Abstract
Cancer cell plasticity drives metastasis and therapy resistance through dynamic transitions between epithelial, mesenchymal, and neural crest stem-like (NCSC) states; however, a unifying mechanism that stabilizes these transitions remains undefined. To address this gap, we introduce a N-cadherin (CDH2)-centered redox–adhesion–exosome (RAX) hub that links oxidative signaling, adhesion dynamics, and exosome-mediated immune communication into a closed-loop framework. Within this network, reactive oxygen species (ROS) pulses license epithelial–mesenchymal transition (EMT), AXL–FAK/Src signaling consolidates mesenchymal adhesion, and selective exosomal cargoes—including miR-21, miR-200, miR-210, and PD-L1—propagate plasticity and immune evasion. Lipid peroxidation acts as a central checkpoint connecting ROS metabolism to PUFA membrane remodeling and ferroptosis vulnerability, buffered by NRF2–GPX4 and FSP1/DHODH axes, thereby converting transient oxidative pulses into persistent malignant states. Mechanistically, the RAX hub synthesizes findings from EMT/CSC biology, ferroptosis defenses, and exosome research into a self-reinforcing system that sustains tumor heterogeneity and stress resilience. Evidence from single-cell and spatial transcriptomics, intravital ROS imaging, and exosome cargo-selector studies supports the feasibility of this model. We further outline validation strategies employing HyPer–EMT–CDH2 tri-reporters, CRISPR perturbation of YBX1/ALIX cargo selectors, and spatial multi-omics in EMT-high tumors. Clinically, tumors enriched in EMT/NCSC programs—such as melanoma, neuroblastoma, small-cell lung cancer, pancreatic ductal adenocarcinoma, and triple-negative breast cancer (TNBC)—represent RAX-dependent contexts. These insights highlight biomarker-guided opportunities to target adhesion switches, ferroptosis defenses, and exosome biogenesis through lipid peroxidation-centered strategies using liquid-biopsy panels (exosomal CDH2, miR-200, miR-210) combined with organoid and xenograft models. By linking lipid peroxidation to ferroptosis defense and oxidative stress adaptation, the RAX hub aligns with the thematic focus of lipid metabolism and redox control in cancer progression. Collectively, the RAX framework may provide a conceptual basis for precision oncology by reframing metastasis and therapy resistance as emergent network properties.
1. Introduction
Cancer progression is increasingly recognized as a process driven not only by genetic alterations but also by profound cellular plasticity, enabling tumor cells to dynamically shift between epithelial, mesenchymal, and stem-like states [1,2,3,4]. This plasticity fuels metastasis, therapy resistance, and tumor recurrence, hallmarks that remain major barriers in oncology [5,6]. Recent studies highlight lipid peroxidation as not merely a byproduct of oxidative stress but a decisive modulator of cancer evolution through ferroptosis sensitivity and membrane remodeling, directly linking oxidative metabolism to metastatic behavior.
Clinically, the persistence of these plastic and therapy-resistant tumor states represents a major unmet need, as recent studies reveal that residual “drug-tolerant persister” cells survive even targeted or immune-checkpoint therapies through non-genetic plasticity programs—marked by slow proliferation, metabolic rewiring, and redox-driven epithelial–mesenchymal transitions—leading to inevitable relapse in aggressive cancers such as triple-negative breast cancer, melanoma, and pancreatic ductal adenocarcinoma [7]. Among these processes, epithelial–mesenchymal transition (EMT) and its reverse, mesenchymal–epithelial transition (MET), stand out as pivotal regulators of malignant progression. EMT confers motility and invasiveness through the repression of E-cadherin and the induction of mesenchymal adhesion molecules such as N-cadherin (CDH2), integrins, and AXL receptor tyrosine kinase (AXL) [8]. Strikingly, these features parallel the migratory behavior of neural crest cells during embryogenesis, suggesting a convergence between developmental and oncogenic programs [9,10].
Neural crest stem cells (NCSCs) are transient, multipotent progenitors with extraordinary migratory capacity, giving rise to diverse lineages, including melanocytes, neurons, and craniofacial mesenchyme [8,10]. Reactivation of neural crest-like transcriptional states has been observed in melanoma and other aggressive tumors, where factors such as MSH homeobox 1 (MSX1) and SRY-box transcription factor 10 (SOX10) drive dedifferentiation and enhance invasiveness [9,11,12]. Indeed, melanoma cells can undergo “neural crest-like reprogramming,” marked by a switch from E-cadherin-high to zinc-finger E-box-binding homeobox 1 (ZEB1)-high states, closely mimicking EMT dynamics [9,13]. Similarly, alternative splicing events and transcriptional rewiring reinforce this neural crest–mesenchymal identity, supporting tumor progression and metastasis [14]. These observations collectively argue that the mesenchymal traits acquired through EMT and the developmental logic of the neural crest converge into a shared cellular program that underlies cancer stemness and dissemination.
CSCs, a subpopulation capable of self-renewal and tumor initiation, embody this convergence. CSCs are sustained by a high degree of phenotypic plasticity, enabling interconversion between stem-like and differentiated states in response to microenvironmental cues [5,15]. Such plasticity is orchestrated not only by transcriptional circuits but also by metabolic and signaling adaptations. For instance, CSCs rely heavily on redox balance and metabolic reprogramming, including lipid metabolism, to withstand oxidative stress and maintain stemness [15]. In parallel, exosomes and microRNA (miR) act as key mediators of intercellular communication, propagating EMT and stemness traits across the tumor microenvironment [6,16]. MicroRNA-21, for example, enhances stemness and EMT in pancreatic cancer, reinforcing CSC traits and therapeutic resistance [16,17,18].
Recent single-cell and spatial transcriptomic studies further demonstrate that CSC plasticity is maintained through hybrid epithelial/mesenchymal (E/M) states, where dynamic adhesion remodeling and redox adaptation coexist. These intermediate states provide a survival advantage under oxidative and immune pressure, underscoring the role of redox signaling as a licensing factor for plastic transitions. These hybrid states couple adhesion switching with a mixed-metabolic, nuclear factor erythroid 2-related factor 2 (NRF2)-high program that buffers reactive oxygen species (ROS) and supports persistence under immune pressure [19,20,21,22].
Importantly, these plastic states are often stabilized by tumor microenvironmental signals, including transforming growth factor-β (TGF-β), cytokines, and hypoxia, which sustain EMT transcription factors and redox adaptation [23,24]. Moreover, therapy-induced stress itself may generate new CSC populations via dedifferentiation. For example, chemotherapy can eradicate leucine-rich repeat-containing G-protein-coupled receptor-5-positive (LGR5+) CSCs but simultaneously drive the emergence of clusterin-expressing “revival stem cells” that replenish the CSC pool and promote relapse [25].
Similar adaptive programs are reinforced by metabolic and redox reprogramming, enabling survival under oxidative pressure [26]. The tumor immune microenvironment further contributes, as tumor-associated macrophages (TAMs) provide cytokine-rich niches that enhance CSC stemness [27] and immune evasion [28]. Despite the wealth of studies on EMT, neural crest biology, and CSC regulation, a unifying framework that integrates these processes is still lacking. Notably, melanoma and its stromal ecology recapitulate developmental programs: neural crest-like EMT/phenotype switching in melanoma cells, and context-dependent reprogramming of stromal fibroblasts that interact with them. This developmental viewpoint motivates our focus on the under-recognized link between cancer-associated fibroblasts (CAFs) and neural crest-derived lineages [29,30].
Here, we propose that EMT/MET-driven mesenchymal traits and NCSC-like programs converge on a CDH2-centered redox–adhesion–exosome (RAX) hub. This hub coordinates adhesion switches, the redox-buffering NRF2–glutathione peroxidase 4 (GPX4) axis, and exosomal miRNA signaling to stabilize CSC states and promote immune evasion. By positioning redox pulses as the upstream licensing event, we highlight how oxidative stress integrates adhesion remodeling and vesicle-mediated communication into a closed circuit of cancer progression [23,27,31,32]. This perspective not only reconciles developmental and oncogenic paradigms but also identifies actionable vulnerabilities for therapeutic intervention.
2. EMT–MET and Neural Crest Convergence
The ability of cancer cells to transition between epithelial and mesenchymal states mirrors developmental processes, particularly those governing neural crest cell migration. EMT and MET endow tumor cells with invasive competence and plasticity reminiscent of NCSCs, suggesting that the molecular logic of embryonic delamination is aberrantly reactivated in tumors. This convergence links adhesion remodeling, transcriptional reprogramming, and migratory potential, establishing a developmental template for tumor progression.
3. Neural Crest Stem Cells (NCSCs) and Developmental Plasticity
NCSCs are transient, multipotent progenitors that delaminate from the dorsal neural tube and migrate extensively to form melanocytes, neurons, glia, smooth muscle, and craniofacial mesenchyme [33]. Their EMT-driven delamination parallels carcinoma invasion [34]. Specification is governed by transcriptional networks: paired box 3 (PAX3)/7 and Msx1 activate forkhead box D3 (FoxD3), snail family transcriptional repressor1 (SNAI1), twist family bHLH transcription factor 1 (TWIST1), and SOX10 [35,36,37]. Snail/Slug and Twist repress cadherins, FoxD3 induces SOX10, and SOX10 maintains multipotency [35,38]. Notably, SOX10 alone can reprogram somatic cells into neural crest-like states, underscoring its role as a master regulator of lineage plasticity [35]. Oncogenic EMT exploits the same circuitry. Twist, Snail, and ZEB1—critical in development—are reactivated in tumors to promote invasion and stemness [38]. In melanoma, ZEB1-driven phenotype switching mimics neural crest-like plasticity, while SOX10 re-expression drives dedifferentiation and therapy resistance [39]. Recent single-cell tracing reveals redox-regulated transcriptional nodes (e.g., SOX10–NRF2) stabilizing trajectories under oxidative stress [40]. These parallels highlight a testable hypothesis: developmental EMT circuits are hijacked to sustain cancer stemness and therapeutic resistance, suggesting that tumor plasticity represents an aberrant recapitulation of embryonic logic.
4. EMT/MET and Mesenchymal Traits in Cancer
EMT dismantles polarity and adhesion while inducing mesenchymal traits—motility, invasiveness, and resistance to apoptosis [41,42]. MET reverses this process, enabling colonization. EMT is orchestrated by transcription factors (Snail, Twist, ZEB1/2), epigenetic remodeling, and non-coding RNAs, converging on a plastic state highly adaptable and therapy-resistant [42,43]. A defining hallmark is the cadherin switch: loss of CDH1 and induction of CDH2, which destabilizes junctions and promotes extracellular matrix (ECM) engagement and migration [44]. Mesenchymal cells further upregulate integrins (e.g., α5β1), AXL, and platelet-derived growth factor receptor beta (PDGFRB), forming survival circuits [42,45]. Among these, AXL—a TAM family receptor tyrosine kinase—emerges as a key EMT driver. Gas6-mediated AXL activation promotes phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase (AKT), mitogen-activated protein kinase (MAPK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, enhancing invasion and therapy resistance [45,46,47]. Elevated AXL correlates with poor prognosis and its inhibition resensitizes tumors [46,47]. Hybrid E/M states identified by single-cell profiling retain partial CDH1 while upregulating CDH2 and integrins. These states exhibit metabolic reprogramming, high NRF2 activity, and PD-L1 expression, allowing persistence under oxidative and immune pressure [19,20]. Thus, EMT is not merely a dissemination mechanism but a licensing step for cancer-stem-cell persistence, linking adhesion remodeling and redox adaptation to immune evasion.
5. Convergence Concept
The parallels between EMT and neural crest delamination underscore a biological convergence. Both involve transcriptional reprogramming, cadherin switching, cytoskeletal remodeling, and migratory competence [48]. Single-cell transcriptomics reveal that EMT/MET plasticity generates hybrid E/M states resembling intermediate neural crest stages [49]. These states are marked by partial adhesion remodeling and stemness, bridging embryonic and oncogenic EMT. At the molecular level, cadherin regulation is central. During neural crest EMT, transient downregulation of CDH2 permits delamination, followed by re-expression for collective migration. In cancer, the CDH1-to-CDH2 switch stabilizes mesenchymal adhesion, directional migration, and anoikis resistance [50,51]. CDH2 thus functions not only as an adhesion molecule but as a hub integrating adhesion dynamics, growth factor signaling (AXL, PDGFRB), cytoskeletal polarity, and redox buffering. Emerging evidence shows CDH2 junctions interact with β-catenin, RhoA/Rac1, and antioxidant pathways, enabling cancer stem cells to buffer ROS [50,52]. Spatial single-cell analyses confirm that hybrid states re-express CDH2 in coordination with ROS-buffering circuits, reinforcing plasticity and resilience [53].
Beyond tumor-intrinsic programs, CAFs—including populations arising from non-fibroblast lineages such as endothelial/perivascular cells, adipocytes, monocytes, and bone marrow-derived mesenchymal stem cells (MSC)—amplify RAX circuitry [54]. CAFs are not a single lineage but a state that can arise from multiple sources; critically, adult fibroblasts—including CAFs—retain a positional ‘HOX code’ that reflects their embryonic origin (mesoderm vs. neural crest-derived ectomesenchyme). This developmental imprint persists across inflammatory states and primary/metastatic tumor stroma, implying that a subset of CAFs in head-and-neck/skin contexts plausibly derives from neural crest lineage fibroblasts [55]. By depositing fibronectin/collagens and activating integrin–FAK signaling, CAFs potentiate CDH2/AXL–FAK-dependent adhesion and migration; by releasing paracrine cues and generating redox bursts, they license EMT/NCSC transitions. In addition, cancer cell-derived extracellular vesicle (EV) can drive chemokine programs (e.g., CXCL1/CXCL8), contributing to stromal heterogeneity and worse outcomes [56]. Table 1. summarizes the distinctions from prior EMT/redox/exosome reviews—covering scope, key gaps, and our added contributions (a CDH2-centered closed loop and EV–immune–ferroptosis coupling). Prior work typically treated EMT/CSC plasticity, ferroptosis defenses, exosomal immune circuits, and biomarker strategies as separate silos; none integrated them into a closed-loop model. In contrast, the RAX hub synthesizes adhesion remodeling (CDH2/FAK/AXL), redox buffering (NRF2–GPX4/FSP1–DHODH), and exosomal PD-L1/miRNA circuits into a single interdependent system. These redox-buffering nodes simultaneously regulate lipid peroxidation intensity and ferroptosis threshold, positioning them as biochemical levers of RAX hub homeostasis.

Table 1.
Translational and mechanistic features distinguishing the CDH2-centered RAX hub from conventional EMT/redox/exosome models.
6. The CDH2-Centered Redox–Adhesion–Exosome (RAX) Hub
Mounting evidence indicates that cancer progression is driven not by isolated pathways but by integrated networks that stabilize plastic, stem-like, and invasive states. At their intersection lies the cadherin switch, linking adhesion remodeling to redox buffering and exosome-mediated communication. We conceptualize this as the CDH2-centered redox–adhesion–exosome (RAX) hub—a dynamic regulatory node synchronizing adhesion, oxidative adaptation, and intercellular signaling. This section dissects the three axes—adhesion/migration, redox buffering, and exosome-mediated communication—and integrates them into a closed feedback circuit.
7. Adhesion and Migration Axis
The cadherin switch, defined by loss of CDH1 and induction of CDH2, dismantles epithelial junctions and establishes mesenchymal adhesions, enhancing motility and invasion [95,96]. CDH2 supports stable collective migration and couple cells to extracellular matrix (ECM) via integrins [96,97]. Integrin–FAK/Src cascades remodel actin cytoskeleton, drive directional motility, and confer anoikis resistance [98,99]. Mesenchymal stability depends on a CDH2–AXL–PDGFRB triad. AXL activation via Gas6 triggers PI3K/AKT, MAPK, and NF-κB signaling to reinforce invasion and drug resistance [99,100], while PDGFRB amplifies PI3K–MAPK pathways and vascular integration [97,101]. Together, this triad forms a positive feedback loop: CDH2 facilitates AXL clustering, AXL enhances mesenchymal gene expression, and PDGFRB stabilizes survival signaling. Hybrid E/M states, defined by partial CDH1 retention with CDH2 and integrin upregulation, display enhanced metastatic efficiency and adaptability [96]. Importantly, tumor-derived exosomes propagate this circuitry. For example, TNBC cells export integrin β4 (ITGB4) to fibroblasts, inducing mitophagy and glycolysis that metabolically prime stroma for invasion; ITGB4 knockdown or exosome blockade disrupts this process [102]. Conversely, exosomal DOC2B can elevate ROS and lipid peroxidation, sensitizing cells to chemotherapy [103]. These findings support the perspective that exosomes act not as byproducts but as context-specific editors of adhesion and migration.
8. Redox-Buffering Axis
Reactive oxygen species (ROS) act both as signaling intermediates and cytotoxic stressors. In cancer, transient ROS pulses license EMT and NCSC-like transitions by coupling to cadherin switching and transcriptional reprogramming [104]. Intravital imaging reveals localized H2O2 surges at invasive fronts temporally aligned with EMT onset [105,106]. To survive oxidative bursts, CSC-like cells activate antioxidant defenses. The NRF2–Kelch-like ECH-associated protein 1 (KEAP1) axis regulates genes including Solute carrier family 7 member 11 (SLC7A11), Glutamate–cysteine ligase catalytic subunit (GCLC), NAD(P)H quinone dehydrogenase 1 (NQO1), heme oxygenase-1 (HO-1), and GPX4 [107,108], thereby sustaining cystine uptake, glutathione (GSH) synthesis, and peroxide detoxification. The GSH–GPX4 circuit reduces phospholipid hydroperoxides and blocks ferroptosis [108,109], while ACSL4 and epigenetic GPX4 regulation fine-tune ferroptosis sensitivity [109,110]. Parallel modules—FSP1-driven CoQ10 and DHODH-driven CoQH2—further trap lipid radicals [111]. These buffering systems stabilize EMT/CSC states but also expose a therapeutic vulnerability: mesenchymal-like cells resist apoptosis yet remain selectively sensitive to ferroptosis triggers [104]. From a perspective standpoint, this paradox defines a new Achilles’ heel—disrupting NRF2–GPX4–FSP1/DHODH axis defenses may collapse redox tolerance and eliminate therapy-resistant CSC pools.
