Metabolic Flux Analysis of Catechin Biosynthesis Pathways Using Nanosensor


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Development of Nanosensor
2.1.1. Designing and Construction
2.1.2. Expression and Purification of Sensor Protein
2.1.3. Characterization of Sensor Protein
2.2. Construction and Expression of (+)-Catechin Biosynthetic Pathway
2.2.1. Construction
2.2.2. Expression
2.2.3. Validation
2.3. Flux Analysis of Metabolites of Catechin Biosynthesis Pathway
2.3.1. Microplate Analysis
2.3.2. Confocal Analysis
3. Results
3.1. Design and Construction of Sensor
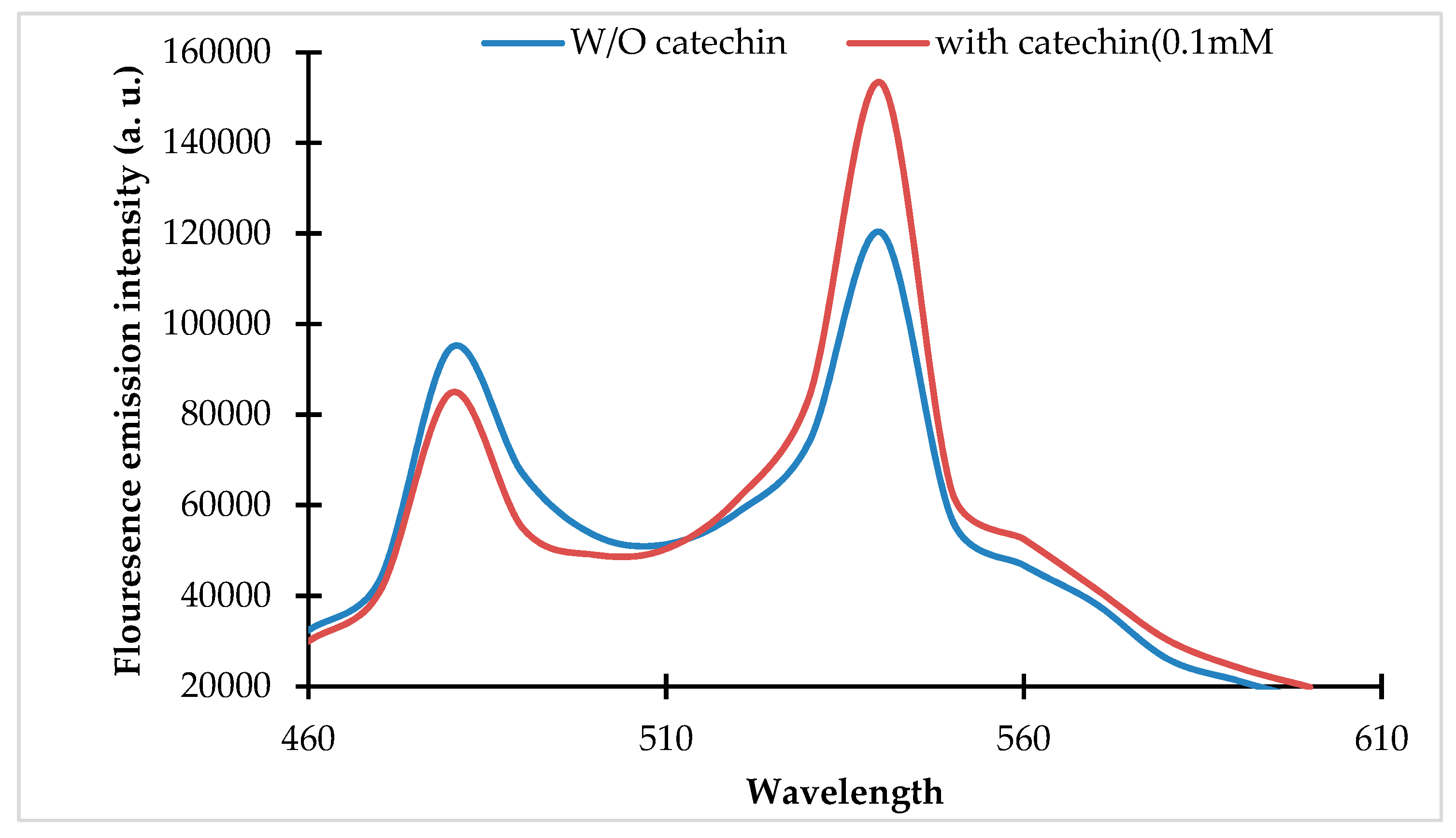
3.2. Characterization of the Sensor
3.3. Validation of the (+)-Catechin Biosynthesis Pathway
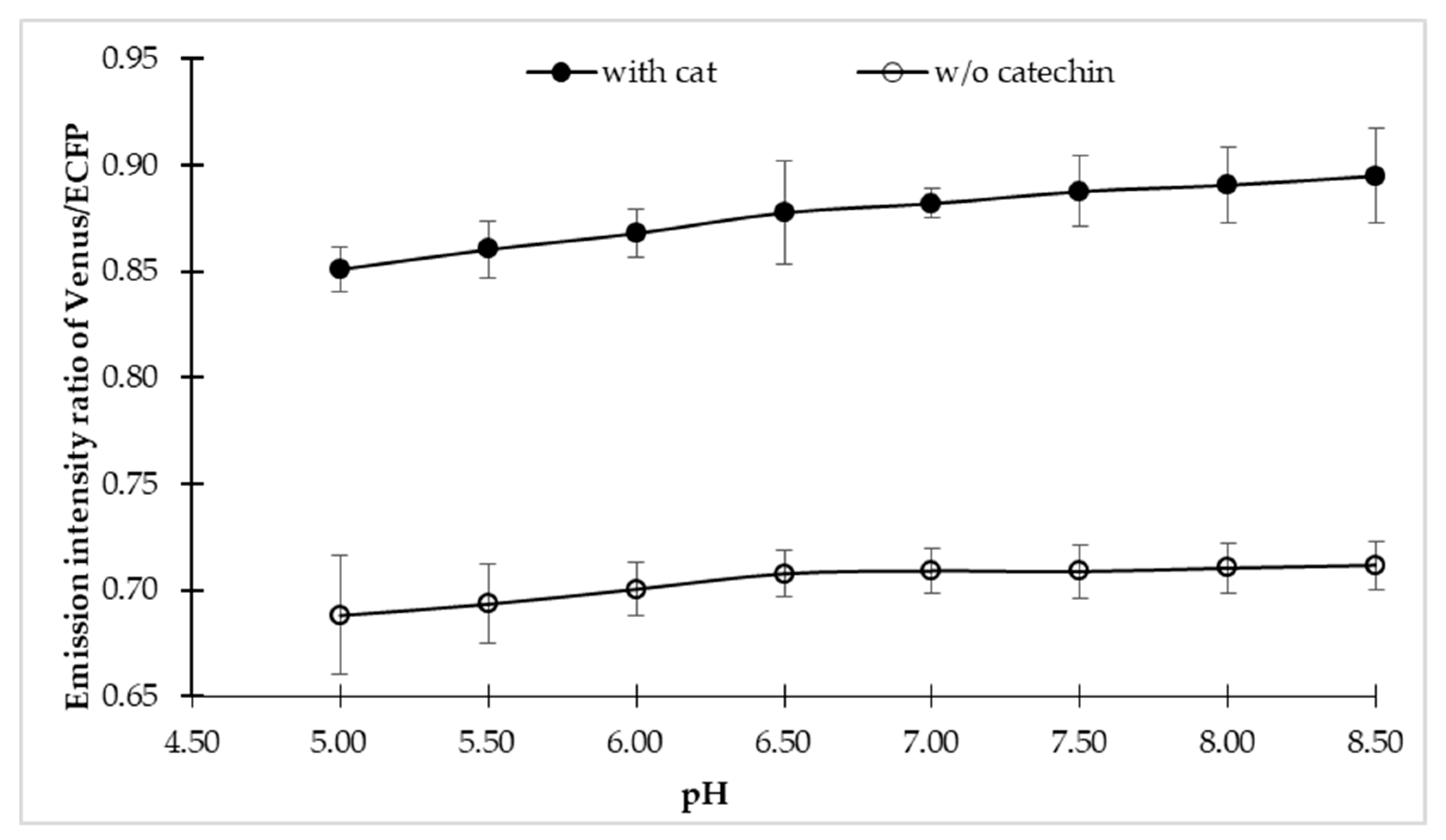
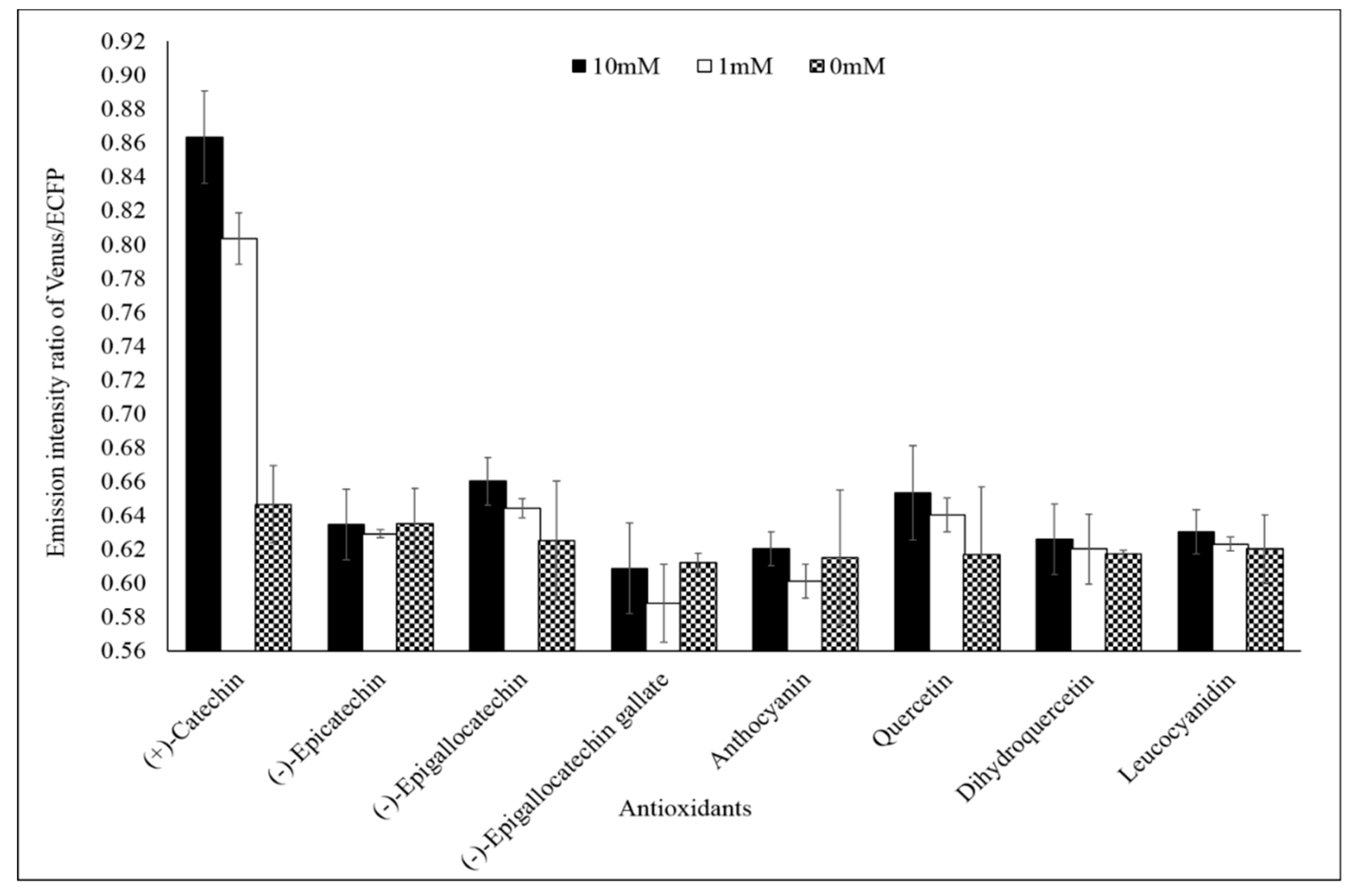
3.4. Fluxomic Analysis of Catechin Biosynthesis Pathway
3.4.1. Microplate Analysis
3.4.2. Confocal Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, G.; Zhang, C.; Wu, M.; Wu, S.; Liu, Q. Research progress of natural antioxidants in foods for the treatment of diseases. Food Sci. Human Wellness. 2014, 3, 110–116. [Google Scholar] [CrossRef]
- Mandal, S.; Yadav, S.; Yadav, S.; Nema, R.K. Antioxidants: A review. J. Chem. Pharm. 2009, 1, 102–104. [Google Scholar]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutri. J. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mukhtar, H. Tea polyphenols for health promotion. Life Sci. 2007, 81, 519–533. [Google Scholar] [CrossRef]
- Dufresne, C.J.; Farnworth, E.R. A review of latest research findings on the health promotion properties of tea. J. Nutr. Biochem. 2001, 12, 404–421. [Google Scholar] [CrossRef]
