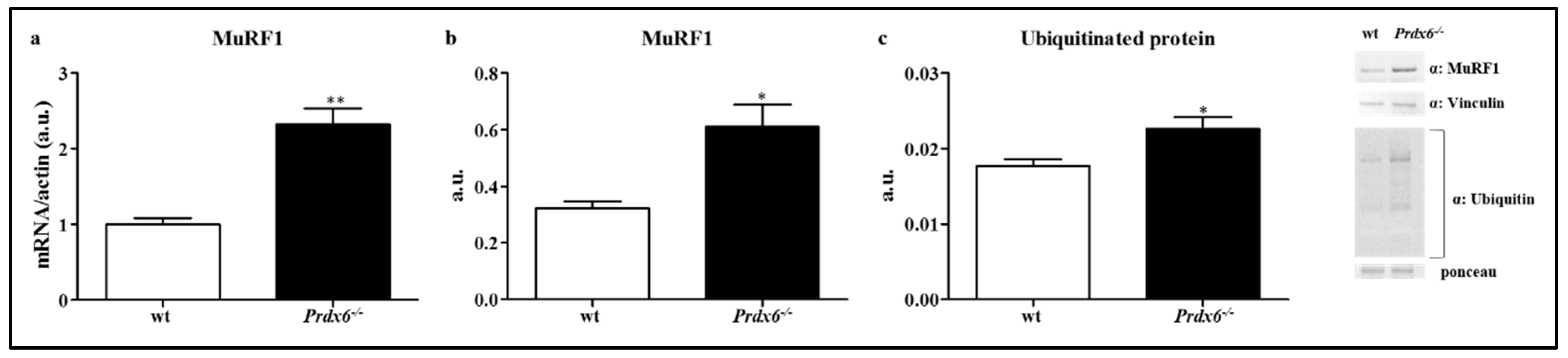
3.5. Loss of Prdx6 Promotes Muscle Atrophy via MuRF1-Ubiquitin Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.M.; Correia, M. Epidemiology of weight loss, malnutrition and sarcopenia: A transatlantic view. Nutrition 2020, 69, 110581. [Google Scholar] [CrossRef] [PubMed]
- Umegaki, H. Sarcopenia and diabetes: Hyperglycemia is a risk factor for age-associated muscle mass and functional reduction. J. Diabetes Investig. 2015, 6, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Bareja, A.; Bartlett, D.B.; White, J.P. Autophagy as a therapeutic target to enhance aged muscle regeneration. Cells 2019, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Borba, V.Z.C.; Costa, T.L.; Moreira, C.A.; Boguszewski, C.L. MECHANISMS OF ENDOCRINE DISEASE: Sarcopenia in endocrine and non-endocrine disorders. Eur. J. Endocrinol. 2019, 180, R185–R199. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. Erratum. Classification and diagnosis of diabetes. Sec. 2. In Standards of Medical Care in Diabetes-2016. Diabetes Care 2016;39(Suppl. 1):S13-S22. Diabetes Care 2016, 39, 1653. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Donadel, G.; Bellia, A.; Caratelli, S.; Coppola, A.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Pacifici, F.; Della Morte, D.; Capuani, B.; Pastore, D.; Bellia, A.; Sbraccia, P.; Di Daniele, N.; Lauro, R.; Lauro, D. Peroxiredoxin6, a Multitask Antioxidant Enzyme Involved in the Pathophysiology of Chronic Noncommunicable Diseases. Antioxid. Redox Signal. 2019, 30, 399–414. [Google Scholar] [CrossRef]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed]
- Sharapov, M.G.; Novoselov, V.I.; Gudkov, S.V. Radioprotective role of Peroxiredoxin 6. Antioxidants (Basel) 2019, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Inglott, F.S.; Alexander, M.Y.; Weston, R. Suppression of SIRT1 in diabetic conditions induces osteogenic differentiation of human vascular smooth muscle cells via RUNX2 signalling. Sci. Rep. 2019, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Curcio, F.; Ferro, G.; Basile, C.; Liguori, I.; Parrella, P.; Pirozzi, F.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Tocchetti, C.G.; et al. Biomarkers in sarcopenia: A multifactorial approach. Exp. Gerontol. 2016, 85, 1–8. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, Y.; Li, L.; Zhao, J.; Qin, W.; Jin, Z.; Liu, W. Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis. 2019, 10, 552. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019, 5, eaau8857. [Google Scholar] [CrossRef]
- QIAGEN. QIAGEN PCR ANALYSIS. Available online: https://dataanalysis.qiagen.com/pcr/arrayanalysis.php (accessed on 4 April 2020).