9. Exosome–miRNA Axis
Exosomes link oxidative stress to adhesion/migration programs. Their secretion involves ESCRT complexes and RAB GTPases (RAB27A/B) [112], with stress pathways such as p53–TSAP6 further enhancing release [113]. Redox stress not only increases vesicle output but also dictates selective cargo loading [114]. The miR-200 family regulates EMT plasticity by suppressing ZEB1/2, while paradoxically aiding metastatic seeding [115]. miR-21 and miR-210 are enriched in hypoxic/redox-stressed exosomes, driving glycolysis, HIF stabilization, and immune evasion [112,116]. Cargo selection is actively controlled: YBX1 directs miR-223 loading, hnRNPA2B1 recognizes EXO-motifs under SUMOylation, [105] and ESCRT adaptors ALIX/TSG101 govern membrane scission and export [117]. Within the RAX hub, these selectors bias cargo toward miR-21/miR-210 and PD-L1 under oxidative stress, amplifying redox–adhesion programs [118]. Oxidative stress—including lipid–aldehyde stress—increases EV release, while hypoxia/ROS signaling elevates exosomal PD-L1 and reshapes immunosuppression; EV miR-21/210 are enriched under redox stress, linking lipid peroxidation to immune checkpoint up-regulation and EMT/CSC support [119]. Lipid peroxidation is initiated by the oxidation of polyunsaturated fatty acids (PUFAs), producing lipid radicals (L•) that propagate chain reactions to form lipid hydroperoxides (LOOH). The subsequent decomposition of unstable LOOH species generates reactive aldehydes, mainly 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA). Recent studies have demonstrated that elevated 4-HNE derived from n-6 PUFA metabolism amplifies oxidative stress-driven signaling cascades such as ASK1–JNK and suppresses AMPK activation, thereby linking lipid peroxidation to redox-dependent transcriptional reprogramming [120]. These electrophilic aldehydes form Michael adducts with cysteine, histidine, and lysine residues on key regulators of epithelial–mesenchymal transition (EMT) and cytoskeletal remodeling, including NF-κB and Nrf2, modulating gene expression associated with cell adhesion and motility. Moreover, long-lived species that maintain lower PUFA content and reduced 4-HNE/MDA burden exhibit enhanced glutathione-S-transferase (GST) activity and resilience to free-radical stress [121], highlighting 4-HNE as a pivotal mediator coupling membrane lipid composition to redox homeostasis. Through this mechanism, lipid–aldehyde stress may directly influence selective exosomal cargo loading—favoring miR-21 and miR-210 packaging—and thereby reinforce EMT plasticity and immune evasion within the RAX hub. Exosomal cargo thus functions as a mobile amplifier. In melanoma, PD-L1+ or FasL+ vesicles impair T cell cytotoxicity and condition pre-metastatic niches [116]. Therapy-induced exosomes further export resistance-promoting miRNAs [74]. Exosomes are active network players, reinforcing malignant states across stromal, vascular, and immune compartments, not passive waste vesicles.
10. Integration as a Closed Circuit
The RAX hub functions as a self-reinforcing feedback loop in which redox, adhesion, and exosomal pathways converge to sustain cellular plasticity. Transient ROS pulses activate EMT and NCSC transcription factors, thereby initiating the cadherin switch from CDH1-to-CDH2 [122]. Subsequent activation of integrin–FAK/Src signaling amplifies migratory and survival programs, consolidating the mesenchymal phenotype. In parallel, EMT reprogramming skews exosomal cargo toward mesenchymal and redox-regulatory microRNAs—notably miR-200, miR-21, and miR-210 [123,124], which propagate cancer stem cell (CSC) traits and reinforce immune suppression. These processes are further stabilized by antioxidant defense modules, including NRF2, GPX4, FSP1, and DHODH [125], which buffer oxidative pressure and preserve redox homeostasis. Together, the CDH2-centered RAX hub integrates adhesion, redox, and exosomal axes into a self-reinforcing circuit that converts transient oxidative cues into durable malignant identities—licensing EMT/NCSC transitions, stabilizing CSC pools, and sustaining immune-evasive, therapy-resistant states. Exosomal PD-L1 and immunosuppressive miRNAs blunt CD8+ T cell activity and promote Treg expansion [79,126]. Therapeutically, concurrent targeting of adhesion (CDH2/AXL–FAK), redox buffering (GPX4/SLC7A11, FSP1, DHODH), and exosomal signaling offers a rational strategy to disrupt this circuit (Figure 1).
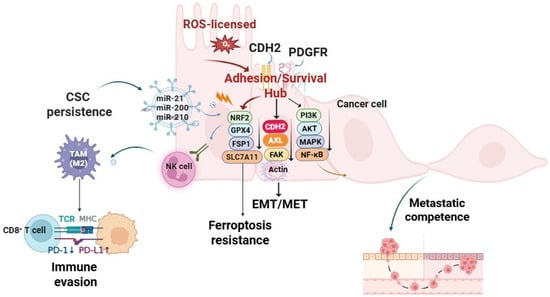
Figure 1.
Transient reactive oxygen species (ROS) pulses are stabilized into persistent malignant states through sequential licensing, lock-in, redox buffering, and broadcasting phases. Licensing: ROS-driven epithelial–mesenchymal transition (EMT) initiates cadherin switching from E-cadherin to N-cadherin (CDH2). Lock-in: CDH2 cooperates with integrins and receptor tyrosine kinases (AXL, PDGFRB) to activate FAK/Src–PI3K/AKT–MAPK signaling, reinforcing adhesion, migration, and mesenchymal stability. Redox buffering: NRF2, GPX4, FSP1, and SLC7A11 restore lipid redox homeostasis and confer ferroptosis-resistance. Broadcasting: selective exosomal cargo—miR-21, miR-200, miR-210, and PD-L1—propagate stemness, remodel the tumor stroma, and mediate immune evasion. Collectively, this closed-loop RAX circuit integrates oxidative signaling, adhesion dynamics, and exosome-mediated communication to sustain cancer cell plasticity and therapy resistance. Arrows indicate direction of regulation: ↓ downregulation, ↑ upregulation. Abbreviations: AKT: protein kinase B; AXL: AXL receptor tyrosine kinase; CDH2: N-cadherin; CSC: cancer stem cell; EMT/MET: epithelial–mesenchymal transition/mesenchymal–epithelial transition; FAK: focal adhesion kinase; FSP1: ferroptosis suppressor protein 1; GPX4: glutathione peroxidase 4; MAPK: mitogen-activated protein kinase; MHC-I: major histocompatibility complex class I; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NRF2: nuclear factor erythroid 2-related factor 2; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; PDGFRB: platelet-derived growth factor receptor beta; SLC7A11: solute carrier family 7 member 11; PI3K: phosphoinositide 3-kinase; TAM: tumor-associated macrophage; TCR: T cell receptor. Created in BioRender. Kim, B. (2025) (License number: YS292KS8UO).
11. Functional Outcomes of the RAX Hub
The integration of adhesion, redox, and exosome–miRNA signaling within the CDH2-centered RAX hub converges on three malignant hallmarks: maintenance of cancer stemness, metastatic competence, and immune evasion [127,128].
12. Cancer Stemness and Plasticity
Transient ROS pulses license E/M switching and NCSC programs. This plasticity is stabilized by CDH2–AXL/FAK signaling and insulated from ferroptosis by nuclear factor erythroid 2-related factor 2 (NRF2)-driven SLC7A11/GPX4 and the ferroptosis suppressor protein 1 (FSP1) axis. Concomitantly, hypoxia/redox stress biases exosomal cargo toward miR-200/210 and programmed death-ligand 1 (PD-L1), exporting plasticity and suppressing cytotoxic immunity [129]. Operational readouts include tumorsphere-formation efficiency, aldehyde dehydrogenase (ALDH) activity, CD44^high/CD24^low fractions, and exosomal PD-L1/miR-200/210 abundance [129,130]. This hybrid circuitry enables dynamic switching between quiescent and invasive states, preserving tumor growth potential [131]. Phenotype switching in melanoma and neuroblastoma illustrates this principle: cells toggle between MITF^high proliferative and MITF^low invasive states, mirroring CSC dormancy versus migratory phenotypes [128,131]. Exosomes reinforce stemness by horizontally transferring miRNAs (miR-21, miR-210) and stemness regulators to non-CSCs, expanding the CSC pool beyond original clones [106,126]. Redox buffering via NRF2 stabilization, GPX4-dependent lipid detoxification, and FSP1-mediated ferroptosis-resistance protects CSCs against ROS surges during EMT/NCSC transitions [126]. Single-cell and spatial transcriptomics confirm that hybrid E/M states are persistent, niche-anchored programs enriched for CSC traits and paracrine signaling [132]. In neuroblastoma, longitudinal profiling identifies cycling between adrenergic and mesenchymal-like states with a discrete “bridge” population, echoing neural crest plasticity [105]. Collectively, these data nominate CSC plasticity as a key outcome of the RAX hub and highlight therapeutic opportunities in ferroptosis sensitization and exosome disruption.
13. Metastatic Competence
The CDH2/integrin/AXL axis operates as a motility engine: CDH1 loss and CDH2 induction destabilize junctions, while integrin β1/β3–ECM binding activates FAK/Src cascades for migration [133,134]. AXL activation further stabilizes mesenchymal states and correlates with poor prognosis [134]. Tumor-derived exosomes extend this program by priming pre-metastatic niches. Integrin-defined exosomal tropism (α6β4/α6β1 for lung; αvβ5 for liver) remodels ECM and recruits stromal/immune cells, generating permissive colonization sites [135]. Exosomal gangliosides and CAMs activate stromal PDGFRB–FAK–ERK signaling, amplifying metastatic support [136]. Thus, intrinsic adhesion dynamics and extrinsic vesicle conditioning jointly fuel metastatic competence. Hybrid E/M states enriched at invasive edges exemplify this coupling.
14. Immune Evasion
The RAX hub subverts immune surveillance through vesicle-mediated signaling. Exosomal PD-L1 acts as a decoy for PD-1, impairing cytotoxic T lymphocytes; PD-L1^+ vesicles are enriched in glioblastoma, HCC, and GI cancers, correlating with checkpoint resistance [137,138]. Vesicular miRNAs (miR-21, miR-146a, miR-210) reprogram macrophages and dendritic cells into tolerogenic states, expanding FOXP3+ Tregs [139,140]. Exosome-conditioned TAMs adopt M2-like phenotypes, secreting IL-10/TGF-β and recruiting Tregs via CXCL12–CXCR4 signaling [141,142]. Collectively, immune evasion emerges as a structurally integrated outcome of the RAX hub, explaining why PD-L1 blockade alone is insufficient and why vesicle biogenesis/cargo inhibition must be combined with checkpoint therapies [143,144].
15. Therapeutic Targeting of the RAX Hub
Because the RAX hub integrates EMT plasticity, CSC persistence, and immune evasion, targeting its nodes offers the possibility of collapsing multiple malignant programs simultaneously [140,145].
16. Adhesion/Signaling Inhibition
Anti-N-cadherin antibodies, notably the monoclonal antibody ADH-1 (Exherin), disrupt CDH2-mediated adhesion and attenuate integrin–FAK/Src signaling, thereby reducing invasion and enhancing chemosensitivity [146]. Focal adhesion kinase (FAK) inhibitors, including Defactinib and GSK2256098, decrease motility, disrupt CSC niches, and attenuate CAF signaling; importantly, they also diminish exosome release [147]. AXL blockade, represented by small-molecule inhibitors (bemcentinib/BGB324, cabozantinib) as well as therapeutic antibodies, interferes with Gas6–AXL signaling, reverses EMT traits, and restores sensitivity to tyrosine kinase inhibitors (TKIs) and checkpoint blockade [148]. Collectively, inhibition of the CDH2–integrin–AXL–FAK axis illustrates how adhesion disruption destabilizes the RAX hub.
17. Redox Vulnerabilities
Covalent GPX4 inhibitors, including RSL3, ML162, and the next-generation compound C18 [149,150], induce ferroptosis in triple-negative breast cancer (TNBC) models [150,151,152]. Agents such as sulfasalazine and erastin deplete cysteine, diminish glutathione (GSH) pools, and trigger ferroptotic death [153]. Dual blockade of GPX4 and the cystine transporter xCT collapses antioxidant defenses and amplifies susceptibility to ferroptosis. Parallel pathways also contribute: FSP1 regenerates ubiquinol to trap lipid radicals, while inhibition of DHODH with brequinar sensitizes GPX4^high tumors, particularly when combined with xCT blockade [153]. Additional pharmacological interventions, including statins, withaferin A, or radiotherapy, intensify lipid peroxidation and synergize with GPX4 or xCT inhibition to overwhelm CSC defenses [149,154]. EMT-associated upregulation of ACSL4 and LPCAT3 promotes PUFA enrichment in membranes, enhancing metastatic adaptability but simultaneously sensitizing cells to ferroptotic stress under GPX4 inhibition. Phosphorylation of ACSL4 by PKCβII and PCK2 further amplifies ferroptotic signaling by increasing its phospholipid remodeling capacity. Conversely, induction of monounsaturated lipid synthesis via SCD1 or LPCAT1 confers ferroptosis resistance in epithelial-like cancer cells [155]. Thus, ferroptosis resistance in EMT/CSC populations represents a dynamic equilibrium between antioxidant buffering (NRF2–GPX4/FSP1/DHODH) and pro-oxidant remodeling (ACSL4–PUFA axis), rather than a fixed phenotype [156].
18. Exosome-Targeting Approaches
Targeting exosomal pathways provides complementary vulnerabilities. Inhibition of Rab27a or ESCRT machinery reduces the export of oncogenic cargo [157]. AntagomiRs or RNA sponges can neutralize oncogenic miRNAs such as miR-21, miR-210, and the miR-200 family, while engineered scaffolds have been developed to sequester these RNAs [158,159]. Moreover, blockade of RNA-binding protein transfer prevents CSC stabilization and therapy resistance [158]. Importantly, combining exosome targeting with ferroptosis inducers or immune checkpoint inhibitors may yield synergistic anti-tumor effects.
19. Multi-Node Targeting Strategy
Preclinical studies indicate that single-axis inhibition can be bypassed by compensatory circuits across adhesion (CDH2/AXL–FAK), redox defenses (GPX4–SLC7A11; ferroptosis-modulating FSP1/DHODH), and exosomal programs (EV cargo/PD-L1). These observations support evaluating multi-node combinations in selected contexts, rather than establishing a universal requirement [159,160]. For example, dual inhibition of GPX4 and xCT enhances ferroptotic cell death in colorectal and ovarian cancer models [161], while suppression of exosome pathways has reduced drug resistance [159,162]. Adhesion blockade through anti-CDH2 antibodies or AXL/FAK inhibitors further limits dissemination and primes tumors for redox collapse [163]. Thus, combining CDH2 interference, GPX4 inhibition, and EV pathway suppression may create synthetic lethality-like vulnerabilities in selected preclinical contexts, warranting prospective testing with endpoints and assay standardization [164]. Nanotechnology and engineered vesicles offer translational platforms to implement such multi-node strategies [163]. CAF ontogeny/positional-code PD: regional HOX gene panels (e.g., HOXA9/HOXD9, MEIS1/PRDM6) in CAF clusters, aligned with neural crest-enriched tumor states and myeloid-chemokine programs [55]. As operational exemplars, Table 2 summarizes axis-pairing combinations with matched PD readouts and early-phase design considerations.
Table 2.
Multi-node therapeutic combinations across adhesion, redox, and exosome axes: pharmacodynamic endpoints and design considerations.
20. Clinical Translation: Targeting Adhesion, Redox, and Exosomal Axes
Emerging clinical trials underscore the translational feasibility of dismantling the RAX hub through multi-node interventions. Adhesion signaling: AXL inhibitors such as bemcentinib (NCT02424617, NCT03184571, NCT03649321) and FAK inhibitors such as defactinib (NCT02758587, NCT03727880) are under evaluation for their ability to suppress EMT-driven dissemination and synergize with immune checkpoint blockade [62,171]. Redox buffering: Pharmacologic induction of ferroptosis through GPX4 inhibitors (e.g., FIN56 analog, NCT04205357) and IKE derivatives (NCT04836577) highlights ferroptosis defense as a tractable therapeutic vulnerability in solid tumors [172,173]. Exosomal circuits: In parallel, strategies targeting exosome secretion or employing engineered exosome carriers are advancing, including plant-derived nanovesicles (NCT03608631) and KRAS siRNA-loaded exosomes (NCT04747574) [174]. Together, these trials reinforce the clinical relevance of targeting adhesion (CDH2/AXL–FAK), redox buffering (GPX4/SLC7A11; FSP1), and exosomal signaling (PD-L1/miRNAs) as a biomarker-guided therapeutic triad to disrupt the malignant RAX hub. Clinically, developmental readouts could be co-opted as stromal stratifiers: positional HOX-code-based annotation of CAF compartments in head/neck and cutaneous tumors, and spatial co-profiling of neural crest-like tumor states with CAF activation and myeloid recruitment. While hypothesis-generating, these proposals are mechanistically grounded in developmental programs observed in human fibroblasts and in vivo melanoma models [55,175]. To contextualize the RAX hub in human tumors, we re-analyzed TCGA and published cohorts. Elevated CDH2 and miR-210 consistently associated with poor survival across colorectal, lung, breast, and PDAC datasets. Notably, patients with CDH2^high/miR-210^high tumors exhibited the worst prognosis (Table 3). These findings reinforce the clinical relevance of the RAX framework and support its evaluation as a prognostic–predictive tool in biomarker-enriched trials.
Table 3.
Clinical cohort evidence linking CDH2 and hypoxia-regulated miR-210 expression to survival outcomes and therapeutic resistance, highlighting their integration into the RAX hub framework.
21. Implications and Future Perspectives
The CDH2-centered RAX hub (redox–adhesion–exosome network) provides tractable axes for diagnosis, patient stratification, and therapy monitoring. Liquid-biopsy-ready exosomal readouts, when coupled with dynamic EMT/NCSC state sensors, could enable personalized disruption of the hub. This view is consistent with recent evidence highlighting biomarkers as central to diagnosis, therapeutic decision-making, and longitudinal monitoring in oncology [181]. In this respect, the RAX framework resonates with the recently proposed ‘ADAPT’ paradigm, advocating feedback-driven, adaptive strategies to counter cancer evolution [182].
