
Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues

 ,
,  ,
, 
 , and
, and 
Abstract
:1. Introduction: Arsenic’s Impacts on Food and Human Health in the USA
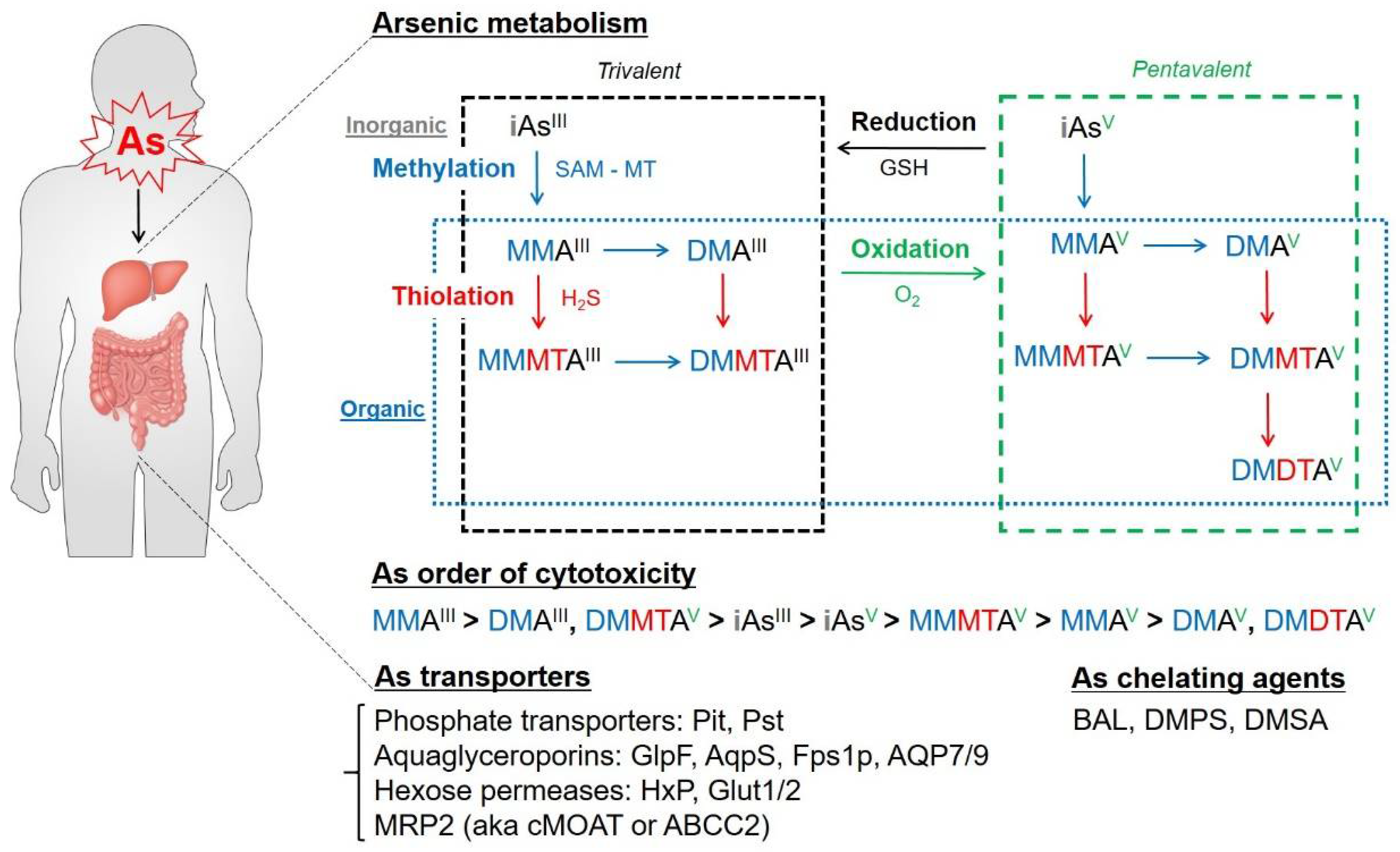
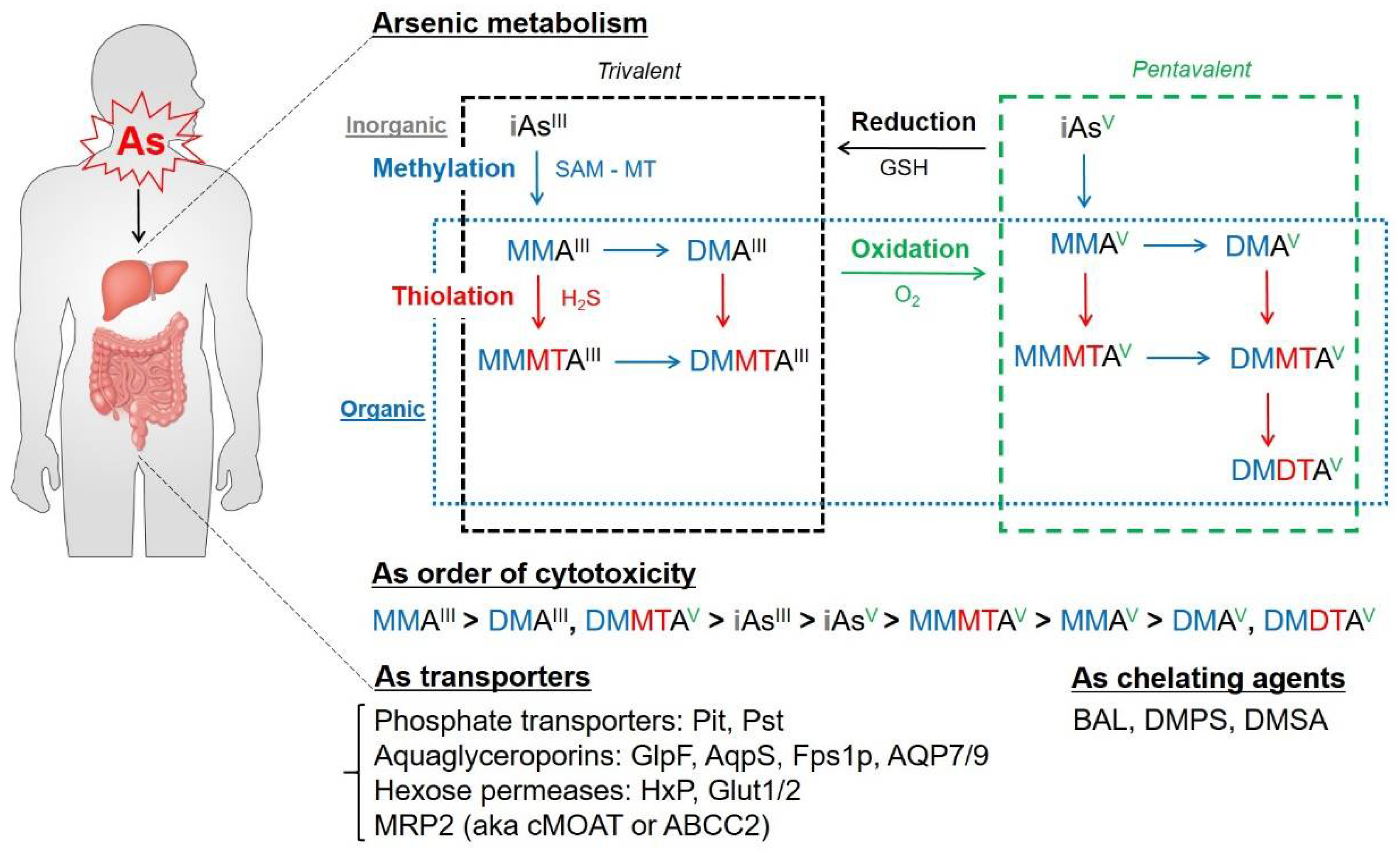
2. As Biochemistry and Metabolism: Biotransformation of As in the Body
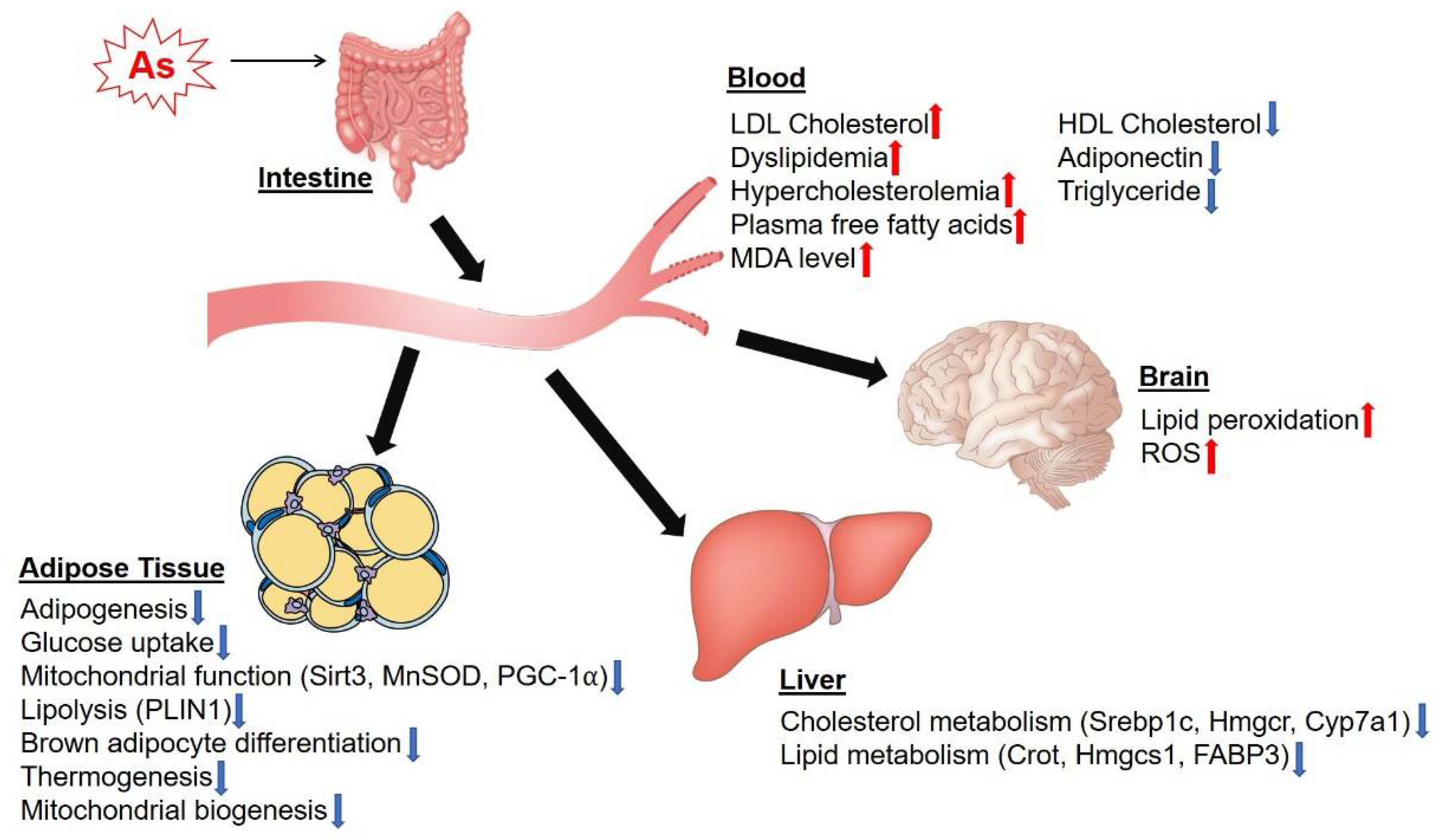