- Higdon, J.V.; Frei, B. Tea catechins and polyphenols: Health effects, metabolism, and antioxidant functions. Crit. Rev. Food Sci. Nutr. 2003, 43, 89–143. [Google Scholar] [CrossRef]
- McKay, D.L.; Blumberg, J.B. The role of tea in human health: An update. J. Am. Coll. Nut. 2002, 21, 1–13. [Google Scholar] [CrossRef]
- Demeule, M.; Michaud-Levesque, J.; Annabi, B.; Gingras, D.; Boivin, D.; Lamy, S.; Bertrand, Y.; Beliveau, R. Green tea catechins as novel antitumor and antiangiogenic compounds. Curr. Med. Chem. 2002, 2, 441–463. [Google Scholar] [CrossRef]
- Chemler, J.A.; Lock, L.T.; Koffas, M.A.; Tzanakakis, E.S. Standardized biosynthesis of flavan-3-ols with effects on pancreatic beta-cell insulin secretion. Appl. Microbiol. Biotechnol. 2007, 77, 797–807. [Google Scholar] [CrossRef]
- Manikandan, R.; Beulaja, M.; Arulvasu, C.; Sellamuthu, S.; Dinesh, D.; Prabhu, D.; Babu, G.; Vaseeharan, B.; Prabhu, N.M. Synergistic anticancer activity of curcumin and catechin: An in vitro study using human cancer cell lines. Microsc. Res. Tech. 2012, 75, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakatsuka, S.; Otani, H.; Kohmoto, K.; Nishimura, S. (+)-Catechin acts as an infection-inhibiting factor in strawberry leaf. Phytopathology 2000, 90, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Kottawa-Arachchi, J.D.; Gunasekare, M.K.; Ranatunga, M.A. Biochemical diversity of global tea [Camellia sinensis (L.) O. Kuntze] germplasm and its exploitation: A review. Genet Resour Crop Ev. 2019, 66, 259–273. [Google Scholar] [CrossRef]