22. Biomarkers: Exosomal CDH2/miR-200/miR-210 Panel
Exosomes are stable carriers of proteins and non-coding RNAs (ncRNAs) in body fluids such as blood, urine, and cerebrospinal fluid, making them highly suitable as non-invasive cancer biomarkers and theranostic tools. Their cargo reflects the dynamic state of the tumor and changes with disease progression [183]. Within this framework, CDH2 protein represents the adhesion and migration arm; exosomal proteomes enriched for adhesion molecules stratify metastatic tropism, and the inclusion of N-cadherin constitutes a testable extension [184]. The miR-200 family functions as a gauge of plasticity and EMT state, since the ZEB1–miR-200 axis governs the EMT/MET balance, and miR-200-based sensors are already employed as live reporters of EMT dynamics [106]. In parallel, miR-210 acts as a sentinel for hypoxia and redox stress, linking hypoxic conditions to EMT-to-NCSC transitions and CSC stabilization, with validation in lung and pancreatic models [185]. Clinical precedent strongly supports this biomarker concept: exosomal proteins including TYRP2, VLA-4, MET, and caveolin-1 predict outcomes in melanoma, while exosomal miRNAs such as miR-21 have been shown to track therapy resistance in lung cancer [112,183]. A practical validation path involves discovery in banked plasma or urine samples using targeted exosome isolation and orthogonal quantification (ELISA or PRM for CDH2, RT-qPCR for miRNAs), followed by the definition of cut-off thresholds in prospective cohorts. Importantly, urine- and plasma-based exosomal panels already distinguish cancer states and monitor therapeutic responses, underscoring the translational feasibility of this approach [183]. Similar integrative strategies combining TCGA datasets with survival data have successfully identified robust progression gene signatures, supporting the feasibility of the RAX biomarker panel [186].
23. Patient Stratification: EMT-High/NCSC-High Tumors as “RAX-Vulnerable”
Tumors with pronounced EMT plasticity and neural crest-like transcriptional programs are predicted to be most dependent on the RAX circuitry and therefore most susceptible to its disruption. In melanoma, CD271 and SOX10 identify highly plastic, therapy-resistant, and metastasis-prone states, making this tumor type an archetype of RAX dependence [39,187]. In neuroblastoma, co-expression of SOX10 and PHOX2B alongside canonical CSC markers such as CD133 and ALDH1 delineates an NCSC-like program that is associated with aggressiveness and poor survival [187]. Lung cancers with hypoxia-driven plasticity also represent promising candidates: exosomal miR-210 derived from lung CSCs promotes metastasis, while neuroendocrine-lineage lung tumors, including subtypes of small-cell lung cancer (SCLC), may likewise be considered for RAX-targeted interventions, although tumor type-specific validation remains essential [185]. Operationally, tissue immunophenotyping (CDH2 ↑/CDH1 ↓; SOX10/CD271 where relevant) should be paired with the liquid-biopsy RAX panel to guide assignment into RAX-targeted combinations.
24. Future Direction: Toward Personalized Therapy by Disrupting the RAX Hub
Future translational strategies should prioritize dynamic state tracking rather than static lineage assessment. The use of Z-cad-style EMT/MET sensors in patient-derived organoids and preclinical models can enable real-time monitoring of plasticity under drug pressure, thereby improving relapse prediction and guiding the rational sequencing of therapies [106]. Complementary liquid-biopsy monitoring, based on serial quantification of exosomal CDH2, miR-200, and miR-210, offers a minimally invasive means of detecting RAX rebound or hub collapse. Circulating miRNAs have already demonstrated feasibility by correlating with tumor burden and normalizing post-resection in multiple cancer contexts [188,189]. An immediate translational implication is that stromal composition and fibroblast ontogeny are not neutral: macroH2A loss in an autochthonous melanoma model increases CAF frequency/activation and promotes neural crest-lineage dedifferentiation with immunosuppressive myeloid influx—features linked to impaired anti-tumor immunity [175]. Moving forward, clinical trial designs should selectively enrich for EMT-high and NCSC-high tumors such as melanoma and neuroblastoma, where RAX dependence is most pronounced. Multi-node therapeutic combinations—targeting the CDH2/AXL–FAK adhesion axis together with ferroptosis vulnerabilities (GPX4/xCT) and exosome biogenesis—can be deployed within biomarker-guided escalation and de-escalation frameworks, as outlined in the therapeutic targeting and clinical translation sections. This approach provides a roadmap for implementing plasticity-aware precision oncology that integrates mechanistic insight with personalized therapy design. Future extensions could integrate AI-enabled mechanistic models of tumor growth, enabling biomarker-enriched personalization of RAX-targeted interventions [190].
25. Discussion
The RAX hub framework offers a mechanistic synthesis of redox signaling, adhesion dynamics, and exosomal communication, and may serve as a conceptual blueprint for precision oncology. From a diagnostic perspective, exosomal cargo—including adhesion molecules such as CDH2, regulatory miRNAs like the miR-200 family and miR-210, together with immune checkpoint proteins such as PD-L1—represents a promising source of liquid-biopsy biomarkers. These vesicle-derived signatures not only reflect EMT status, hypoxic adaptation, and therapy resistance but also provide minimally invasive tools for monitoring tumor plasticity and predicting patient response to immune checkpoint blockade [191,192].
Lipid peroxidation generates reactive aldehydes such as 4-HNE and MDA, which covalently modify cysteine, histidine, or lysine residues in membrane and cytosolic proteins, forming stable Michael adducts that alter adhesion dynamics and transcriptional control. 4-HNE accumulates at the invasive front of tumors, where it promotes E-to-N-cadherin switching and FAK/Src activation, driving EMT and CAF reprogramming through NF-κB signaling [193,194]. Through Keap1 adduction and GSK3β inhibition, 4-HNE activates Nrf2 and stabilizes mesenchymal persistence within the RAX hub [195]. Conversely, MDA-derived adducts serve as neoantigenic determinants that induce T cell exhaustion and immune tolerance, further reinforcing redox-driven immune evasion [196]. Clinically, plasma levels of 4-HNE and MDA correlate with tumor stage and oxidative burden, suggesting their potential utility as liquid-biopsy biomarkers of lipid peroxidation intensity and RAX hub activity [197]. Moreover, oxidative stress enhances 4-HNE-dependent exosomal export of PD-L1 and miR-21/210, providing a molecular bridge between lipid peroxidation and immune escape pathways [198].
These biomarkers are measurable in plasma and urine, offering real-time assessment of cancer stemness, therapy resistance, and metastatic risk. While CAFs are widely recognized by clinicians, their developmental provenance is often overlooked; converging evidence indicates that CAF states interweave with neural crest biology—both via persistent positional codes in adult fibroblasts and via tumor-driven re-engagement of neural crest-like programs in melanoma [29,55]. Rather than proposing a new lineage origin, this emerging concept highlights that fibroblast plasticity and neural crest-linked transcriptional memory may share a conserved developmental logic. This developmental parallel may help explain the remarkable heterogeneity, migratory capacity, and therapy resistance of CAF, emphasizing the importance of re-examining stromal biology through a neural crest-inspired lens. By contextualizing CAF behavior within broader principles of developmental reprogramming, this framework aligns with established literature yet opens a safer, mechanistically grounded path for understanding tumor–stroma evolution.
On the therapeutic side, the closed-circuit nature of the RAX hub underscores the necessity for multi-pronged interventions. Concurrent inhibition of adhesion-driven plasticity (CDH2 and AXL–FAK signaling) [109], ferroptosis-resistance circuits (the GPX4–xCT antioxidant axis and the FSP1–DHODH mitochondrial pathway) [199,200,201], and exosome biogenesis or selective cargo loading (Rab27a, ALIX, YBX1) could synergistically dismantle the adaptive resilience of cancer stem-like cells [200]. Through covalent Keap1 modification and GSK3β phosphorylation, 4-HNE creates a feed-forward Nrf2 loop that reinforces the RAX-dependent ferroptosis-defense network [195]. Such integrated targeting strategies may disrupt both metabolic and EV-mediated defenses, thereby enhancing the efficacy of conventional and immunotherapeutic modalities. This strategy provides a rational framework for designing multi-node therapies that could overcome resistance to immune checkpoint blockade, ferroptosis inducers, and targeted agents.
Clinically, tumors enriched in EMT and NCSC-like signatures—including melanoma, neuroblastoma, small-cell lung cancer (SCLC), and defined subsets of pancreatic ductal adenocarcinoma (PDAC) and triple-negative breast cancer (TNBC)—represent high-priority candidates for biomarker-driven clinical trials. In patients, plasma MDA (or MDA-protein adducts) levels strongly correlate with tumor stage and oxidative load, validating its potential as a circulating redox biomarker [202]. Elevated MDA-protein adducts in patient plasma positively correlate with oxidative load and advanced tumor stage [197]. Such tumors display pronounced cellular plasticity, therapeutic resistance, and immune-evasive traits, underscoring the translational potential of stratifying patients on the basis of EMT/NCSC transcriptional program [203,204,205]. Incorporating RAX hub signatures into patient stratification could refine therapeutic decision-making and identify subsets most likely to benefit from plasticity-aware interventions. In neuroblastoma, stromal neural crest progenitors exhibit marked radioresistance, undergo α-SMA-positive CAF-like differentiation after irradiation, and support regrowth of tumor neuroblasts—illustrating how neural crest-derived stroma can shape treatment response in vivo [206]. Collectively, these data argue that parts of the CAF compartment are developmentally imprinted and, in head–neck/skin settings, plausibly neural crest-derived; in melanoma, stromal–tumor crosstalk further re-engages neural crest programs with consequences for immunity and therapy response [55,175].
Ultimately, the convergence of liquid-biopsy biomarkers—including circulating tumor cells, cfDNA/ctDNA, and EV—with state-aware preclinical models such as organoids and patient-derived xenografts (PDXs) provides a robust translational framework. These platforms not only preserve tumor heterogeneity and therapeutic vulnerabilities but also enable parallel testing of rational drug combinations, thereby establishing a roadmap that bridges biomarker discovery with precision clinical intervention [207,208,209]. This approach may bridge the gap between mechanistic insight and clinical application, positioning the RAX hub as an actionable target in next-generation oncology. Integrating spatial transcriptomics and exosomal miRNA profiling would further strengthen mechanistic insight into redox-driven plasticity and intercellular communication. Recent studies utilizing single-cell and spatially resolved technologies have revealed how tumor-derived exosomes reprogram immune cells through specific miRNA cargo, such as miR-424 (322)/503, miR-21, and miR-155, thereby reinforcing therapy resistance and niche remodeling in cancer stem cell populations [94,210,211]. Oxidative–aldehyde stress enhances exosomal PD-L1 and miR-21/210 release, linking lipid peroxidation to immune checkpoint up-regulation [198]. Particularly, spatial transcriptomics combined with exosome-tracking approaches [211] enable precise localization of exosome–immune interactions in the tumor microenvironment, which could be applied to validate CDH2-centered redox licensing and EMT–CSC coupling. Furthermore, profiling of exosomal miRNAs from hypoxia- or ROS-conditioned cancer cells has provided functional evidence of redox-regulated vesicle content reprogramming [94,210,211].
26. Positioning and Strengths
The CDH2-centered RAX hub provides an integrative framework unifying three previously disconnected axes of cancer biology: redox dynamics, adhesion signaling, and exosome-mediated communication. Whereas EMT, CSC, and EV studies have historically advanced in parallel, this model synthesizes them into a closed circuit in which transient ROS pulses license EMT/NCSC transitions, CDH2 switching anchors mesenchymal persistence, and selective exosomal cargo propagates these programs across the tumor microenvironment. A key strength is explanatory breadth. Intravital imaging and HyPer probe studies demonstrate that EMT is punctuated by localized ROS bursts coinciding with N-cadherin upregulation and invasiveness, validating ROS pulses as in vivo “licensing signals” [212]. On the vesicular side, biochemical genetics shows that exosomal cargo loading is actively regulated by RNA-binding proteins (YBX1, hnRNPA2B1) and ESCRT adaptors (ALIX, TSG101), with functional consequences for migration and invasion [213]. Single-cell and spatial transcriptomics further confirm that hybrid E/M states are stable, niche-organized programs enriched for CSC markers and paracrine signaling [214]. Together, these converging data support the RAX hub as a multi-axis driver of cancer stemness, invasion, and resistance. This multi-axis interaction is schematized, illustrating how ROS-licensed CDH2 switching, redox buffering, and exosomal cargo selection are integrated into a self-reinforcing RAX hub driving hybrid E/M and NCSC programs (Figure 2). 4-HNE-induced FAK phosphorylation and cytoskeletal remodeling further sustain mesenchymal motility within the adhesion arm of the RAX hub [193,194]. Through covalent KEAP1 cysteine adduction and Akt-dependent GSK3β Ser9 phosphorylation, 4-HNE promotes NRF2 stabilization and a feed-forward antioxidant loop that reinforces the RAX-dependent ferroptosis–defense network [196,198].
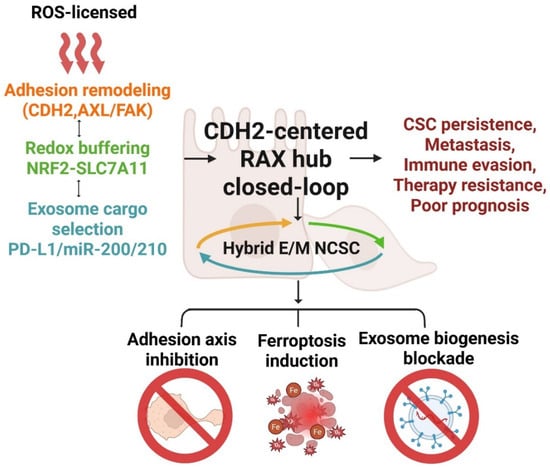
Figure 2.
Schematic model of the CDH2-centered redox–adhesion–exosome (RAX) hub illustrating its closed-loop architecture in cancer stemness and therapy resistance. Transient reactive oxygen species (ROS) pulses trigger adhesion remodeling through coordinated activation of CDH2, AXL, and FAK, which in turn couple with NRF2–SLC7A11-driven redox buffering and selective exosomal cargo enrichment (PD-L1, miR-200/210). These interlinked mechanisms form a self-reinforcing circuit that maintains hybrid epithelial/mesenchymal (E/M) and neural crest stem-like (NCSC) states, thereby promoting cancer-stem-cell (CSC) persistence, metastasis, immune evasion, and therapeutic resistance. The bottom panel highlights three therapeutic avenues—adhesion axis inhibition, ferroptosis induction, and exosome biogenesis blockade—as potential interventions to disrupt this adaptive plasticity network. Abbreviations: AXL: AXL receptor tyrosine kinase; CDH2: Cadherin-2; FAK: focal adhesion kinase; NRF2: nuclear factor erythroid 2–related factor 2; SLC7A11: solute carrier family 7 member 11; PD-L1: programmed death-ligand 1; E/M: epithelial/mesenchymal; NCSC: neural crest stem cell; CSC: cancer stem cell; ROS: reactive oxygen species. Created in BioRender. Kim, B. (2025) (License number: SU291KFI04).
27. Limitations
Despite converging evidence, the framework remains largely preclinical. ROS pulsing has been studied mainly with live-cell probes in xenografts and organoids, leaving spatiotemporal dynamics in patient tumors unresolved. Similarly, while YBX1/hnRNPA2B1/ALIX are validated cargo selectors in vitro, there in vivo role in shaping metastatic niches is not fully proven. Single-cell EMT trajectories robustly identify hybrid states, but caveats remain—batch effects, sequencing depth, and trajectory inference may distort their prevalence across datasets [215]. Moreover, most evidence derives from neural crest-derived tumors (melanoma, neuroblastoma, SCLC); generalizability to pancreatic, colorectal, and breast cancers requires further validation.
Furthermore, tumor-type heterogeneity must be acknowledged. While the RAX circuitry captures core features of redox–adhesion–exosome crosstalk, EMT and adhesion programs vary across malignancies, and certain tumors exhibit mesenchymal plasticity independent of N-cadherin or canonical EMT markers. Likewise, technical inconsistencies in EV isolation and characterization—ranging from ultracentrifugation to polymer-based precipitation—can confound comparative analyses of exosomal PD-L1 or miRNA content between studies. Standardization of EV handling and inclusion of multiple adhesion markers (e.g., vimentin, integrin-β1) will be necessary to ensure reproducibility and translational relevance.
Three mechanistic gaps deserve focused resolution: First, causal imaging of ROS pulses, EMT entry, and CDH1-to-CDH2 switching in the same cells in vivo. Second, genetic validation that YBX1/hnRNPA2B1/ALIX govern EV cargo choice and control metastasis/immune tone. Third, harmonized atlases quantifying EMT state transitions and exosomal PD-L1/miR-210 signals in patient tissues across disease stages. There is a clear need for integrated single-cell, spatial, and liquid-biopsy validation. To mitigate the risk of overextension, we propose a staged validation roadmap with pre-specified Go/No-Go thresholds. In the preclinical tier, ≥2 pharmacodynamic (PD) axes (e.g., CDH2–FAK phosphorylation decrease; lipid ROS increase; EV-PD-L1 suppression) must be activated without dose-limiting toxicity to proceed. In the translational tier, biomarker stratification (CDH2^high/miR-210^high or EV-PD-L1 dynamics) must reproducibly segregate survival outcomes across at least two independent cohorts. Only upon fulfilling both tiers should RAX-targeted interventions progress to biomarker-enriched early-phase clinical trials. These criteria prevent premature escalation and anchor the ambitious scope of the RAX framework to measurable decision points.
28. Outstanding Questions and Cross-Disease Relevance
Several critical questions remain unresolved. Orthotopic tumors engineered to express HyPer (H2O2 sensor) could capture ROS pulses, while a Z-cad-style EMT/MET reporter and a CDH2 knock-in tag would read out TF activation and the CDH1-to-CDH2 switch in the same cell [106]. HyPer probes have been validated for dynamic H2O2 imaging in living cells and in vivo models [216,217,218,219], while single-cell transcriptomic analysis confirm that EMT trajectories generate hybrid EM states enriched for CSC traits and paracrine signaling [215]. Such integrated tri-reporter systems, coupled with intravital microscopy, would allow causal relationships between redox dynamics, transcriptional reprogramming, and adhesion remodeling to be established in vivo.
Another unresolved question is what encodes exosome cargo choice. Tumor-specific CRISPR knockouts or point mutants of YBX1, HNRNPA2B1, or ALIX (e.g., SUMO-site variants) could test their roles in exosomal miR-21, miR-200, miR-210 and PD-L1 loading, and the resulting effects on migration, T cell cytotoxicity, and TAM polarization. YBX1 has been identified as a key RNA-binding protein that selectively sorts miRNAs into exosomes, while HNRNPA2B1 directly regulates RNA cargo loading through post-translational modifications and has been validated by CRISPR knockout approaches in tumor models. In addition, ALIX functions within the ESCRT machinery to coordinate exosome biogenesis and cargo specificity [220,221]. Together, these mechanisms provide a strong genetic framework to test how exosome-mediated communication drives CSC plasticity and immune remodeling within the RAX hub.