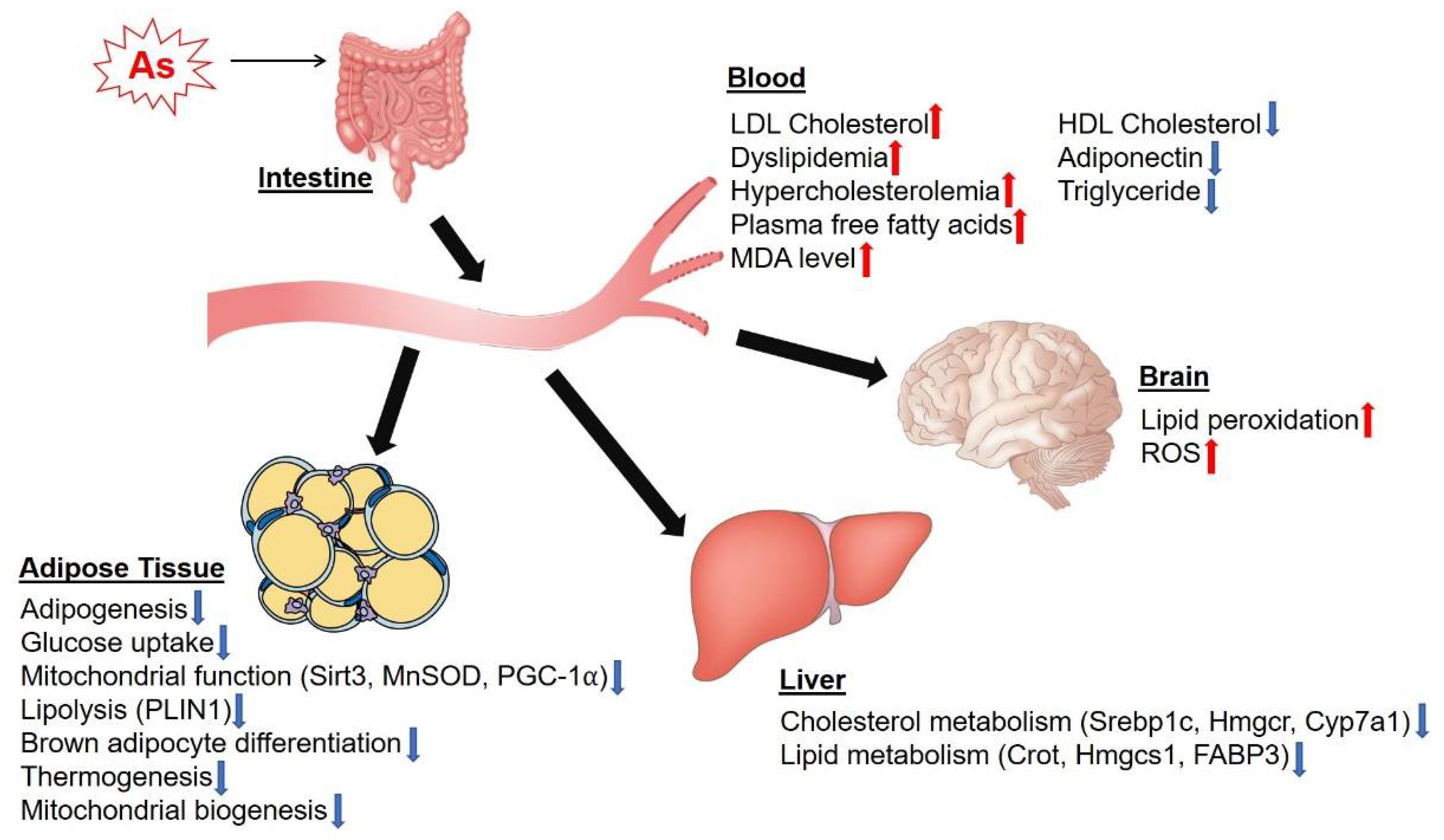
3. Effects of as on Lipid Metabolism and Adipocyte Function
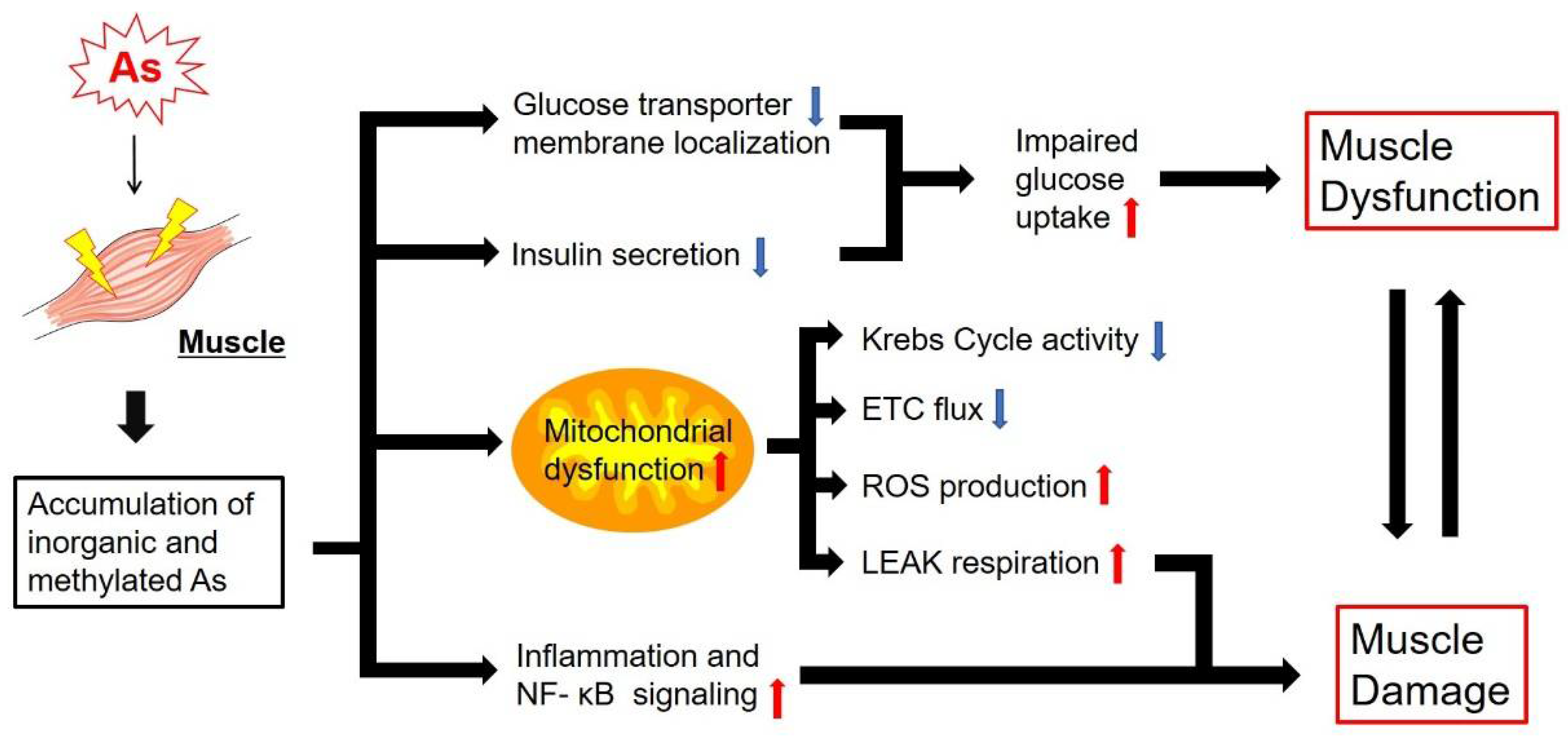
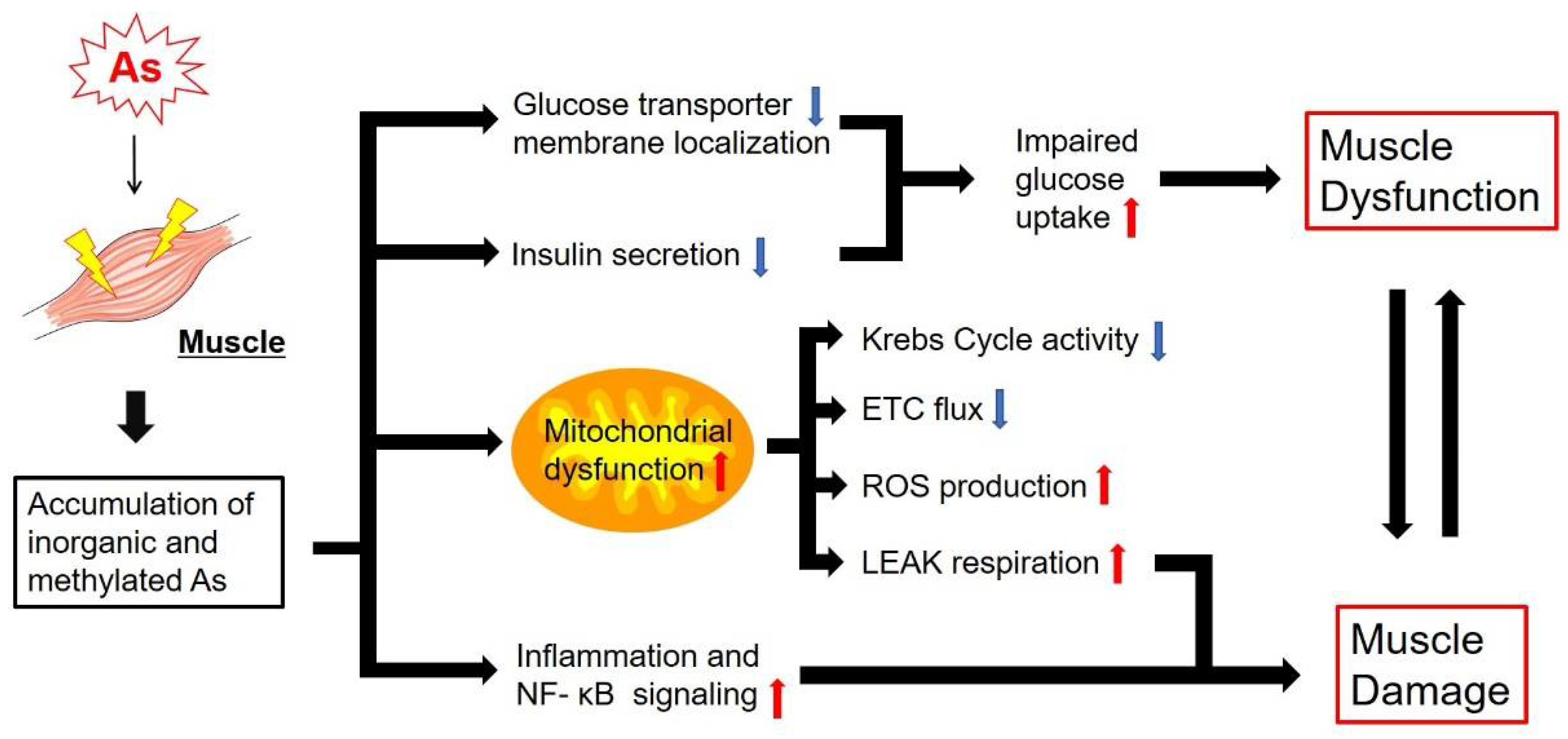
4. Effects of As on Glucose Metabolism, Mitochondrial Energy Metabolism and Muscle Function
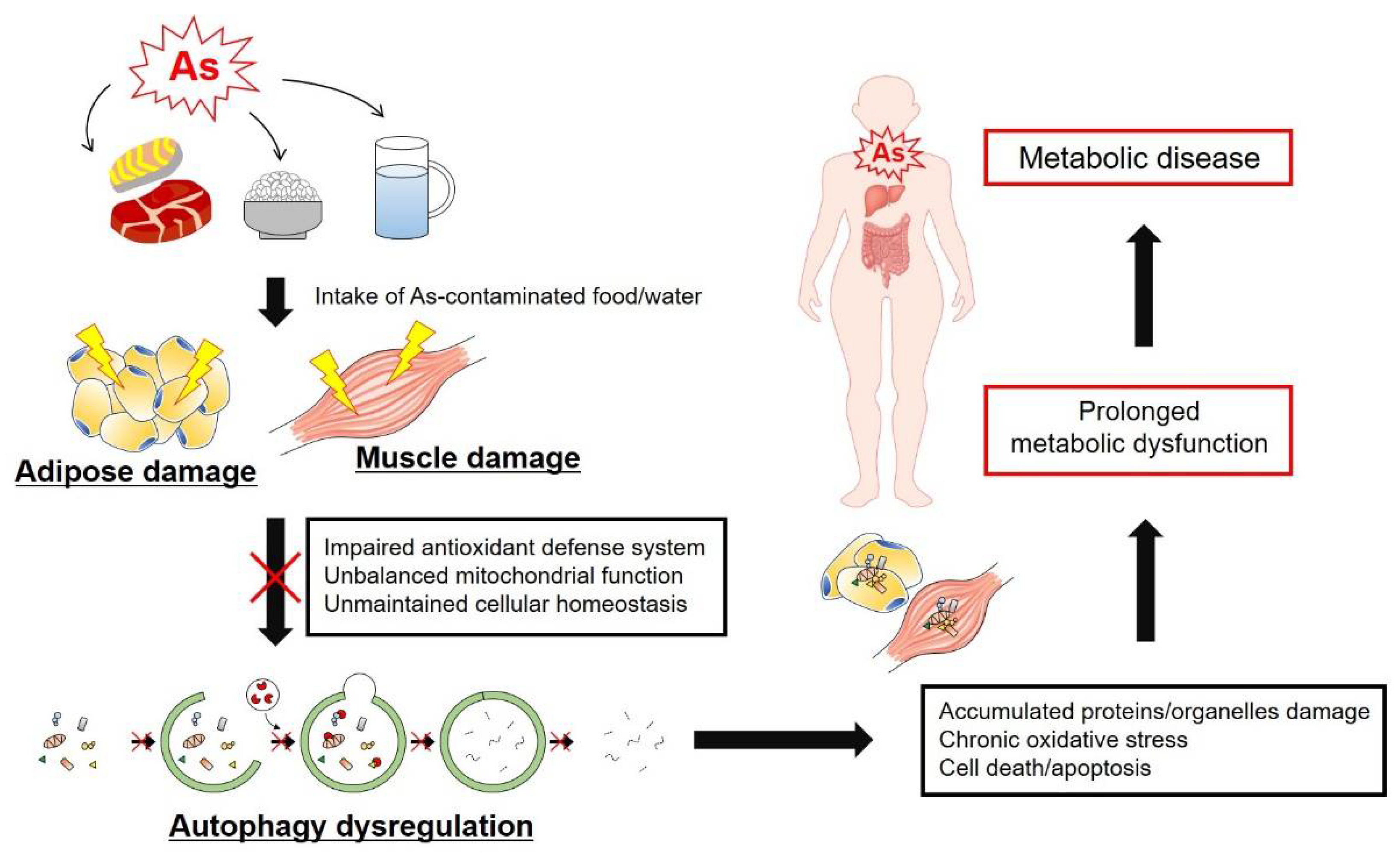
5. Effects of as on Autophagy in Adipose and Muscle Tissues