- Callicott, R.J.; Womack, J.E. Real-time PCR assay for measurement of mouse telomeres. Comp. Med. 2006, 56, 17–22. [Google Scholar]
- Ross, J.L.; Queme, L.F.; Lamb, J.E.; Green, K.J.; Jankowski, M.P. Sex differences in primary muscle afferent sensitization following ischemia and reperfusion injury. Biol. Sex. Differ. 2018, 9, 2. [Google Scholar] [CrossRef]
- Shang, G.K.; Han, L.; Wang, Z.H.; Liu, Y.P.; Yan, S.B.; Sai, W.W.; Wang, D.; Li, Y.H.; Zhang, W.; Zhong, M. Sarcopenia is attenuated by TRB3 knockout in aging mice via the alleviation of atrophy and fibrosis of skeletal muscles. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, X.; Chen, H.Z. Sirtuins and insulin resistance. Front. Endocrinol. (Lausanne) 2018, 9, 748. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.L.C.; Ramasamy, T.S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 2018, 43, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Candau, R.B.; Bernardi, H. FoxO transcription factors: Their roles in the maintenance of skeletal muscle homeostasis. Cell Mol. Life Sci. 2014, 71, 1657–1671. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Chao, L.; Chao, J. Pivotal role of JNK-dependent FOXO1 activation in downregulation of kallistatin expression by oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1048–H1054. [Google Scholar] [CrossRef]
- Dey, D.; Goldhamer, D.J.; Yu, P.B. Contributions of Muscle-Resident Progenitor Cells to Homeostasis and Disease. Curr. Mol. Biol. Rep. 2015, 1, 175–188. [Google Scholar] [CrossRef][Green Version]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef]
- Wu, Z.; Woodring, P.J.; Bhakta, K.S.; Tamura, K.; Wen, F.; Feramisco, J.R.; Karin, M.; Wang, J.Y.; Puri, P.L. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell Biol. 2000, 20, 3951–3964. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef]
- Morley, J.E. Diabetes and aging: Epidemiologic overview. Clin. Geriatr. Med. 2008, 24, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Miyazawa, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Development- and age-associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech. Ageing Dev. 2006, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Singh, P.; Chhunchha, B.; Kubo, E.; Shinohara, T.; Bhargavan, B.; Singh, D.P. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C954–C967. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Fatma, N.; Singh, D.P. Sumoylation-deficient Prdx6 gains protective function by amplifying enzymatic activity and stability and escapes oxidative stress-induced aberrant Sumoylation. Cell Death Dis. 2017, 8, e2525. [Google Scholar] [CrossRef]
- Ozkosem, B.; Feinstein, S.I.; Fisher, A.B.; O’Flaherty, C. Advancing age increases sperm chromatin damage and impairs fertility in peroxiredoxin 6 null mice. Redox Biol. 2015, 5, 15–23. [Google Scholar] [CrossRef]
- Rizvi, S.; Raza, S.T.; Mahdi, F. Telomere length variations in aging and age-related diseases. Curr. Aging Sci. 2014, 7, 161–167. [Google Scholar] [CrossRef]
- Lu, J.; Vallabhaneni, H.; Yin, J.; Liu, Y. Deletion of the major peroxiredoxin Tsa1 alters telomere length homeostasis. Aging Cell 2013, 12, 635–644. [Google Scholar] [CrossRef]