A third question is whether hybrid E/M hubs are universal. Combined RNAscope and immunofluorescence for CDH2, AXL, FAK, PD-L1, and miR-210, integrated with spatial transcriptomics, could map hybrid E/M niches and EV-linked immune states across tumor stages. Recent spatial and imaging studies indicated that hybrid E/M phenotypes persist as niche-organized states enriched for CSC traits and immune-evasive programs; in colon cancer, phenotypic plasticity and niche-driven EMT dynamics are highlighted in comprehensive reviews of metastatic formation [218]. These findings underscore the feasibility of mapping hybrid niches across cancers to test the generalizability of RAX-driven plasticity.
They exhibit intrinsic plasticity, NCSC-like programs, and EV-mediated immune editing, together with tolerogenic macrophage states and checkpoint signaling, converging on exosomal PD-L1/miRNA immunomodulation [39,187]. Beyond these, PDAC demonstrates cadherin switching—loss of CDH1 with gain of CDH2—AXL/integrin/FAK signaling, reliance on GPX4/xCT redox buffering, and exosome-driven stromal and immune remodeling [222]. Similarly, colorectal and gastric cancers exploit exosomal miR-200 to reinforce epithelial plasticity, while miR-21 and miR-210 mediate hypoxic adaptation and immune suppression, correlating with metastasis and poor prognosis [223,224,225,226]. In TNBC, CDH2, AXL, and FAK signaling sustain invasive traits, while GPX4 and xCT protect CSC-like cells from ferroptosis; exosome cargo carrying PD-L1 and miRNAs further transfer drug resistance [216,227]. Importantly, systemic comorbidities such as diabetes converge on redox regulators including GPX4, linking metabolic stress to enhanced RAX reliance across multiple cancers.
29. Translational Outlook and Validation Roadmap
The RAX model yields biomarker–intervention loops that connect mechanistic discovery to translational application. Circulating exosomal CDH2, miR-200, and miR-210 emerge as minimally invasive biomarkers for hub activity. EMT-high/NCSC-high tumors such as melanoma, neuroblastoma, and subsets of lung cancers represent prime stratification candidates. Rational therapeutic strategies will require simultaneous disruption of adhesion (CDH2/AXL–FAK), redox buffering (GPX4/SLC7A11, FSP1, DHODH), and exosomal signaling to overcome redundancy and collapse CSC resilience.
To operationalize this framework, we propose a five-tier validation roadmap. First, causality can be interrogated using orthotopic tumors engineered with HyPer H2O2 sensors, EMT reporters, and CDH2 fluorescent knock-ins, enabling dynamic visualization of ROS-driven EMT/NCSC transitions. HyPer probes have been validated for dynamic H2O2 imaging in living cells and in vivo models [216,217,218,219], while single cell transcriptomic analysis confirm that EMT trajectories generate hybrid EM states enriched for CSC traits and paracrine signaling [215]. Intravital microscopy could integrate these reporters to resolve causality in vivo.
Second, pharmacologic buffering of ROS pulses using catalase mimetics or myeloperoxidase (MPO) inhibitors has been shown to modulate oxidative bursts in vivo [228]. Conversely, blockade of FAK/AXL signaling disrupts cadherin–integrin coupling and EMT-associated adhesion remodeling. Integrating these approaches, we propose that applying catalase mimetics versus adhesion pathway inhibitors in the same model would reveal whether pulse-to-adhesion coupling is redox-gated, directly testing the causal role of oxidative dynamics in EMT/NCSC stabilization [141,142,143,144,145].
Tumor-specific CRISPR knockouts or point mutants of YBX1, HNRNPA2B1, or ALIX (e.g., SUMO-site variants) could test their roles in exosomal miR-21, miR-200, miR-210 and PD-L1 loading, and the resulting effects on migration, T cell cytotoxicity, and TAM polarization [220]. YBX1 has been identified as a key RNA-binding protein that selectively sorts miRNAs into exosomes, while HNRNPA2B1 directly regulates RNA cargo loading through post-translational modifications and has been validated by CRISPR knockout approaches in tumor models. In addition, ALIX functions within the ESCRT machinery to coordinate exosome biogenesis and cargo specificity [221]. Together, these findings provide a mechanistic basis for testing how cargo selectors drive exosome-mediated plasticity and immune remodeling in the RAX hub.
Combined RNAscope and immunofluorescence for CDH2, AXL, FAK, PD-L1, and miR-210, integrated with spatial transcriptomics, could map hybrid E/M niches and EV-linked immune states across tumor stages [229]. Recent spatial transcriptomic and imaging studies confirm that hybrid E/M phenotypes are not transient but persist as niche-organized states enriched for CSC traits and immune-evasive programs. For example, Sahoo et al. demonstrated that hybrid E/M states sustain high PD-L1 levels and immune suppression, while Teeuwssen and colleagues highlighted the role of phenotypic plasticity and niche-driven EMT plasticity in metastatic progression in colon cancer models [230,231].
Finally, therapeutic stress tests should combine inhibition of CDH2/AXL–FAK axis (e.g., AXL ± FAK inhibitors) together with ferroptosis defenses (GPX4 or xCT blockade), and optionally suppress EV biogenesis (Rab27a, ESCRT). Readouts should include lipid-ROS accumulation/ferroptosis markers, CSC frequency, EV cargo flux (PD-L1, miR-21, miR-200, miR-210), and intratumoral T/NK cell function. This design is supported by evidence that FAK/AXL targeting disrupts mesenchymal programs and can improve response to anti-PD-1, while GPX4, xCT inhibition sensitizes mesenchymal/CSC-like cells to ferroptosis; EV output (Rab27a, ESCRT) sustains immune evasion via PD-L1 cargo, providing complementary leverage points [65,231,232].
Preclinical studies substantiate this concept: genetic or pharmacologic inhibition of the FAK axis reduces mesenchymal stability and significantly enhances anti-PD-1 efficacy with greater CD8+ T cell infiltration in vivo [233,234]. Similarly, blockade of TAM/AXL signaling suppresses EMT-associated invasion and confers immuno-oncologic benefit in animal models [233,235]. On the redox arm, disabling GPX4 or SLC7A11 elevates lipid peroxides and selectively triggers ferroptotic death in mesenchymal/CSC-like populations, effects that are reversed by ferrostatin-1 rescue. Finally, vesicle biogenesis via Rab27a, ESCRT machinery is essential for the export of exosomal PD-L1 and miRNA, sustaining T cell suppression; blocking these nodes diminishes immune evasion and sensitizes tumors to checkpoint blockade [220,221,236,237,238].
This roadmap integrates causality testing, temporal inhibition, genetic dissection of cargo selection, spatial mapping of hybrid niches, and therapeutic stress-testing. Together, these strategies directly link mechanistic validation to biomarker development and clinical translation. In parallel, a composite liquid-biopsy panel—exosomal CDH2, miR-200, and miR-210—combined with tissue immunophenotyping (CDH2 ↑/CDH1 ↓; lineage markers as appropriate) enables longitudinal tracking and patient stratification (Figure 3A,B). Building on preclinical strategies (Table 2) and patient-cohort analyses (Table 3), we outline a staged validation roadmap (Table 4) that links mechanistic dissection to biomarker-guided clinical translation. This roadmap aligns with broader bench-to-bedside lessons from cancer nanomedicine, underscoring the need for stepwise validation [239]. Therapeutic targeting of lipid peroxidation dynamics—via ferroptosis inducers or antioxidant modulators—represents a direct translational extension of the RAX framework within the redox oncology paradigm.
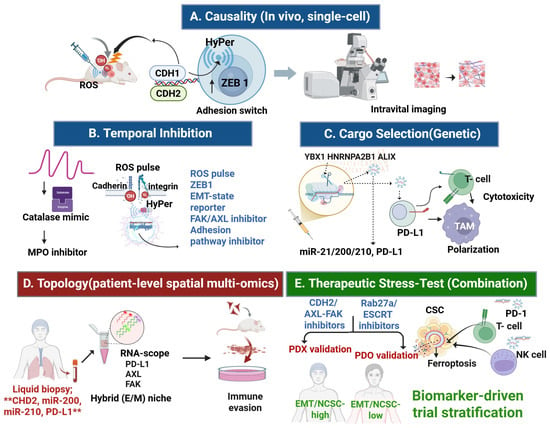
Figure 3.
Experimental and translational validation roadmaps for the CDH2-centered redox–adhesion–exosome (RAX) hub. (A) Causality (in vivo, single-cell): Orthotopic tumors engineered with HyPer (H2O2 sensor), a ZEB1 EMT-state reporter, and a CDH2 knock-in tag enable real-time visualization of ROS pulses, transcription factor activation, and CDH1→CDH2 adhesion switching, linking oxidative bursts to CSC enrichment. (B) Temporal inhibition: ROS buffering with catalase mimetics or myeloperoxidase (MPO) inhibitors suppresses oxidative pulses, while FAK/AXL inhibition disrupts cadherin–integrin coupling, jointly testing whether ROS dynamics gate adhesion remodeling. (C) Cargo selection (genetic): CRISPR-mediated knockout of exosomal cargo selectors (YBX1, HNRNPA2B1, ALIX) alters loading of miRNAs (miR-21, miR-200, miR-210) and PD-L1, thereby reshaping tumor migration, T cell cytotoxicity, and macrophage polarization (TAM). (D) Topology (patient-level spatial multi-omics): Integration of RNAscope and multiplex immunofluorescence for CDH2, AXL, FAK, PD-L1, and miR-210 with spatial transcriptomics maps hybrid epithelial/mesenchymal (E/M) niches enriched for CSC traits and immune evasion signatures. (E) Therapeutic stress test (combination): In EMT-high tumors, concurrent inhibition of CDH2/AXL–FAK adhesion, GPX4 and xCT-mediated ferroptosis defenses, and Rab27a/ESCRT-dependent EV biogenesis reduces CSC frequency, enhances lipid–ROS accumulation, depletes exosomal PD-L1/miRNA cargo, and augments cytotoxic T lymphocyte (CTL) and natural killer (NK) cell infiltration. ** indicate key biomarkers included in the liquid-biopsy RAX panel. Arrows indicate direction of regulation: ↓ downregulation, ↑ upregulation. Abbreviations: AXL: AXL receptor tyrosine kinase; ALIX: ALG-2 interacting protein X; CDH2: cadherin 2; also known as N-cadherin; CSC: cancer stem cell; E/M: epithelial/mesenchymal; FAK: focal adhesion kinase; GPX4: glutathione peroxidase 4; HNRNPA2B1: heterogeneous nuclear ribonucleoprotein A2/B1; HyPer: hydrogen peroxide sensor probe; miR: microRNA; MPO: myeloperoxidase; NK cell: natural killer cell; PD-L1: programmed death-ligand 1; PDO: patient-derived organoid; PDX: patient-derived xenograft; ROS: reactive oxygen species; RNAscope: RNA in situ hybridization technology; SLC7A11: solute carrier family 7 member 11, also known as xCT, TAM: tumor-associated macrophage; ZEB1: zinc finger E-box binding homeobox 1; YBX1: Y-box binding protein 1. Created in BioRender. Kim, B. (2025) (License number: TC292KY13P).
Table 4.
Redefining cancer plasticity: A translational and paradigm-shifting framework.
Although our analysis highlights melanoma, neuroblastoma, and SCLC as prototypic EMT/NCSC-driven malignancies, similar associations are observed in colorectal and gastric cancers. For instance, miR-210 overexpression correlates with poor overall survival in CRC cohorts, while CDH2 upregulation predicts inferior prognosis in gastric cancer patients. These convergent signals across diverse epithelial tumors underscore the cross-disease generalizability of the RAX hub framework and support its broader translational potential. Patients stratified by combined CDH2^high/miR-210^high expression could be prioritized for RAX-targeted intervention trials, analogous to PD-L1 or tumor mutational burden (TMB)-based enrichment in immunotherapy trials [176,177,241,242,243,244,245]. Across melanoma patient cohorts, exosomal PD-L1 levels consistently predicted inferior outcomes under ICIs, where higher EV PD-L1 abundance was linked to shorter overall survival and reduced therapeutic efficacy. Serial monitoring of EV PD-L1 further enabled discrimination between durable responders and early progressors [246,247,248], supporting its role as a putative predictive biomarker for biomarker-guided RAX-targeted interventions.
Tissue/EV miR-200 low/ZEB1 high EMT axis correlates with adverse survival across multiple cancers, as supported by meta-analysis and NSCLC cohort data [249]. Exosomal miR-210 reflects hypoxia- and redox-driven adaptation, and its elevation has been associated with tumor progression and poorer clinical outcomes in NSCLC and PDAC cohorts [250]. On the adhesion arm, CDH2 expression consistently associates with aggressive behavior and reduced survival across tumor types; moreover, genetic reduction in CDH2 prolongs survival in a KPC pancreatic cancer model, underscoring its prognostic and therapeutic relevance [251,252].
Clinically, activation of the AXL/FAK signaling pathway has been repeatedly linked to poor prognosis and therapeutic resistance and is currently the focus of multiple translational studies and clinical trials [253,254]. Together, these literature-derived clinical associations support biomarker-guided enrichment strategies—particularly panels combining exosomal CDH2, miR-200, miR-210, and EV-PD-L1—for precision RAX-targeted interventions [255]. Pan-cancer meta-analyses demonstrate that low miR-200 and high ZEB1 expression correlate with EMT activation, enhanced invasion, and significantly worse overall and disease-free survival across multiple tumor types, supported by TCGA-based pan-cancer datasets [256]. Emerging clinical evidence further highlights the translational relevance of EV-associated microRNAs in RAX-dependent cancers. In particular, EV miR-210 has been associated with hypoxia-conditioned redox adaptation, metastatic progression, and unfavorable outcomes in observational analyses of PDAC and non-small-cell lung cancer (NSCLC) cohorts. In PDAC, serum-derived EV miR-210 levels are significantly elevated compared with controls and associate with early detection as well as adverse prognosis [257]. Complementary mechanistic and clinical analyses confirm that miR-210 functions as a central hypoxia/redox-responsive oncomiR that promotes invasion, angiogenesis, and therapy resistance [258]. Serial changes in EV PD-L1 (±miR-210) were associated with response status in exploratory NSCLC cohorts, supporting their potential predictive utility; however, prospective validation with standardized assays and predefined thresholds is required [249,250]. Standardized assays and pre-specified thresholds will be essential to determine whether these EV markers function as predictive rather than purely prognostic indicators. Collectively, these findings support the integration of EV miR-210 quantification into liquid-biopsy panels, in combination with exosomal CDH2 and miR-200 family members, to refine patient stratification for RAX-targeted therapeutic strategies.
Across cancers, N-cadherin (CDH2) upregulation is consistently linked to aggressive disease biology and poor survival outcomes. A large meta-analysis confirmed that elevated CDH2 predicts inferior overall survival and higher risk of metastasis in epithelial-derived tumors [251,259]. Importantly, genetic reduction in Cdh2 in a KPC pancreatic cancer model prolonged survival by ~25%, providing in vivo evidence that targeting CDH2 can improve prognosis [252]. Clinically, the AXL–FAK axis represents a conserved driver of resistance and poor prognosis across multiple tumor types [260]. AXL overexpression promotes epithelial–mesenchymal transition (EMT), enhances cell migration/invasion, and sustains therapy resistance, while its crosstalk with FAK amplifies integrin signaling and adhesion remodeling [260,261]. Evidence from NSCLC, TNBC, RCC, and PDAC demonstrates that AXL activation correlates with adverse survival and immune evasion, positioning it as both a biomarker of poor prognosis and a therapeutic target [260]. Preclinical studies show that combined AXL and FAK inhibition disrupts mesenchymal stability, restores drug sensitivity, and synergizes with anti-PD-1 therapy, leading to enhanced CD8+ T cell infiltration [261]. Ongoing clinical trials of AXL inhibitors (e.g., bemcentinib, cabozantinib) underscore the translational relevance of this pathway [262]. Together, integrating exosomal CDH2, miR-200/miR-210, and EV-PD-L1 with AXL–FAK activation provides a composite stratification framework for identifying RAX hub-dependent tumors most likely to benefit from multi-node therapeutic disruption (Figure 3; Table 5). This aligns with emerging perspectives that move beyond the genetic paradigm of cancer, emphasizing cell-state plasticity and tissue-level field effects as central drivers of malignancy [240]. As summarized in Table 5, recent landmark studies emphasize the clinical feasibility of biomarker-guided strategies, the conceptual shift beyond the genetic paradigm [240], and the necessity of dynamic, patient-specific modeling approaches [182,190]. Collectively, these findings substantiate the RAX hub as a field-shaping and translationally actionable framework.
Table 5.
Clinical associations of RAX-hub biomarkers across cancers: evidence and paradigm-shifting implications.
30. Conclusions
Cancer progression is driven less by static hierarchies than by dynamic plasticity. Here, we integrate developmental and oncogenic paradigms into a single framework: an EMT/MET–neural crest convergence anchored by a CDH2-centered redox–adhesion–exosome (RAX) hub. In this model, transient ROS pulses license EMT/NCSC transcriptional switches; cadherin switching and integrin–FAK/Src signaling establish migratory adhesion states; exosomes propagate mesenchymal, redox, and immunoregulatory programs; and redox-buffering circuits—NRF2–SLC7A11–GPX4 and FSP1–CoQ10/DHODH–CoQH2 axes—maintain oxidative balance. The outcome is stabilization of cancer stemness, metastatic competence, and immune evasion as a self-reinforcing circuit.
This perspective yields three translational opportunities. First, it nominates liquid-biopsy biomarkers—exosomal CDH2, miR-200, and miR-210—as minimally invasive, real-time reporters of hub activity. Second, it defines a biomarker-driven stratification schema: EMT-high/NCSC-high tumors, including melanoma, neuroblastoma, small-cell lung cancer, and subsets of pancreatic and triple-negative breast cancers, are predicted to be most RAX-dependent and thus therapeutically targetable. Third, it proposes a therapeutic blueprint: rather than single-node inhibition, rational multi-node combinations (e.g., CDH2, AXL–FAK inhibitors plus GPX4, xCT ferroptosis blockade and exosome suppression) may collapse both intrinsic buffering and extrinsic niche programming.
Operationalizing this strategy requires translational pipelines: state-aware preclinical models using EMT sensors, organoids, and patient-derived xenografts, prospective validation of the exosomal CDH2/miR-200/miR-210 biomarker panel in liquid-biopsy cohorts, and early-phase clinical trials enriched for EMT/NCSC-high patients, with biomarker-guided escalation across adhesion, redox, and exosome axes. Key open questions include the quantitative rules of ROS pulsing, the logic of exosomal cargo selection, and resistance trajectories under multi-node therapeutic pressure.