- Lambert, J.D.; Lee, M.J.; Lu, H.; Meng, X.; Hong, J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. Nutr. J. 2003, 133, 4172–4177. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, J.B.; Hahn, B.S.; Kim, K.H.; Ha, S.H.; Kim, J.B.; Kim, Y.H. EST analysis of genes involved in secondary metabolism in Camellia sinensis (tea), using suppression subtractive hybridization. Plant. Sci. 2004, 166, 953–961. [Google Scholar] [CrossRef]
- Hwang, E.I.; Kaneko, M.; Ohnishi, Y.; Horinouchi, S. Production of plant-specific flavanones by Escherichia coli containing an artificial gene cluster. Appl. Environ. Microbiol. 2003, 69, 2699–2706. [Google Scholar] [CrossRef]
- Farr’e, G.; Blancquaert, D.; Capell, T.; Van Der Straeten, D.; Christou, P.; Zhu, C. Engineering complex metabolic pathways in plants. Annu. Rev. Plant Biol. 2014, 65, 187–223. [Google Scholar] [CrossRef]
- Stephanopoulos, G. Metabolic fluxes and metabolic engineering. Metab. Eng. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- Lalonde, S.; Ehrhardt, D.W.; Frommer, W.B. Shining light on signaling and metabolic networks by genetically encoded biosensors. Curr. Opin. Plant. Biol. 2005, 8, 574–581. [Google Scholar] [CrossRef][Green Version]
- Ameen, S.; Ahmad, M.; Mohsin, M.; Qureshi, M.I.; Ibrahim, M.M.; Abdin, M.Z.; Ahmad, A. Designing, construction and characterization of genetically encoded FRET-based nanosensor for real time monitoring of lysine flux in living cells. J. Nanobiotechnol. 2016, 14, 49. [Google Scholar] [CrossRef]
- Besnard, J.; Okumoto, S. Glutamine flux imaging using genetically encoded sensors. J. Vis. Exp. 2014, 89, e51657. [Google Scholar] [CrossRef]
- Fehr, M.; Frommer, W.B.; Lalonde, S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. USA 2002, 99, 9846–9851. [Google Scholar] [CrossRef]