- Paules, C.; Dantas, A.P.; Miranda, J.; Crovetto, F.; Eixarch, E.; Rodriguez-Sureda, V.; Dominguez, C.; Casu, G.; Rovira, C.; Nadal, A.; et al. Premature placental aging in term small-for-gestational-age and fetal-growth-restricted fetuses. Ultrasound Obstet. Gynecol. 2018. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; McDonagh, B.; Porter, H.; Giakoumaki, I.I.; Earl, K.E.; Nye, G.A.; Vasilaki, A.; Brooks, S.V.; Richardson, A.; Van Remmen, H.; et al. Comparison of whole body SOD1 knockout with muscle-specific SOD1 knockout mice reveals a role for nerve redox signaling in regulation of degenerative pathways in skeletal muscle. Antioxid. Redox Signal. 2018, 28, 275–295. [Google Scholar] [CrossRef]
- Cooper, C.; Fielding, R.; Visser, M.; van Loon, L.J.; Rolland, Y.; Orwoll, E.; Reid, K.; Boonen, S.; Dere, W.; Epstein, S.; et al. Tools in the assessment of sarcopenia. Calcif. Tissue Int. 2013, 93, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, A.; Radu, B.M.; Radu, M.; Cretoiu, S.M. Muscle changes during atrophy. Adv. Exp. Med. Biol. 2018, 1088, 73–92. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic, N.; Lim, J.Y.; Miljkovic, I.; Frontera, W.R. Aging of skeletal muscle fibers. Ann. Rehabil. Med. 2015, 39, 155–162. [Google Scholar] [CrossRef]
- Giovannini, S.; Marzetti, E.; Borst, S.E.; Leeuwenburgh, C. Modulation of GH/IGF-1 axis: Potential strategies to counteract sarcopenia in older adults. Mech. Ageing Dev. 2008, 129, 593–601. [Google Scholar] [CrossRef]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Fulco, M.; Ladurner, A.; Sartorelli, V.; Rosenthal, N. SirT1 in muscle physiology and disease: Lessons from mouse models. Dis. Model. Mech. 2010, 3, 298–303. [Google Scholar] [CrossRef]
- Wu, X.; Ji, P.; Zhang, L.; Bu, G.; Gu, H.; Wang, X.; Xiong, Y.; Zuo, B. The Expression of Porcine Prdx6 Gene Is Up-Regulated by C/EBPbeta and CREB. PLoS ONE 2015, 10, e0144851. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacifici, F.; Della-Morte, D.; Piermarini, F.; Arriga, R.; Scioli, M.G.; Capuani, B.; Pastore, D.; Coppola, A.; Rea, S.; Donadel, G.; et al. Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia. Antioxidants 2020, 9, 329. https://doi.org/10.3390/antiox9040329
Pacifici F, Della-Morte D, Piermarini F, Arriga R, Scioli MG, Capuani B, Pastore D, Coppola A, Rea S, Donadel G, et al. Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia. Antioxidants. 2020; 9(4):329. https://doi.org/10.3390/antiox9040329
Chicago/Turabian StylePacifici, Francesca, David Della-Morte, Francesca Piermarini, Roberto Arriga, Maria Giovanna Scioli, Barbara Capuani, Donatella Pastore, Andrea Coppola, Silvia Rea, Giulia Donadel, and et al. 2020. "Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia" Antioxidants 9, no. 4: 329. https://doi.org/10.3390/antiox9040329
APA StylePacifici, F., Della-Morte, D., Piermarini, F., Arriga, R., Scioli, M. G., Capuani, B., Pastore, D., Coppola, A., Rea, S., Donadel, G., Andreadi, A., Abete, P., Sconocchia, G., Bellia, A., Orlandi, A., & Lauro, D. (2020). Prdx6 Plays a Main Role in the Crosstalk between Aging and Metabolic Sarcopenia. Antioxidants, 9(4), 329. https://doi.org/10.3390/antiox9040329