Collectively, the RAX hub may redefine metastasis and therapy resistance as emergent network properties, proposing a biomarker-guided roadmap for precision oncology. By measuring and dismantling this network rather than chasing single effectors, we anticipate a path toward translational frameworks for multi-target redox and exosome modulation in metastatic cancers, in which metastatic and immune-evasive phenotypes become not only predictable but also clinically actionable—and ultimately preventable.
Author Contributions
M.N.P. conceived and designed the study, performed methodology design, data curation, and validation, and was responsible for resource management, visualization, and manuscript writing and revision. She also supervised the overall research project. J.C. contributed to formal analysis, software development, and data investigation, and assisted in visualization and manuscript editing. R.I.M.d.A.R. contributed to visualization and critically revised the manuscript. D.V.D. provided critical manuscript review and scientific editing. S.-G.K. participated in project administration, supervision, and manuscript revision. B.K. oversaw the project, provided conceptual and administrative supervision, and contributed to manuscript review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1I1A2066868); the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2020-NR049559, RS-2024-00350362); the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2020-KH087790); and the TIPS Program (No. RS-2024-00507224) funded by the Ministry of SMEs and Startups (MSS, Republic of Korea).
Institutional Review Board Statement
Not applicable. This review did not involve human participants or animal experiments.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| protein kinase B | (AKT) |
| ALG-2 interacting protein X | (ALIX) |
| AXL receptor tyrosine kinase | (AXL) |
| cancer stem cell | (CSC) |
| cell-free DNA | (cfDNA) |
| circulating tumor cell | (CTC) |
| clustered regularly interspaced short palindromic repeats | (CRISPR) |
| cadherin 2, also known as N-cadherin | (CDH2) |
| epithelial–mesenchymal transition | (EMT) |
| epithelial/mesenchymal | (E/M) |
| extracellular vesicle | (EV) |
| extracellular matrix | (ECM) |
| focal adhesion kinase | (FAK) |
| ferroptosis suppressor protein 1 | (FSP1) |
| forkhead box D3 | (FoxD3) |
| glutamate–cysteine ligase catalytic subunit | (GCLC) |
| glutathione | (GSH) |
| glutathione peroxidase 4 | (GPX4) |
| heterogeneous nuclear ribonucleoprotein A2/B1 | (hnRNPA2B1) |
| heme oxygenase-1 | (HO-1) |
| HyPer hydrogen peroxide sensor probe | (HyPer) |
| kelch-like ECH-associated protein 1 | (KEAP1) |
| leucine-rich repeat-containing G-protein coupled receptor 5 positive | (LGR5+) |
| mitogen-activated protein kinase | (MAPK) |
| myeloperoxidase | (MPO) |
| Msh homeobox 1 | (MSX1) |
| NAD(P)H quinone dehydrogenase 1 | (NQO1) |
| neural crest stem cell | (NCSC) |
| nuclear factor erythroid 2–related factor 2 | (NRF2) |
| nuclear factor kappa-light-chain-enhancer of activated B cells | (NF-κB) |
| pancreatic ductal adenocarcinoma | (PDAC) |
| platelet-derived growth factor receptor beta | (PDGFRB) |
| programmed death-ligand 1 | (PD-L1) |
| proto-oncogene tyrosine-protein kinase Src | (Src) |
| reactive oxygen species | (ROS) |
| RNA in situ hybridization technology | (RNAscope) |
| SRY-box transcription factor 10 | (SOX10) |
| solute carrier family 7 member 11, also known as xCT | (SLC7A11, xCT) |
| small-cell lung cancer | (SCLC) |
| snail family transcriptional repressor 1 | (SNAI1, Snail protein) |
| twist family bHLH transcription factor 1 | (TWIST1, Twist protein) |
| transforming growth factor-β | (TGF-β) |
| Thioredoxin | (TRX) |
| triple-negative breast cancer | (TNBC) |
| Y-box binding protein 1 | (YBX1) |
| zinc finger E-box binding homeobox 1 | (ZEB1) |
References
- Moon, H.; Park, H.; Ro, S.W. c-Myc-driven Hepatocarcinogenesis. Anticancer Res. 2021, 41, 4937–4946. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G. Molecular Target and Action Mechanism of Anti-Cancer Agents. Int. J. Mol. Sci. 2023, 24, 8259. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Shim, H.S.; Kim, S.; Lee, I.H.; Kim, J.; Yoon, S.; Kim, H.D.; Park, I.; Jeong, J.H.; Yoo, C.; et al. Clinical practice recommendations for the use of next-generation sequencing in patients with solid cancer: A joint report from KSMO and KSP. J. Pathol. Transl. Med. 2024, 58, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-Z.; Ren, Y.-W.; Liu, Y.; Zhao, T.; Huang, Z.-P.; Lu, K. METTL3-mediated m6A modification of FoxP4 promotes HCC metastasis. Adv. Tradit. Med. 2025, 1–8. [Google Scholar] [CrossRef]
- BharathwajChetty, B.; Sajeev, A.; Vishwa, R.; Aswani, B.S.; Alqahtani, M.S.; Abbas, M.; Kunnumakkara, A.B. Dynamic interplay of nuclear receptors in tumor cell plasticity and drug resistance: Shifting gears in malignant transformations and applications in cancer therapeutics. Cancer Metastasis Rev. 2024, 43, 321–362. [Google Scholar] [CrossRef]
- Kharkar, P.S. Cancer Stem Cell (CSC) Inhibitors in Oncology—A Promise for a Better Therapeutic Outcome: State of the Art and Future Perspectives. J. Med. Chem. 2020, 63, 15279–15307. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent cancer cells: The deadly survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Javali, A.; Lakshmanan, V.; Palakodeti, D.; Sambasivan, R. Modulation of β-catenin levels regulates cranial neural crest patterning and dispersal into first pharyngeal arch. Dev. Dyn. 2020, 249, 1347–1364. [Google Scholar] [CrossRef]
- Heppt, M.V.; Wang, J.X.; Hristova, D.M.; Wei, Z.; Li, L.; Evans, B.; Beqiri, M.; Zaman, S.; Zhang, J.; Irmler, M.; et al. MSX1-Induced Neural Crest-Like Reprogramming Promotes Melanoma Progression. J. Investig. Dermatol. 2018, 138, 141–149. [Google Scholar] [CrossRef]
- Zhou, Y.; Snead, M.L. Derivation of cranial neural crest-like cells from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2008, 376, 542–547. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Fukunaga-Kalabis, M.; Yu, H.; Xu, X.; Kong, J.; Lee, J.T.; Herlyn, M. Human dermal stem cells differentiate into functional epidermal melanocytes. J. Cell Sci. 2010, 123 Pt 6, 853–860. [Google Scholar] [CrossRef]
- Lee, B.W.L.; Ghode, P.; Ong, D.S.T. Redox regulation of cell state and fate. Redox Biol. 2019, 25, 101056. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [PubMed]
- Kazantseva, J.; Sadam, H.; Neuman, T.; Palm, K. Targeted alternative splicing of TAF4: A new strategy for cell reprogramming. Sci. Rep. 2016, 6, 30852. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, S.; Lee, J.; Lee, C.; Kang, H.; Kang, J.; Lee, J.S.; Shin, E.; Youn, H.; Youn, B. Lipid metabolism in cancer stem cells: Reprogramming, mechanisms, crosstalk, and therapeutic approaches. Cell. Oncol. 2025, 48, 1181–1201. [Google Scholar] [CrossRef] [PubMed]
- Mortoglou, M.; Miralles, F.; Arisan, E.D.; Dart, A.; Jurcevic, S.; Lange, S.; Uysal-Onganer, P. microRNA-21 Regulates Stemness in Pancreatic Ductal Adenocarcinoma Cells. Int. J. Mol. Sci. 2022, 23, 1275. [Google Scholar] [CrossRef]
- Tesfaye, A.A.; Azmi, A.S.; Philip, P.A. miRNA and Gene Expression in Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2019, 189, 58–70. [Google Scholar] [CrossRef]
- Tao, S.-C.; Guo, S.-C. Role of extracellular vesicles in tumour microenvironment. Cell Commun. Signal. 2020, 18, 163. [Google Scholar] [CrossRef]
- Canciello, A.; Cerveró-Varona, A.; Peserico, A.; Mauro, A.; Russo, V.; Morrione, A.; Giordano, A.; Barboni, B. “In medio stat virtus”: Insights into hybrid E/M phenotype attitudes. Front. Cell Dev. Biol. 2022, 10, 1038841. [Google Scholar] [CrossRef]
- Davies, A.; Zoubeidi, A.; Beltran, H.; Selth, L.A. The transcriptional and epigenetic landscape of cancer cell lineage plasticity. Cancer Discov. 2023, 13, 1771–1788. [Google Scholar] [CrossRef]
- Bocci, F.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Deciphering the dynamics of epithelial-mesenchymal transition and cancer stem cells in tumor progression. Curr. Stem Cell Rep. 2019, 5, 11–21. [Google Scholar] [CrossRef]
- Mir, R.; Javid, J.; Ullah, M.F.; Alrdahe, S.; Altedlawi, I.A.; Mustafa, S.K.; Jalal, M.M.; Altayar, M.A.; Albalawi, A.D.; Abunab, M.K. Metabolic reprogramming and functional crosstalk within the tumor microenvironment (TME) and A Multi-omics anticancer approach. Med. Oncol. 2025, 42, 373. [Google Scholar] [CrossRef]
- Zheng, X.; Yu, C.; Xu, M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front. Oncol. 2021, 11, 678333. [Google Scholar] [CrossRef] [PubMed]
- Fico, F.; Santamaria-Martínez, A. The Tumor Microenvironment as a Driving Force of Breast Cancer Stem Cell Plasticity. Cancers 2020, 12, 3863. [Google Scholar] [CrossRef] [PubMed]
- Hlavca, S.; Chan, W.H.; Engel, R.M.; Abud, H.E. Clusterin: A marker and mediator of chemoresistance in colorectal cancer. Cancer Metastasis Rev. 2024, 43, 379–391. [Google Scholar] [CrossRef]
- Lee, Y. Role of Vitamin C in Targeting Cancer Stem Cells and Cellular Plasticity. Cancers 2023, 15, 5657. [Google Scholar] [CrossRef]
- Verona, F.; Di Bella, S.; Schirano, R.; Manfredi, C.; Angeloro, F.; Bozzari, G.; Todaro, M.; Giannini, G.; Stassi, G.; Veschi, V. Cancer stem cells and tumor-associated macrophages as mates in tumor progression: Mechanisms of crosstalk and advanced bioinformatic tools to dissect their phenotypes and interaction. Front. Immunol. 2025, 16, 1529847. [Google Scholar] [CrossRef]
- Mestiri, S.; Sami, A.; Sah, N.; El-Ella, D.M.A.; Khatoon, S.; Shafique, K.; Raza, A.; Mathkor, D.M.; Haque, S. Cellular plasticity and non-small cell lung cancer: Role of T and NK cell immune evasion and acquisition of resistance to immunotherapies. Cancer Metastasis Rev. 2025, 44, 27. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Vandyck, H.H.; Hillen, L.M.; Bosisio, F.M.; van den Oord, J.; Zur Hausen, A.; Winnepenninckx, V. Rethinking the biology of metastatic melanoma: A holistic approach. Cancer Metastasis Rev. 2021, 40, 603–624. [Google Scholar] [CrossRef]
- Zheng, X.; Dai, F.; Feng, L.; Zou, H.; Feng, L.; Xu, M. Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression. Front. Oncol. 2021, 11, 617597. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, J.; Li, Y.; Zhang, K.; Wang, T.; Bo, Z.; Lu, S.; Rodríguez, R.A.; Wei, R.; Zhu, M.; et al. EMT and cancer stem cells: Drivers of therapy resistance and promising therapeutic targets. Drug Resist. Updates 2025, 83, 101276. [Google Scholar] [CrossRef]
- Ji, Y.; Hao, H.; Reynolds, K.; McMahon, M.; Zhou, C.J. Wnt Signaling in Neural Crest Ontogenesis and Oncogenesis. Cells 2019, 8, 1173. [Google Scholar] [CrossRef] [PubMed]
- Duband, J.L. Diversity in the molecular and cellular strategies of epithelium-to-mesenchyme transitions: Insights from the neural crest. Cell Adh. Migr. 2010, 4, 458–482. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, T.; Kawamura, N.; Watanabe, N.; Kitagawa, D.; Goshima, N.; Kunisada, T. Sox10 Functions as an Inducer of the Direct Conversion of Keratinocytes Into Neural Crest Cells. Stem Cells Dev. 2020, 29, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Monsoro-Burq, A.H. PAX transcription factors in neural crest development. Semin. Cell Dev. Biol. 2015, 44, 87–96. [Google Scholar] [CrossRef]
- Mayanil, C.S. Transcriptional and epigenetic regulation of neural crest induction during neurulation. Dev. Neurosci. 2013, 35, 361–372. [Google Scholar] [CrossRef]
- Aljouda, N.A.; Shrestha, D.; DeVaux, C.; Olsen, R.R.; Alleboina, S.; Walker, M.; Cheng, Y.; Freeman, K.W. Transcription factor 4 is a key mediator of oncogenesis in neuroblastoma by promoting MYC activity. Mol. Oncol. 2025, 19, 808–824. [Google Scholar] [CrossRef]
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533. [Google Scholar] [CrossRef]
- Boda, E.; Lorenzati, M.; Parolisi, R.; Harding, B.; Pallavicini, G.; Bonfanti, L.; Moccia, A.; Bielas, S.; Di Cunto, F.; Buffo, A. Molecular and functional heterogeneity in dorsal and ventral oligodendrocyte progenitor cells of the mouse forebrain in response to DNA damage. Nat. Commun. 2022, 13, 2331. [Google Scholar] [CrossRef]
- Dela Cruz, M.C.P.; Medina, P.M.B. Epithelial-mesenchymal transition (EMT) and its role in acquired epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) chemoresistance in non-small cell lung cancer (NSCLC). Cancer Pathog. Ther. 2025, 3, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M.; Haley, J.D. A systems view of epithelial–mesenchymal transition signaling states. Clin. Exp. Metastasis 2011, 28, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 25, 843–854. [Google Scholar] [CrossRef]
- Shenoy, A.K.; Jin, Y.; Luo, H.; Tang, M.; Pampo, C.; Shao, R.; Siemann, D.W.; Wu, L.; Heldermon, C.D.; Law, B.K. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J. Clin. Investig. 2016, 126, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818. [Google Scholar] [CrossRef] [PubMed]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Bria, E.; Carbognin, L.; Malaguti, P.; Ferretti, G.; Cognetti, F.; Milella, M.; Ciuffreda, L. AXL receptor in breast cancer: Molecular involvement and therapeutic limitations. Int. J. Mol. Sci. 2020, 21, 8419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef]
- Zhang, A.; Aslam, H.; Sharma, N.; Warmflash, A.; Fakhouri, W.D. Conservation of epithelial-to-mesenchymal transition process in neural crest cells and metastatic cancer. Cells Tissues Organs 2021, 210, 151–172. [Google Scholar] [CrossRef]
- Zhao, R.; Moore, E.L.; Gogol, M.M.; Unruh, J.R.; Yu, Z.; Scott, A.R.; Wang, Y.; Rajendran, N.K.; Trainor, P.A. Identification and characterization of intermediate states in mammalian neural crest cell epithelial to mesenchymal transition and delamination. Elife 2024, 13, RP92844. [Google Scholar] [CrossRef]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef]
- Schuhwerk, H.; Brabletz, T. Mutual regulation of TGFβ-induced oncogenic EMT, cell cycle progression and the DDR. Semin. Cancer Biol. 2023, 97, 86–103. [Google Scholar] [CrossRef]
- László, Z.I.; Lele, Z. Flying under the radar: CDH2 (N-cadherin), an important hub molecule in neurodevelopmental and neurodegenerative diseases. Front. Neurosci. 2022, 16, 972059. [Google Scholar] [CrossRef]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: The two sides of the same coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef] [PubMed]
- Pfeiferová, L.; Španko, M.; Šáchová, J.; Hradilová, M.; Pienta, K.J.; Valach, J.; Machoň, V.; Výmolová, B.; Šedo, A.; Bušek, P. The HOX code of human adult fibroblasts reflects their ectomesenchymal or mesodermal origin. Histochem. Cell Biol. 2025, 163, 38. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Thadani, N.; Gallagher, E.E.; Azzaro, I.; Bodnar, C.M.; McCarty, C.P.; Romano, G.; Webster, M.R.; Capparelli, C. Plasticity and Functional Heterogeneity of Cancer-Associated Fibroblasts. Cancer Res. 2025, 85, 3378–3398. [Google Scholar] [CrossRef]
- Garg, M. Emerging roles of epithelial-mesenchymal plasticity in invasion-metastasis cascade and therapy resistance. Cancer Metastasis Rev. 2022, 41, 131–145. [Google Scholar] [CrossRef]
- Waryah, C.; Alves, E.; Mazzieri, R.; Dolcetti, R.; Thompson, E.W.; Redfern, A.; Blancafort, P. Unpacking the Complexity of Epithelial Plasticity: From Master Regulator Transcription Factors to Non-Coding RNAs. Cancers 2023, 15, 3152. [Google Scholar] [CrossRef]
- Wang, X.; Xue, X.; Pang, M.; Yu, L.; Qian, J.; Li, X.; Tian, M.; Lyu, A.; Lu, C.; Liu, Y. Epithelial–mesenchymal plasticity in cancer: Signaling pathways and therapeutic targets. MedComm 2024, 5, e659. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Jiao, S.; Han, G.; Zhu, J.; Liu, T. Functional and clinical characteristics of focal adhesion kinases in cancer progression. Front. Cell Dev. Biol. 2022, 10, 1040311. [Google Scholar] [CrossRef]