- Hu, H.; Gu, Y.; Xu, L.; Zou, Y.; Wang, A.; Tao, R.; Chen, X.; Zhao, Y.; Yang, Y. A genetically encoded toolkit for tracking live-cell histidine dynamics in space and time. Sci. Rep. 2017, 7, 43479. [Google Scholar] [CrossRef]
- Whitfield, J.H.; Zhang, W.H.; Herde, M.K.; Clifton, B.E.; Radziejewski, J.; Janovjak, H.; Hanneberger, C.; Jackson, C.J. Construction of a robust and sensitive arginine biosensor through ancestral protein reconstruction. Protein Sci. 2015, 24, 1412–1422. [Google Scholar] [CrossRef]
- Ahmad, M.; Mohsin, M.; Iqrar, S.; Manzoor, O.; Siddiqi, T.O.; Ahmad, A. Live cell imaging of vitamin B12 dynamics by genetically encoded fluorescent nanosensor. Sens. Actuator B-Chem. 2018, 257, 866–874. [Google Scholar] [CrossRef]
- Sandmann, G. Combinatorial biosynthesis of carotenoids in a heterologous host: A powerful approach for the biosynthesis of novel structures. Chem. Bio. Chem. 2002, 3, 629–635. [Google Scholar] [CrossRef]
- Casanal, A.; Zander, U.; Munoz, C.; Dupeux, F.; Luque, I.; Botella, M.A.; Schwab, W.; Valpuesta, V.; Marquez, J.A. The strawberry pathogenesis-related 10 (PR-10) Fra a proteins control flavonoid biosynthesis by binding to metabolic intermediates. J. Biol. Chem. 2013, 288, 35322–35332. [Google Scholar] [CrossRef]
- Umar, K.M.; Abdulkarim, S.M.; Radu, S.; Abdul Hamid, A.; Saari, N. Engineering the production of major catechins by Escherichia coli carrying metabolite genes of Camellia sinensis. Sci. World J. 2012, 2012, 529031. [Google Scholar] [CrossRef]
- Kaneko, M.; Ohnishi, Y.; Horinouchi, S. Cinnamate: Coenzyme A ligase from the filamentous bacterium Streptomyces coelicolor A3 (2). J. Bacteriol. 2003, 185, 20–27. [Google Scholar] [CrossRef]
- Chemler, J.A.; Fowler, Z.L.; McHugh, K.P.; Koffas, M.A. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab. Eng. 2010, 12, 96–104. [Google Scholar] [CrossRef]