- Wium, M.; Ajayi-Smith, A.F.; Paccez, J.D.; Zerbini, L.F. The role of the receptor tyrosine kinase Axl in carcinogenesis and development of therapeutic resistance: An overview of molecular mechanisms and future applications. Cancers 2021, 13, 1521. [Google Scholar] [CrossRef]
- Sang, Y.B.; Kim, J.-H.; Kim, C.-G.; Hong, M.H.; Kim, H.R.; Cho, B.C.; Lim, S.M. The development of AXL inhibitors in lung cancer: Recent progress and challenges. Front. Oncol. 2022, 12, 811247. [Google Scholar] [CrossRef]
- Goyette, M.-A.; Côté, J.-F. AXL receptor tyrosine kinase as a promising therapeutic target directing multiple aspects of cancer progression and metastasis. Cancers 2022, 14, 466. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bo, Y.; Xue, D.; Qin, L. Ferroptosis-immune crosstalk in cervical cancer: Mechanisms and therapeutic implications. Front. Immunol. 2025, 16, 1657905. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuang, H.; Yang, X.; Guo, Z.; Zhou, K.; Liu, N.; An, Y.; Chen, Y.; Zhang, Z.; Wang, M.; et al. Exploring the Mechanism of Ferroptosis Induction by Sappanone A in Cancer: Insights into the Mitochondrial Dysfunction Mediated by NRF2/xCT/GPX4 Axis. Int. J. Biol. Sci. 2024, 20, 5145–5161. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Li, Z.; Sun, K.; Liu, Y.; Fang, Z.; Sun, S.; Li, C.; Wang, Z. Mitochondrial Regulation of Ferroptosis in Cancer Cells. Int. J. Biol. Sci. 2025, 21, 2179–2200. [Google Scholar] [CrossRef]
- She, W.; Su, J.; Ma, W.; Ma, G.; Li, J.; Zhang, H.; Qiu, C.; Li, X. Natural products protect against spinal cord injury by inhibiting ferroptosis: A literature review. Front. Pharmacol. 2025, 16, 1557133. [Google Scholar] [CrossRef]
- Song, T.; Yu, Z.; Shen, Q.; Xu, Y.; Hu, H.; Liu, J.; Zeng, K.; Lei, J.; Yu, L. Pharmacodynamic and Toxicity Studies of 6-Isopropyldithio-2’-guanosine Analogs in Acute T-Lymphoblastic Leukemia. Cancers 2024, 16, 1614. [Google Scholar] [CrossRef]
- Chen, I.P.; Henning, S.; Bender, M.; Degenhardt, S.; Mhamdi Ghodbani, M.; Bergmann, A.K.; Volkmer, B.; Brockhoff, G.; Wege, A.K.; Greinert, R. Detection of Human Circulating and Extracellular Vesicle-Derived miRNAs in Serum of Humanized Mice Transplanted with Human Breast Cancer (HER2(+) and TNBC) Cells-A Proof of Principle Investigation. Int. J. Mol. Sci. 2025, 26, 3629. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Liu, C.; Zhao, Y. Extracellular vesicles in cancers: Mechanisms, biomarkers, and therapeutic strategies. MedComm 2024, 5, e70009. [Google Scholar] [CrossRef]
- Li, Z.; Gao, Y.; Cao, Y.; He, F.; Jiang, R.; Liu, H.; Cai, H.; Zan, T. Extracellular RNA in melanoma: Advances, challenges, and opportunities. Front. Cell Dev. Biol. 2023, 11, 1141543. [Google Scholar] [CrossRef]
- Rahimian, S.; Mirkazemi, K.; Kamalinejad, A.; Doroudian, M. Exosome-based advances in pancreatic cancer: The potential of mesenchymal stem cells. Crit. Rev. Oncol. Hematol. 2025, 207, 104594. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, A.K.; Moglad, E.; Afzal, M.; Babu, M.A.; Goyal, K.; Roopashree, R.; Kaur, I.; Kumar, S.; Kumar, M.; Chauhan, A.S.; et al. Liquid biopsies and exosomal ncRNA: Transforming pancreatic cancer diagnostics and therapeutics. Clin. Chim. Acta 2025, 567, 120105. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhong, L.; Momeni, M.R. MicroRNA-155 and its exosomal form: Small pieces in the gastrointestinal cancers puzzle. Cell Biol. Toxicol. 2024, 40, 77. [Google Scholar] [CrossRef] [PubMed]
- El-Moaty, Z.A.; Wasef, M.W.; Elsayed, Y.A.; Shafiq, A.S.; Elnagar, M.A.; Mohamed, H.R.; Elesawyf, A.E.; Abdel Mageedg, S.S.; Mohammed, O.A.; Abdel-Reheim, M.A.; et al. Unraveling the therapeutic landscape of miRNAs in pancreatic cancer. Naunyn Schmiedebergs Arch. Pharmacol. 2025, 398, 14941–14959. [Google Scholar] [CrossRef]
- Pani, G.; Giannoni, E.; Galeotti, T.; Chiarugi, P. Redox-based escape mechanism from death: The cancer lesson. Antioxid. Redox Signal. 2009, 11, 2791–2806. [Google Scholar] [CrossRef]
- Chang, Y.; Li, G.; Zhai, Y.; Huang, L.; Feng, Y.; Wang, D.; Zhang, W.; Hu, H. Redox regulator GLRX is associated with tumor immunity in glioma. Front. Immunol. 2020, 11, 580934. [Google Scholar] [CrossRef]
- Kennel, K.B.; Greten, F.R. Immune cell-produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Li, Q.; He, G.; Yu, Y.; Li, X.; Peng, X.; Yang, L. Exosome crosstalk between cancer stem cells and tumor microenvironment: Cancer progression and therapeutic strategies. Stem Cell Res. Ther. 2024, 15, 449. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, C.; Zeng, C.; Sun, C.; Xia, L.; Wang, G.; Chen, X.; Zhang, B.; Liu, J.; Ding, Z.-y. Cancer stem cells and niches: Challenges in immunotherapy resistance. Mol. Cancer 2025, 24, 52. [Google Scholar] [CrossRef]
- Liu, H.; Liang, Z.; Wang, F.; Zhou, C.; Zheng, X.; Hu, T.; He, X.; Wu, X.; Lan, P. Exosomes from mesenchymal stromal cells reduce murine colonic inflammation via a macrophage-dependent mechanism. JCI Insight 2019, 4, e131273. [Google Scholar] [CrossRef]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021, 42, 101928. [Google Scholar] [CrossRef]
- Scheinberg, T.; Mak, B.; Butler, L.; Selth, L.; Horvath, L.G. Targeting lipid metabolism in metastatic prostate cancer. Ther. Adv. Med. Oncol. 2023, 15, 17588359231152839. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Mantovani, A.; Targher, G.; Baffy, G. Nonalcoholic fatty liver disease and chronic kidney disease: Epidemiology, pathogenesis, and clinical and research implications. Int. J. Mol. Sci. 2022, 23, 13320. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulou, E.; Thymis, J.; Lampsas, S.; Pavlidis, G.; Katogiannis, K.; Vlachomitros, D.; Katsanaki, E.; Kostelli, G.; Pililis, S.; Pliouta, L. The triad of risk: Linking MASLD, cardiovascular disease and type 2 diabetes; from pathophysiology to treatment. J. Clin. Med. 2025, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Huo, K.-G.; D’Arcangelo, E.; Tsao, M.-S. Patient-derived cell line, xenograft and organoid models in lung cancer therapy. Transl. Lung Cancer Res. 2020, 9, 2214. [Google Scholar] [CrossRef]
- Hu, M.; Cui, Y.; Huang, Q.; Chu, K.; McKinzie, S.; Patrick, M.; Iyengar, S.; Abuduli, M.; Spatz, M.; Joshi, N. SPACE: Spatially resolved multiomic analysis for high-throughput CRISPR screening in 3D models. bioRxiv 2025. [Google Scholar] [CrossRef]
- Liu, L.; Chen, A.; Li, Y.; Mulder, J.; Heyn, H.; Xu, X. Spatiotemporal omics for biology and medicine. Cell 2024, 187, 4488–4519. [Google Scholar] [CrossRef]
- Hui, T.; Zhou, J.; Yao, M.; Xie, Y.; Zeng, H. Advances in Spatial Omics Technologies. Small Methods 2025, 9, 2401171. [Google Scholar] [CrossRef]
- Ge, Y.; Weng, H.; Sun, Y.; Wu, M. Integrated single-cell and spatial transcriptomic analysis reveals YBX1 drives immune regulation in GBM progression. Heliyon 2024, 10, e29451. [Google Scholar] [CrossRef]
- Ali, H.; Zhou, N.; Chen, L.; van Hijfte, L.; Karri, V.; Zhou, Y.; Habashy, K.; Arrieta, V.A.; Kim, K.-S.; Duffy, J.; et al. Targeting CHEK2-YBX1&YBX3 regulatory hub to potentiate immune checkpoint blockade response in gliomas. bioRxiv 2025. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Q.; Ni, K.; Zhang, P.; Liu, Y.; Xie, J.; Ji, W.; Cheng, C.; Zhou, Q. Combining single-cell sequencing and spatial transcriptome sequencing to identify exosome-related features of glioblastoma and constructing a prognostic model to identify BARD1 as a potential therapeutic target for GBM patients. Front. Immunol. 2023, 14, 1263329. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Xu, P.; Ye, Z.; Liu, W. STmiR: A Novel XGBoost-based framework for spatially resolved miRNA activity prediction in cancer transcriptomics. PLoS ONE 2025, 20, e0322082. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, H.; Wang, X.; Yang, Y.; Zhong, K.; Zhang, X.; Li, H. MSC-EVs and UCB-EVs promote skin wound healing and spatial transcriptome analysis. Sci. Rep. 2025, 15, 4006. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef]
- Sinha, D.; Saha, P.; Samanta, A.; Bishayee, A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules 2020, 10, 1561. [Google Scholar] [CrossRef]
- Hu, K.; Chen, F. Identification of significant pathways in gastric cancer based on protein-protein interaction networks and cluster analysis. Genet. Mol. Biol. 2012, 35, 701–708. [Google Scholar] [CrossRef]
- Neuendorf, H.M.; Simmons, J.L.; Boyle, G.M. Therapeutic targeting of anoikis resistance in cutaneous melanoma metastasis. Front. Cell Dev. Biol. 2023, 11, 1183328. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Huang, S.; Qin, X.; Fu, S.; Hu, J.; Jiang, Z.; Hu, M.; Zhang, B.; Liu, J.; Chen, Y.; Wang, M.; et al. STAMBPL1/TRIM21 Balances AXL Stability Impacting Mesenchymal Phenotype and Immune Response in KIRC. Adv. Sci. 2025, 12, e2405083. [Google Scholar] [CrossRef]
- Hänze, J.; Kessel, F.; Di Fazio, P.; Hofmann, R.; Hegele, A. Effects of multi and selective targeted tyrosine kinase inhibitors on function and signaling of different bladder cancer cells. Biomed. Pharmacother. 2018, 106, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, S.; Bhat, S.; Upadhya, D.; Mascarenhas, R.; Kabekkodu, S.P. Biological functions of extracellular vesicle double C2-like domain beta in cervical cancer. Sci. Rep. 2025, 15, 477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, N.; Ding, C.; Zhang, H.; Liu, D.; Liu, S. Ferroptosis and EMT resistance in cancer: A comprehensive review of the interplay. Front. Oncol. 2024, 14, 1344290. [Google Scholar] [CrossRef]
- Xu, Y.; Lou, D.; Chen, P.; Li, G.; Usoskin, D.; Pan, J.; Li, F.; Huang, S.; Hess, C.; Tang, R.; et al. Single-cell MultiOmics and spatial transcriptomics demonstrate neuroblastoma developmental plasticity. Dev. Cell 2025, 60, 2248–2263.e11. [Google Scholar] [CrossRef]
- Toneff, M.; Sreekumar, A.; Tinnirello, A.; Hollander, P.D.; Habib, S.; Li, S.; Ellis, M.; Xin, L.; Mani, S.; Rosen, J. The Z-cad dual fluorescent sensor detects dynamic changes between the epithelial and mesenchymal cellular states. BMC Biol. 2016, 14, 47. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R. NFE2L2 and ferroptosis resistance in cancer therapy. Cancer Drug Resist. 2024, 7, 41. [Google Scholar] [CrossRef]
- Hou, J.; Wang, B.; Li, J.; Liu, W. Ferroptosis and its role in gastric and colorectal cancers. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2024, 28, 183–196. [Google Scholar] [CrossRef]
- Veglia Tranchese, R.; Battista, S.; Cerchia, L.; Fedele, M. Ferroptosis in Cancer: Epigenetic Control and Therapeutic Opportunities. Biomolecules 2024, 14, 1443. [Google Scholar] [CrossRef]
- Pei, Y.; Qian, Y.; Wang, H.; Tan, L. Epigenetic Regulation of Ferroptosis-Associated Genes and Its Implication in Cancer Therapy. Front. Oncol. 2022, 12, 771870. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Z.; Ruan, S.; Yan, Q.; Chen, Y.; Cui, J.; Wang, X.; Huang, S.; Hou, B. Regulation of iron metabolism and ferroptosis in cancer stem cells. Front. Oncol. 2023, 13, 1251561. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Jena, B.C.; Mandal, M. The emerging roles of exosomes in anti-cancer drug resistance and tumor progression: An insight towards tumor-microenvironment interaction. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188488. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Ochiya, T. Interactions between cancer cells and normal cells via miRNAs in extracellular vesicles. Cell Mol. Life Sci. 2015, 72, 1849–1861. [Google Scholar] [CrossRef]
- Lattmann, E.; Levesque, M.P. The Role of Extracellular Vesicles in Melanoma Progression. Cancers 2022, 14, 3086. [Google Scholar] [CrossRef]
- Lin, W.; Fang, J.; Wei, S.; He, G.; Liu, J.; Li, X.; Peng, X.; Li, D.; Yang, S.; Li, X.; et al. Extracellular vesicle-cell adhesion molecules in tumours: Biofunctions and clinical applications. Cell Commun. Signal. 2023, 21, 246. [Google Scholar] [CrossRef]
- Thong, T.; Wang, Y.; Brooks, M.D.; Lee, C.T.; Scott, C.; Balzano, L.; Wicha, M.S.; Colacino, J.A. Hybrid Stem Cell States: Insights Into the Relationship Between Mammary Development and Breast Cancer Using Single-Cell Transcriptomics. Front. Cell Dev. Biol. 2020, 8, 288. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Liu, H.-M.; Xiang, J.-Y.; Zhou, X.-C.; Wang, D.; Chen, R.-Y.; Tan, W.-L.; Liang, L.-Q.; Liu, L.-L.; Shi, M.-J. Alpha lipoamide inhibits diabetic kidney fibrosis via improving mitochondrial function and regulating RXRα expression and activation. Acta Pharmacol. Sin. 2023, 44, 1051–1065. [Google Scholar] [CrossRef]
- Fu, A.; Li, J.; Ding, Q.; Guo, R.; Pi, A.; Yang, W.; Chen, Y.; Dou, X.; Song, Z.; Li, S. Upregulation of 4-hydroxynonenal contributes to the negative effect of n-6 polyunsaturated fatty acid on alcohol-induced liver injury and hepatic steatosis. J. Agric. Food Chem. 2022, 70, 6418–6428. [Google Scholar] [CrossRef]
- Salehi, F.; Kavoosi, G.; Jacobs, P.; Bennett, N.C.; Ahmadian, S.; Bastani, B.; Gholami, M. The road to a long lifespan in the Persian squirrel, a natural model for extended longevity: Resisting free radical stress and healthy phospholipids. GeroScience 2025, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Liu, Q.; Wong, P.P.; Song, E. Cancer stem cell mimicry for immune evasion and therapeutic resistance. Cell Stem Cell 2024, 31, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Paul, S. Exosome-based cancer stem cell communication: Implication for detecting and eliminating cancer stem cells. MedComm—Future Med. 2022, 1, e23. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Nag, S.; Mukherjee, S.; Kulkarni, M.; Chandane, P.; Mandal, D.; Mukerjee, N.; Mirgh, D.; Anand, K.; Adhikari, M.D.; et al. Role of Exosomes in Epithelial-Mesenchymal Transition. ACS Appl. Bio Mater. 2024, 7, 44–58. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef]
- Hardy, K.; Wu, F.; Tu, W.; Zafar, A.; Boulding, T.; McCuaig, R.; Sutton, C.R.; Theodoratos, A.; Rao, S. Identification of chromatin accessibility domains in human breast cancer stem cells. Nucleus 2016, 7, 50–67. [Google Scholar] [CrossRef][Green Version]
- Schick, B.; Pillong, L.; Wenzel, G.; Wemmert, S. Neural Crest Stem Cells in Juvenile Angiofibromas. Int. J. Mol. Sci. 2022, 23, 1932. [Google Scholar] [CrossRef]
- Tang, Y.; Durand, S.; Dalle, S.; Caramel, J. EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers 2020, 12, 2154. [Google Scholar] [CrossRef]
- Tomei, S.; Ibnaof, O.; Ravindran, S.; Ferrone, S.; Maccalli, C. Cancer stem cells are possible key players in regulating anti-tumor immune responses: The role of immunomodulating molecules and microRNAs. Cancers 2021, 13, 1674. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, L.; Liu, H.; Luo, X. Cancer stem cell-targeted therapeutic approaches for overcoming trastuzumab resistance in HER2-positive breast cancer. Stem Cells 2021, 39, 1125–1136. [Google Scholar] [CrossRef]
- Ferreres, J.R.; Vinyals, A.; Campos-Martin, R.; Espín, R.; Podlipnik, S.; Ramos, R.; Bertran, E.; Carrera, C.; Marcoval, J.; Malvehy, J.; et al. PRRX1 silencing is required for metastatic outgrowth in melanoma and is an independent prognostic of reduced survival in patients. Mol. Oncol. 2024, 18, 2471–2494. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Honda, M.; Tomita, M.; Li, L.; Adawy, A.; Xue, W.; Hibi, T. Intravital Imaging of Immune Responses in the Cancer Microenvironment. Cancer Med. 2025, 14, e70899. [Google Scholar] [CrossRef] [PubMed]
- Greer, S.E.; Haller, S.J.; Lee, D.; Dudley, A.T. N-cadherin and β1 integrin coordinately regulate growth plate cartilage architecture. Mol. Biol. Cell 2024, 35, ar49. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Schinelli, S. Integrins and Exosomes, a Dangerous Liaison in Cancer Progression. Cancers 2017, 9, 95. [Google Scholar] [CrossRef]
- Vakili-Ghartavol, Z.; Deli, H.; Shadboorestan, A.; Sahebnasagh, R.; Motevaseli, E.; Ghahremani, M.H. Exosomes and their distinct integrins transfer the characteristics of oxaliplatin- and 5-FU-resistant behaviors in colorectal cancer cells. Mol. Med. 2025, 31, 49. [Google Scholar] [CrossRef]
- Hasnat, M.A.; Ohmi, Y.; Yesmin, F.; Kaneko, K.; Kambe, M.; Kitaura, Y.; Ito, T.; Imao, Y.; Kano, K.; Mishiro-Sato, E.; et al. Action Mechanisms of Exosomes Derived from GD3/GD2-Positive Glioma Cells in the Regulation of Phenotypes and Intracellular Signaling: Roles of Integrins. Int. J. Mol. Sci. 2024, 25, 12752. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ishikawa, E.; Sugii, N.; Matsuda, M. Therapeutic Strategies for Overcoming Immunotherapy Resistance Mediated by Immunosuppressive Factors of the Glioblastoma Microenvironment. Cancers 2020, 12, 1960. [Google Scholar] [CrossRef]
- Currenti, J.; Mishra, A.; Wallace, M.; George, J.; Sharma, A. Immunosuppressive mechanisms of oncofetal reprogramming in the tumor microenvironment: Implications in immunotherapy response. Biochem. Soc. Trans. 2023, 51, 597–612. [Google Scholar] [CrossRef]
- Wilczyński, M.; Wilczyński, J.; Nowak, M. MiRNAs as Regulators of Immune Cells in the Tumor Microenvironment of Ovarian Cancer. Cells 2024, 13, 1343. [Google Scholar] [CrossRef]
- Xing, Y.; Ruan, G.; Ni, H.; Qin, H.; Chen, S.; Gu, X.; Shang, J.; Zhou, Y.; Tao, X.; Zheng, L. Tumor Immune Microenvironment and Its Related miRNAs in Tumor Progression. Front. Immunol. 2021, 12, 624725. [Google Scholar] [CrossRef]
- Liu, P.; Sun, Z. Chemokines and their receptors in the esophageal carcinoma tumor microenvironment: Key factors for metastasis and progression. Front. Oncol. 2025, 15, 1523751. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, B.; Huang, B.; Zhang, K.; Guo, F.; Wang, Z.; Shang, D. A stumbling block in pancreatic cancer treatment: Drug resistance signaling networks. Front. Cell Dev. Biol. 2025, 12, 1462808. [Google Scholar] [CrossRef] [PubMed]
- Zare, E.; Yaghoubi, S.M.; Khoshnazar, M.; Jafari Dargahlou, S.; Machhar, J.S.; Zheng, Z.; Duijf, P.H.G.; Mansoori, B. MicroRNAs in Cancer Immunology: Master Regulators of the Tumor Microenvironment and Immune Evasion, with Therapeutic Potential. Cancers 2025, 17, 2172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiang, Z.; Tang, D. The value of exosome-derived noncoding RNAs in colorectal cancer proliferation, metastasis, and clinical applications. Clin. Transl. Oncol. 2022, 24, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Sharma, A.; Patne, K.; Tabasum, S.; Suryavanshi, J.; Rawat, L.; Machaalani, M.; Eid, M.; Singh, R.P.; Choueiri, T.K.; et al. AXL signaling in cancer: From molecular insights to targeted therapies. Signal Transduct. Target. Ther. 2025, 10, 37. [Google Scholar] [CrossRef]
- Zuo, X.; Zhang, H.; Zhou, T.; Duan, Y.; Shou, H.; Yu, S.; Gao, C. Spheroids of Endothelial Cells and Vascular Smooth Muscle Cells Promote Cell Migration in Hyaluronic Acid and Fibrinogen Composite Hydrogels. Research 2020, 2020, 8970480. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, S.; Fu, J.-L.; Deng, X.-X.; Guo, Y.; Guo, Z.-W.; Han, S.-Y. AXL promotes malignant phenotypes in tumor cells and might be a potential antitumor target of natural products. Adv. Chin. Med. 2024, 1, 179–189. [Google Scholar] [CrossRef]
- Koeberle, S.C.; Kipp, A.P.; Stuppner, H.; Koeberle, A. Ferroptosis-modulating small molecules for targeting drug-resistant cancer: Challenges and opportunities in manipulating redox signaling. Med. Res. Rev. 2023, 43, 614–682. [Google Scholar] [CrossRef]
- Lee, J.; Seo, Y.; Roh, J.-L. Emerging Therapeutic Strategies Targeting GPX4-Mediated Ferroptosis in Head and Neck Cancer. Int. J. Mol. Sci. 2025, 26, 6452. [Google Scholar] [CrossRef]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef]
- Chen, T.; Leng, J.; Tan, J.; Zhao, Y.; Xie, S.; Zhao, S.; Yan, X.; Zhu, L.; Luo, J.; Kong, L. Discovery of novel potent covalent glutathione peroxidase 4 inhibitors as highly selective ferroptosis inducers for the treatment of triple-negative breast cancer. J. Med. Chem. 2023, 66, 10036–10059. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.L. SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer. Antioxidants 2022, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, Z.; Chen, S.; Liang, Y.; Zhu, K.; Su, N.; Liu, T.; Zhao, B. Enhancing Ferroptosis-Mediated Radiosensitization via Synergistic Disulfidptosis Induction. ACS Nano 2025, 19, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, T.; Zhao, C.; Li, G. The Regulation of Exosome Generation and Function in Physiological and Pathological Processes. Int. J. Mol. Sci. 2023, 25, 255. [Google Scholar] [CrossRef]
- Luo, Y.W.; Liu, C.G.; Kirby, J.A.; Chu, C.; Zang, D.; Chen, J. The Emerging Role of Extracellular Vesicle-Derived lncRNAs and circRNAs in Tumor and Mesenchymal Stem Cells: The Biological Functions and Potential for Clinical Application. Cancers 2025, 17, 2186. [Google Scholar] [CrossRef]
- Chuang, Y.T.; Shiau, J.P.; Tang, J.Y.; Farooqi, A.A.; Chang, F.R.; Tsai, Y.H.; Yen, C.Y.; Chang, H.W. Connection of Cancer Exosomal LncRNAs, Sponging miRNAs, and Exosomal Processing and Their Potential Modulation by Natural Products. Cancers 2023, 15, 2215. [Google Scholar] [CrossRef]
- Pavić, J.N.; Živanović, M.N.; Virijević, K.D.; Tanasković, I.D.; Stanković, V.D.; Marić, N.T.; Cvetković, D.M.; Filipović, N.D. INFLUENCE OF CYTOSTATICS ON RELATIVE GENE EXPRESSION IN REDOX STATUS, APOPTOSIS AND MIGRATION COLORECTAL CARCINOMA MODEL SYSTEM. Kragujev. J. Sci. 2023, 45, 159–177. [Google Scholar] [CrossRef]
- Pavic, J.; Zivanovic, M.; Virijević, K.; Tanasković, I.; Stanković, V.; Maric, N.; Cvetkovic, D.; Filipovic, N. Influence of cytostatics on relative gene expression in redox status, apoptosis and migration colorectal carcinoma model system. Kragujev. J. Sci. 2023, 45, 159–177. [Google Scholar] [CrossRef]
- Li, C.; Xing, S.; Zhang, D.; Li, R.; Li, Q.; Luo, H.; Liu, F. Exosomal long non-coding RNAs in gastrointestinal cancer: Chemoresistance mediators and therapeutic targets. J. Transl. Med. 2025, 23, 889. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, M.; Zheng, X.; He, Y.; Ma, X.; Li, J.; Pu, K. Application of exosome engineering modification in targeted delivery of therapeutic drugs. Biochem. Pharmacol. 2023, 215, 115691. [Google Scholar] [CrossRef] [PubMed]
- Shirani, N.; Abdi, N.; Chehelgerdi, M.; Yaghoobi, H.; Chehelgerdi, M. Investigating the role of exosomal long non-coding RNAs in drug resistance within female reproductive system cancers. Front. Cell Dev. Biol. 2025, 13, 1485422. [Google Scholar] [CrossRef]
- Lucarini, V.; Angiolini, V.; Nardozi, D.; Benvenuto, M.; Focaccetti, C.; Mancini, P.; Splendiani, E.; Autilio, T.M.; Cortese, C.; Bei, R.; et al. In vitro synergistic effect of AXL, FAK and ErbB receptors inhibitors for head and neck cancer. Biol. Direct 2025, 20, 77. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, X.; Wang, Y.; Yuan, J.; Li, J.; Hang, W.; Meng, H. SLC7A11 inhibits ferroptosis and downregulates PD-L1 levels in lung adenocarcinoma. Front. Immunol. 2024, 15, 1372215. [Google Scholar] [CrossRef]
- Yang, Y.C.; Jiang, Q.; Yang, K.P.; Wang, L.; Sethi, G.; Ma, Z. Extracellular vesicle-mediated ferroptosis, pyroptosis, and necroptosis: Potential clinical applications in cancer therapy. Cell Death Discov. 2024, 10, 23. [Google Scholar] [CrossRef]
- Wang, L.; He, J.; Hu, H.; Tu, L.; Sun, Z.; Liu, Y.; Luo, F. Lung CSC-derived exosomal miR-210-3p contributes to a pro-metastatic phenotype in lung cancer by targeting FGFRL1. J. Cell Mol. Med. 2020, 24, 6324–6339. [Google Scholar] [CrossRef]
- Chen, Q.; Xie, X. Association of Exosomal miR-210 with Signaling Pathways Implicated in Lung Cancer. Genes 2021, 12, 1248. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. (Landmark Ed.) 2014, 19, 687. [Google Scholar] [CrossRef] [PubMed]
- Koenders, S.T.; Wijaya, L.S.; Erkelens, M.N.; Bakker, A.T.; van der Noord, V.E.; van Rooden, E.J.; Burggraaff, L.; Putter, P.C.; Botter, E.; Wals, K. Development of a retinal-based probe for the profiling of retinaldehyde dehydrogenases in cancer cells. ACS Cent. Sci. 2019, 5, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sakamoto, S.; Saito, S.; Pae, S.; Yamada, Y.; Kanaoka, S.; Wei, J.; Goto, Y.; Sazuka, T.; Imamura, Y. The Regulation and Function of the Amino Acid Transporters LAT1, ASCT2, xCT in Urological Cancers. Receptors 2024, 3, 474–493. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, Y.; Xu, W.; Zhang, X.; Jiang, J. Engineered exosome-mediated delivery of circDIDO1 inhibits gastric cancer progression via regulation of MiR-1307-3p/SOCS2 Axis. J. Transl. Med. 2022, 20, 326. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Carcamo, S.; Agarwal, A.; Tung, N.; Humblin, É.; Goldberg, M.S.; Vyas, N.S.; Beaumont, K.G.; Demircioglu, D.; Sridhar, S. MacroH2A restricts inflammatory gene expression in melanoma cancer-associated fibroblasts by coordinating chromatin looping. Nat. Cell Biol. 2023, 25, 1332–1345. [Google Scholar] [CrossRef]
- Qin, Q.; Furong, W.; Baosheng, L. Multiple functions of hypoxia-regulated miR-210 in cancer. J. Exp. Clin. Cancer Res. 2014, 33, 50. [Google Scholar] [CrossRef]
- Lian, M.; Mortoglou, M.; Uysal-Onganer, P. Impact of Hypoxia-Induced miR-210 on Pancreatic Cancer. Curr. Issues Mol. Biol. 2023, 45, 9778–9792. [Google Scholar] [CrossRef]
- Wendling, D.; Verhoeven, F.; Chouk, M.; Prati, C. Le SARS-CoV-2 peut-il induire une arthrite réactionnelle? Rev. Rhum. Ed. Fr. 2021, 88, 326–328. [Google Scholar] [CrossRef]
- Schlangen, J.; Petko, C.; Hansen, J.H.; Michel, M.; Hart, C.; Uebing, A.; Fischer, G.; Becker, K.; Kramer, H.-H. Two-Dimensional Global Longitudinal Strain Rate Is a Preload Independent Index of Systemic Right Ventricular Contractility in Hypoplastic Left Heart Syndrome Patients After Fontan Operation. Circ. Cardiovasc. Imaging 2014, 7, 880–886. [Google Scholar] [CrossRef]
- Fernandes-Pontes, F.; Lobo, J.; Jeronimo, C.; Henrique, R. Identification of novel biomarkers in renal cell carcinoma. Expert Rev. Mol. Diagn. 2025, 25, 465–477. [Google Scholar] [CrossRef]
- Passaro, A.; Al Bakir, M.; Hamilton, E.G.; Diehn, M.; André, F.; Roy-Chowdhuri, S.; Mountzios, G.; Wistuba, I.I.; Swanton, C.; Peters, S. Cancer biomarkers: Emerging trends and clinical implications for personalized treatment. Cell 2024, 187, 1617–1635. [Google Scholar] [CrossRef] [PubMed]
- Strobl, M.; Gallaher, J.; Robertson-Tessi, M.; West, J.; Anderson, A. Treatment of evolving cancers will require dynamic decision support. Ann. Oncol. 2023, 34, 867–884. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; Srikanth, S.; Mukherjee, A.G.; Gopalakrishnan, A.V.; Wanjari, U.R.; Vellingiri, B.; Renu, K.; Madhyastha, H. Biogenesis and functional implications of extracellular vesicles in cancer metastasis. Clin. Transl. Oncol. 2025, 27, 2913–2935. [Google Scholar] [CrossRef]
- Chen, M.; Wu, C.; Fu, Z.; Liu, S. ICAM1 promotes bone metastasis via integrin-mediated TGF-β/EMT signaling in triple-negative breast cancer. Cancer Sci. 2022, 113, 3751–3765. [Google Scholar] [CrossRef] [PubMed]
- Zabeti Touchaei, A.; Norollahi, S.E.; Najafizadeh, A.; Babaei, K.; Bakhshalipour, E.; Vahidi, S.; Samadani, A.A. Therapeutic combinations of exosomes alongside cancer stem cells (CSCs) and of CSC-derived exosomes (CSCEXs) in cancer therapy. Cancer Cell Int. 2024, 24, 334. [Google Scholar] [CrossRef]
- Sheng, K.L.; Kang, L.; Pridham, K.J.; Dunkenberger, L.E.; Sheng, Z.; Varghese, R.T. An integrated approach to biomarker discovery reveals gene signatures highly predictive of cancer progression. Sci. Rep. 2020, 10, 21246. [Google Scholar] [CrossRef]
- Khan, S.; Alson, D.; Sun, L.; Maloney, C.; Sun, D. Leveraging Neural Crest-Derived Tumors to Identify NF1 Cancer Stem Cell Signatures. Cancers 2024, 16, 3639. [Google Scholar] [CrossRef]
- Liu, X.; Papukashvili, D.; Wang, Z.; Liu, Y.; Chen, X.; Li, J.; Li, Z.; Hu, L.; Li, Z.; Rcheulishvili, N.; et al. Potential utility of miRNAs for liquid biopsy in breast cancer. Front. Oncol. 2022, 12, 940314. [Google Scholar] [CrossRef]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer: Current status, challenges and future prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef]
- Lorenzo, G.; Ahmed, S.R.; Hormuth, D.A.; Vaughn, B.; Kalpathy-Cramer, J.; Solorio, L.; Yankeelov, T.E.; Gomez, H. Patient-specific, mechanistic models of tumor growth incorporating artificial intelligence and big data. Annu. Rev. Biomed. Eng. 2024, 26, 529–560. [Google Scholar] [CrossRef]
- Seo, H.A.; Moeng, S.; Sim, S.; Kuh, H.J.; Choi, S.Y.; Park, J.K. MicroRNA-based combinatorial cancer therapy: Effects of MicroRNAs on the efficacy of anti-cancer therapies. Cells 2019, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Abdel Halim, A.S.; Rudayni, H.A.; Chaudhary, A.A.; Ali, M.A. MicroRNAs: Small molecules with big impacts in liver injury. J. Cell. Physiol. 2023, 238, 32–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gambino, F., Jr.; Algenio, C.S.; Wu, C.; Gao, Y.; Bouchard, C.S.; Qiao, L.; Bu, P.; Zhao, S. Inflammation and oxidative stress induced by lipid peroxidation metabolite 4-hydroxynonenal in human corneal epithelial cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Čipak Gašparović, A.; Milković, L.; Dandachi, N.; Stanzer, S.; Pezdirc, I.; Vrančić, J.; Šitić, S.; Suppan, C.; Balic, M. Chronic oxidative stress promotes molecular changes associated with epithelial mesenchymal transition, NRF2, and breast cancer stem cell phenotype. Antioxidants 2019, 8, 633. [Google Scholar] [CrossRef]
- Zhong, T.; Li, Y.; Jin, M.; Liu, J.; Wu, Z.; Zhu, F.; Zhao, L.; Fan, Y.; Xu, L.; Ji, J. Downregulation of 4-HNE and FOXO4 collaboratively promotes NSCLC cell migration and tumor growth. Cell Death Dis. 2024, 15, 546. [Google Scholar] [CrossRef]
- Galam, L.; Failla, A.; Soundararajan, R.; Lockey, R.F.; Kolliputi, N. 4-hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget 2015, 6, 41508. [Google Scholar] [CrossRef]
- Dupuy, J.; Cogo, E.; Fouché, E.; Guéraud, F.; Pierre, F.; Plaisancié, P. Epithelial-mesenchymal interaction protects normal colonocytes from 4-HNE-induced phenotypic transformation. PLoS ONE 2024, 19, e0302932. [Google Scholar] [CrossRef]
- Reyes-Jiménez, E.; Ramírez-Hernández, A.A.; Santos-Álvarez, J.C.; Velázquez-Enríquez, J.M.; Pina-Canseco, S.; Baltiérrez-Hoyos, R.; Vásquez-Garzón, V.R. Involvement of 4-hydroxy-2-nonenal in the pathogenesis of pulmonary fibrosis. Mol. Cell. Biochem. 2021, 476, 4405–4419. [Google Scholar] [CrossRef]
- Wang, F.; Min, J. DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduct. Target. Ther. 2021, 6, 244. [Google Scholar] [CrossRef]
- Wang, D.; Tang, L.; Zhang, Y.; Ge, G.; Jiang, X.; Mo, Y.; Wu, P.; Deng, X.; Li, L.; Zuo, S. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022, 13, 544. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Y.; Sun, Q.; Hong, Y.; Liang, S.; Jiang, H.; Zhang, X.; Zhang, S.; Chen, Q. Targeting ferroptosis opens new avenues in gliomas. Int. J. Biol. Sci. 2024, 20, 4674. [Google Scholar] [CrossRef]
- Ma, W.; Kantarjian, H.; Yeh, C.H.; Zhang, Z.J.; Cortes, J.; Albitar, M. BCR-ABL truncation due to premature translation termination as a mechanism of resistance to kinase inhibitors. Acta Haematol. 2009, 121, 27–31. [Google Scholar] [CrossRef]
- Vegliante, R.; Pastushenko, I.; Blanpain, C. Deciphering functional tumor states at single-cell resolution. Embo J. 2022, 41, e109221. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, T.; Jin, Y.; Huang, B.; Zhang, Y. Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research. Front. Oncol. 2020, 10, 593085. [Google Scholar] [CrossRef] [PubMed]