- Hotze, M.; Schroder, G.; Schroder, J. Cinnamate 4-hydroxylase from Catharanthus roseus and a strategy for the functional expression of plant cytochrome P450 proteins as translational fusions with P450 reductase in Escherichia coli. FEBS Lett. 1995, 374, 345–350. [Google Scholar]
- Pompon, D.; Louerat, B.; Bronine, A.; Urban, P. Yeast expression of animal and plant P450s in optimized redox environments. Methods Enzymol. 1996, 272, 51–64. [Google Scholar]
- Fischer, D.; Stich, K.; Britsch, L.; Grisebach, H. Purification and characterization of (+) dihydroflavonol (3-hydroxyflavanone) 4-reductase from flowers of Dahlia variabilis. Arch. Biochem. Biophys. 1988, 264, 40–47. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kausar, H.; Ambrin, G.; Okla, M.K.; Soufan, W.; Al-Ghamdi, A.A.; Ahmad, A. Metabolic Flux Analysis of Catechin Biosynthesis Pathways Using Nanosensor. Antioxidants 2020, 9, 288. https://doi.org/10.3390/antiox9040288
Kausar H, Ambrin G, Okla MK, Soufan W, Al-Ghamdi AA, Ahmad A. Metabolic Flux Analysis of Catechin Biosynthesis Pathways Using Nanosensor. Antioxidants. 2020; 9(4):288. https://doi.org/10.3390/antiox9040288
Chicago/Turabian StyleKausar, Habiba, Ghazala Ambrin, Mohammad K. Okla, Walid Soufan, Abdullah A. Al-Ghamdi, and Altaf Ahmad. 2020. "Metabolic Flux Analysis of Catechin Biosynthesis Pathways Using Nanosensor" Antioxidants 9, no. 4: 288. https://doi.org/10.3390/antiox9040288
APA StyleKausar, H., Ambrin, G., Okla, M. K., Soufan, W., Al-Ghamdi, A. A., & Ahmad, A. (2020). Metabolic Flux Analysis of Catechin Biosynthesis Pathways Using Nanosensor. Antioxidants, 9(4), 288. https://doi.org/10.3390/antiox9040288