- Huertas-Castaño, C.; Martínez-López, L.; Cabrera-Roldán, P.; Pastor, N.; Mateos, J.C.; Mateos, S.; Pardal, R.; Domínguez, I.; Orta, M.L. Influence of stromal neural crest progenitor cells on neuroblastoma radioresistance. Int. J. Radiat. Biol. 2025, 101, 153–163. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, E.; Shen, H.; Tan, J.; Zeng, S. The functional and clinical roles of liquid biopsy in patient-derived models. J. Hematol. Oncol. 2023, 16, 36. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.-O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Lyu, P.; Gu, X.; Wang, F.; Sun, H.; Zhou, Q.; Yang, S.; Yuan, W. Advances in targeting cancer-associated fibroblasts through single-cell spatial transcriptomic sequencing. Biomark. Res. 2024, 12, 73. [Google Scholar] [CrossRef]
- Park, M.N.; Kim, M.; Lee, S.; Kang, S.; Ahn, C.-H.; Tallei, T.E.; Kim, W.; Kim, B. Targeting Redox Signaling Through Exosomal MicroRNA: Insights into Tumor Microenvironment and Precision Oncology. Antioxidants 2025, 14, 501. [Google Scholar] [CrossRef]
- Malagoli Tagliazucchi, G.; Wiecek, A.J.; Withnell, E.; Secrier, M. Genomic and microenvironmental heterogeneity shaping epithelial-to-mesenchymal trajectories in cancer. Nat. Commun. 2023, 14, 789. [Google Scholar] [CrossRef]
- Ma, L.; Singh, J.; Schekman, R. Two RNA-binding proteins mediate the sorting of miR223 from mitochondria into exosomes. Elife 2023, 12, e85878. [Google Scholar] [CrossRef]
- Bocci, F.; Zhou, P.; Nie, Q. Single-Cell RNA-Seq Analysis Reveals the Acquisition of Cancer Stem Cell Traits and Increase of Cell-Cell Signaling during EMT Progression. Cancers 2021, 13, 5726. [Google Scholar] [CrossRef] [PubMed]
- Hamraoui, A.; Jourdren, L.; Thomas-Chollier, M. AsaruSim: A single-cell and spatial RNA-Seq Nanopore long-reads simulation workflow. Bioinformatics 2025, 41, btaf087. [Google Scholar] [CrossRef] [PubMed]
- Malla, R.; Bhamidipati, P.; Samudrala, A.S.; Nuthalapati, Y.; Padmaraju, V.; Malhotra, A.; Rolig, A.S.; Malhotra, S.V. Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer. Cancers 2025, 17, 1167. [Google Scholar] [CrossRef] [PubMed]
- Schuhwerk, H.; Kleemann, J.; Gupta, P.; van Roey, R.; Armstark, I.; Kreileder, M.; Feldker, N.; Ramesh, V.; Hajjaj, Y.; Fuchs, K.; et al. The EMT transcription factor ZEB1 governs a fitness-promoting but vulnerable DNA replication stress response. Cell Rep. 2022, 41, 111819. [Google Scholar] [CrossRef]
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A genetically encoded H2O2 probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem. Biol. 2013, 8, 535–542. [Google Scholar] [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Hao, W.; Chen, Z.; Tang, J.; Yang, R.; Gao, W.Q.; Xu, H. hnRNPA2B1 promotes the occurrence and progression of hepatocellular carcinoma by downregulating PCK1 mRNA via a m6A RNA methylation manner. J. Transl. Med. 2023, 21, 861. [Google Scholar] [CrossRef]
- Singh, R.; Gupta, S.C.; Peng, W.X.; Zhou, N.; Pochampally, R.; Atfi, A.; Watabe, K.; Lu, Z.; Mo, Y.Y. Regulation of alternative splicing of Bcl-x by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. 2016, 7, e2262. [Google Scholar] [CrossRef]
- Xu, S.; Cao, B.; Xuan, G.; Xu, S.; An, Z.; Zhu, C.; Li, L.; Tang, C. Function and regulation of Rab GTPases in cancers. Cell Biol. Toxicol. 2024, 40, 28. [Google Scholar] [CrossRef] [PubMed]
- Danac, J.M.C.; Uy, A.G.G.; Garcia, R.L. Exosomal microRNAs in colorectal cancer: Overcoming barriers of the metastatic cascade (Review). Int. J. Mol. Med. 2021, 47, 112. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, S.; Moeinafshar, A.; Rezaei, N. The multifaceted role of extracellular vesicles (EVs) in colorectal cancer: Metastasis, immune suppression, therapy resistance, and autophagy crosstalk. J. Transl. Med. 2024, 22, 452. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhong, X.; Hu, Z.; Zhao, S.; Wei, P.; Li, D. An insight into small extracellular vesicles: Their roles in colorectal cancer progression and potential clinical applications. Clin. Transl. Med. 2020, 10, e249. [Google Scholar] [CrossRef]
- Shakerian, N.; Darzi-Eslam, E.; Afsharnoori, F.; Bana, N.; Noorabad Ghahroodi, F.; Tarin, M.; Mard-Soltani, M.; Khalesi, B.; Hashemi, Z.S.; Khalili, S. Therapeutic and diagnostic applications of exosomes in colorectal cancer. Med. Oncol. 2024, 41, 203. [Google Scholar] [CrossRef]
- Das, K.; Paul, S.; Ghosh, A.; Gupta, S.; Mukherjee, T.; Shankar, P.; Sharma, A.; Keshava, S.; Chauhan, S.C.; Kashyap, V.K.; et al. Extracellular Vesicles in Triple-Negative Breast Cancer: Immune Regulation, Biomarkers, and Immunotherapeutic Potential. Cancers 2023, 15, 4879. [Google Scholar] [CrossRef]
- Hassanpour, H.; Mojtahed, M.; Nasiri, L.; Vaez-Mahdavi, M.R.; Fallah, A.A. Association of sulfur mustard toxicity with oxidant/antioxidant system in veterans: A meta-analysis of case-control studies. Int. Immunopharmacol. 2025, 147, 114007. [Google Scholar] [CrossRef]
- Sahoo, S.; Nayak, S.P.; Hari, K.; Purkait, P.; Mandal, S.; Kishore, A.; Levine, H.; Jolly, M.K. Immunosuppressive Traits of the Hybrid Epithelial/Mesenchymal Phenotype. Front. Immunol. 2021, 12, 797261. [Google Scholar] [CrossRef]
- Teeuwssen, M.; Fodde, R. Cell Heterogeneity and Phenotypic Plasticity in Metastasis Formation: The Case of Colon Cancer. Cancers 2019, 11, 1368. [Google Scholar] [CrossRef]
- Engelsen, A.S.T.; Lotsberg, M.L.; Abou Khouzam, R.; Thiery, J.P.; Lorens, J.B.; Chouaib, S.; Terry, S. Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front. Immunol. 2022, 13, 869676. [Google Scholar] [CrossRef]
- Li, X.; Si, W.; Zhang, Y.; Yang, P.; Ruan, L.; Ba, H.; Chen, Z. Alpha-Heredin targets CAMKII/DRP1-Mediated mitochondrial fission to trigger ferroptosis in pancreatic ductal adenocarcinoma. Phytomedicine 2025, 145, 157048. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.-L.; Khanal, P.; Kelly, T.; Hao, Y.; Yang, X. Kinome-Wide Screening Identifies FAK as a Novel Post-Translational Regulator of PD-L1 Stability and Immune Evasion in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2025, 26, 10108. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, X.; Zhu, X.; Li, L.; Zeng, X.; Ren, J.; Wang, L.; Wu, J.; Zhang, Q.; Wang, S.; et al. CD248 induces PD-L1 expression on cancer-associated fibroblasts to promote NSCLC immune escape. Front. Cell Dev. Biol. 2025, 13, 1635915. [Google Scholar] [CrossRef] [PubMed]
- Zgeib, K. Molecular Characterization of Dedifferentiation-Induced Oncogenic Stemness in the Intestinal Epithelium. Ph.D. Thesis, Stevens Institute of Technology, Hoboken, NJ, USA, 2025. [Google Scholar]
- Tang, J.; Chen, Z.; Wang, Q.; Hao, W.; Gao, W.-Q.; Xu, H. hnRNPA2B1 promotes colon cancer progression via the MAPK pathway. Front. Genet. 2021, 12, 666451. [Google Scholar] [CrossRef]
- Gupta, A.; Yadav, S.; Pt, A.; Mishra, J.; Samaiya, A.; Panday, R.K.; Shukla, S. The HNRNPA2B1–MST1R–Akt axis contributes to epithelial-to-mesenchymal transition in head and neck cancer. Lab. Investig. 2020, 100, 1589–1601. [Google Scholar] [CrossRef]
- Yu, M.; Fei, B.; Chu, S. Targeting HNRNPA2B1 to overcome chemotherapy resistance in gastric cancer stem cells: Mechanisms and therapeutic potential. J. Biol. Chem. 2025, 301, 108234. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Li, H.; Hu, T.Y. Recent advances in the bench-to-bedside translation of cancer nanomedicines. Acta Pharm. Sin. B 2025, 15, 97–122. [Google Scholar] [CrossRef]
- Huang, S.; Soto, A.M.; Sonnenschein, C. The end of the genetic paradigm of cancer. PLoS Biol. 2025, 23, e3003052. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, J.; Shi, M.; Ding, Y.; Sun, H.; Yuan, F.; Zou, Z. Elevated expression of miR-210 predicts poor survival of cancer patients: A systematic review and meta-analysis. PLoS ONE 2014, 9, e89223. [Google Scholar] [CrossRef]
- Luo, S.; Lin, R.; Liao, X.; Li, D.; Qin, Y. Identification and verification of the molecular mechanisms and prognostic values of the cadherin gene family in gastric cancer. Sci. Rep. 2021, 11, 23674. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wu, W.; Liang, L.; Han, S.; Chen, T.; Pan, S.; Xue, M.; Li, S. Prognostic role of microRNA-210 in various carcinomas: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 15283–15289. [Google Scholar] [PubMed]
- Liao, T.T.; Cheng, W.C.; Yang, C.Y.; Chen, Y.Q.; Su, S.H.; Yeh, T.Y.; Lan, H.Y.; Lee, C.C.; Lin, H.H.; Lin, C.C.; et al. The microRNA-210-Stathmin1 Axis Decreases Cell Stiffness to Facilitate the Invasiveness of Colorectal Cancer Stem Cells. Cancers 2021, 13, 1833. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, N.; Hu, X.; Wang, H. Tumor-associated exosomes promote lung cancer metastasis through multiple mechanisms. Mol. Cancer 2021, 20, 117. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef]
- Martínez-Vila, C.; González-Navarro, E.A.; Teixido, C.; Martin, R.; Aya, F.; Juan, M.; Arance, A. Lymphocyte T Subsets and Outcome of Immune Checkpoint Inhibitors in Melanoma Patients: An Oncologist’s Perspective on Current Knowledge. Int. J. Mol. Sci. 2024, 25, 9506. [Google Scholar] [CrossRef]
- van den Ende, T.; de Clercq, N.C.; van Berge Henegouwen, M.I.; Gisbertz, S.S.; Geijsen, E.; Verhoeven, R.; Meijer, S.L.; Schokker, S.; Dings, M.; Bergman, J.J. Neoadjuvant chemoradiotherapy combined with atezolizumab for resectable esophageal adenocarcinoma: A single-arm phase II feasibility trial (PERFECT). Clin. Cancer Res. 2021, 27, 3351–3359. [Google Scholar] [CrossRef]
- Pantano, F.; Zalfa, F.; Iuliani, M.; Simonetti, S.; Manca, P.; Napolitano, A.; Tiberi, S.; Russano, M.; Citarella, F.; Foderaro, S.; et al. Large-Scale Profiling of Extracellular Vesicles Identified miR-625-5p as a Novel Biomarker of Immunotherapy Response in Advanced Non-Small-Cell Lung Cancer Patients. Cancers 2022, 14, 2435. [Google Scholar] [CrossRef]
- Cui, Q.; Li, W.; Wang, D.; Wang, S.; Yu, J. Prognostic significance of blood-based PD-L1 analysis in patients with non-small cell lung cancer undergoing immune checkpoint inhibitor therapy: A systematic review and meta-analysis. World J. Surg. Oncol. 2023, 21, 318. [Google Scholar] [CrossRef]
- Dou, X.; Hua, Y.; Chen, Z.; Chao, F.; Li, M. Extracellular vesicles containing PD-L1 contribute to CD8+ T-cell immune suppression and predict poor outcomes in small cell lung cancer. Clin. Exp. Immunol. 2022, 207, 307–317. [Google Scholar] [CrossRef]
- Genova, C.; Tasso, R.; Rosa, A.; Rossi, G.; Reverberi, D.; Fontana, V.; Marconi, S.; Croce, M.; Dal Bello, M.G.; Dellepiane, C. Prognostic role of soluble and extracellular vesicle-associated PD-L1, B7-H3 and B7-H4 in non-small cell lung cancer patients treated with immune checkpoint inhibitors. Cells 2023, 12, 832. [Google Scholar] [CrossRef] [PubMed]
- de Miguel-Perez, D.; Russo, A.; Arrieta, O.; Ak, M.; Barron, F.; Gunasekaran, M.; Mamindla, P.; Lara-Mejia, L.; Peterson, C.B.; Er, M.E.; et al. Extracellular vesicle PD-L1 dynamics predict durable response to immune-checkpoint inhibitors and survival in patients with non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2022, 41, 186. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, D.; Ghidotti, P.; Proto, C.; Brambilla, M.; De Toma, A.; Ferrara, R.; Galli, G.; Ganzinelli, M.; Lo Russo, G.; Prelaj, A.; et al. Circulating CD81-expressing extracellular vesicles as biomarkers of response for immune-checkpoint inhibitors in advanced NSCLC. Front. Immunol. 2022, 13, 987639. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-L.; Sun, J.; Lu, Y.; Liu, Y.; Cao, H.; Zhang, H.; Calin, G.A. MiR-200 family and cancer: From a meta-analysis view. Mol. Asp. Med. 2019, 70, 57–71. [Google Scholar] [CrossRef]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef]
- Trifylli, E.M.; Kriebardis, A.G.; Koustas, E.; Papadopoulos, N.; Fortis, S.P.; Tzounakas, V.L.; Anastasiadi, A.T.; Sarantis, P.; Vasileiadi, S.; Tsagarakis, A. A current synopsis of the emerging role of extracellular vesicles and micro-RNAs in pancreatic cancer: A forward-looking plan for diagnosis and treatment. Int. J. Mol. Sci. 2024, 25, 3406. [Google Scholar] [CrossRef]
- Hussain, Z.; Nigri, J.; Tomasini, R. The cellular and biological impact of extracellular vesicles in pancreatic cancer. Cancers 2021, 13, 3040. [Google Scholar] [CrossRef]
- Zhang, Z.; Xue, B.; Chen, Y.; Shao, Y.; Wang, D. A systematic review and meta-analysis combined with bioinformatic analysis on the predictive value of E-cadherin in patients with renal cell carcinoma. Expert Rev. Mol. Diagn. 2024, 24, 859–871. [Google Scholar] [CrossRef]
- Albarrán, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Román, M.; Calvo, J.C.; Pérez de Aguado, P.; Moreno, J.; Guerrero, P. Receptor tyrosine kinase inhibitors for the treatment of recurrent and unresectable bone sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef]
- Grundy, M.; Narendran, A. The hepatocyte growth factor/mesenchymal epithelial transition factor axis in high-risk pediatric solid tumors and the anti-tumor activity of targeted therapeutic agents. Front. Pediatr. 2022, 10, 910268. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor tyrosine kinases and cancer: Oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.K. Analysis of Immune Cell Functions to Improve the Therapeutic Efficacy of a Novel Cell-based Therapy of Combined Autologous Monocytes and Dual-Interferons for Ovarian Cancer. Ph.D. Thesis, University of Maryland, Baltimore, MD, USA, 2023. [Google Scholar]
- Chen, T. Determinants of Treatment Outcome Heterogeneity for Antibody and Antibody Drug Conjugates in Solid Tumors. Ph.D. Thesis, State University of New York at Buffalo, New York, NY, USA, 2021. [Google Scholar]
- Shimada, Y.; Matsubayashi, J.; Kudo, Y.; Maehara, S.; Takeuchi, S.; Hagiwara, M.; Kakihana, M.; Ohira, T.; Nagao, T.; Ikeda, N. Serum-derived exosomal PD-L1 expression to predict anti-PD-1 response and in patients with non-small cell lung cancer. Sci. Rep. 2021, 11, 7830. [Google Scholar] [CrossRef] [PubMed]
- Sychowski, G.; Romanowicz, H.; Ciesielski, W.; Hogendorf, P.; Durczyński, A.; Smolarz, B. Diagnostic and Therapeutic Potential of Selected microRNAs in Colorectal Cancer: A Literature Review. Cancers 2025, 17, 2135. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Yue, X.; Xia, T. Exosomal microRNA-210 is a potentially non-invasive biomarker for the diagnosis and prognosis of glioma. Oncol. Lett. 2020, 19, 1967–1974. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Khaboushan, A.S.; Salimian, S.N.; Mehraban, S.; Bahramy, A.; Zafari, N.; Kajbafzadeh, A.-M.; Johnson, J.; Zolbin, M.M. Prognostic significance of non-coding RNAs related to the tumorigenic epithelial-mesenchymal transition (EMT) process among ovarian cancer patients: A systematic review and meta-analysis. Heliyon 2024, 10, e35202. [Google Scholar] [CrossRef]
- Chen, S.-l.; Hu, D.; Chen, T.-z.; Shen, S.-y.; Zhao, C.-f.; Wang, C.; Tong, S.-y.; Liu, Z.; Lin, S.-h.; Jin, L.-x. Pan-cancer screening and validation of CALU’s role in EMT regulation and tumor microenvironment in triple-negative breast cancer. J. Inflamm. Res. 2024, 17, 6743–6764. [Google Scholar] [CrossRef]
- Wang, H.; Yu, T.; Mao, L. Placental-cadherin, a biomarker for local immune status and poor prognosis among patients with tongue squamous cell carcinoma. Eur. Arch. Oto-Rhino-Laryngol. 2022, 279, 3597–3609. [Google Scholar] [CrossRef]
- Su, Y.; Li, J.; Witkiewicz, A.K.; Brennan, D.; Neill, T.; Talarico, J.; Radice, G.L. N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene 2012, 31, 4484–4489. [Google Scholar] [CrossRef][Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).