6. Conclusion: Perspective on the Connection between As Exposure and Metabolic Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Dose and Duration of As | Reference | |
|---|---|---|---|
| Cell culture | 3T3-L1 preadipocytes, mouse-adipose-derived stromal vascular fraction cells and human adipose tissue-derived stem cells | 5 μM (sodium arsenite), 1 μM (trivalent monomethylated arsenic), or 2 μM (trivalent dimethylated arsenic) in medium, 2 days with DMI (differentiation media) | [75] |
| 3T3-L1 adipocytes | 1 mM, sodium arsenite (NaAsO2), methylarsine oxide and iododimethylarsine in medium, until full differentiation for several days | [102] | |
| 3T3-L1 | 3 μM, arsenic trioxide (As2O3) in medium, during differentiation for 24 h | [119] | |
| 3T3-F442A | 0.5 μM/L, arsenic trioxide in medium, 3 days | [120] | |
| Adipose-tissue-derived primary human mesenchymal stem cells (hMSCs) | 1 μM, sodium arsenite in medium, 72 h | [78] | |
| C2C12 myoblast cells and 3T3-L1 | 2 μM, sodium arsenite in medium, 8 weeks | [76] | |
| C2C12 myotubules | 1 μM, arsenic trioxide in medium, 48 h | [90] | |
| C3H 10T1/2 adipocytes | 6 μM, sodium arsenite in medium, 2 months | [77,121] | |
| EJ-1, human bladder cancer cells | Up to 1 mM (iASIII, iASV, MMAV, MMMTAV, DMAV, DMAIII,DMMTAV or DMDTAV) in medium, 24 h | [58] | |
| H9c2 rat cardiomyoblastoma cells | 50 μM, arsenic trioxide in medium, 24 h | [142] | |
| HaCaT human keratinocytes | 25 μM, sodium arsenite in medium, 24 h | [35] | |
| HEK293, NIH3T3, BEAS-2B | 1 μM, sodium arsenite in medium, 24 h | [144] | |
| HIB1B adipocytes | 10 μM, sodium arsenite in medium, 6 days | [79] | |
| Human–hamster hybrid AL cells | 0.25 μg/mL, sodium arsenite in medium, 60 days | [110] | |
| INS-1 832/13 beta cells | 2 μM (sodium arsenite, dimethyl arsenite), 0.5 μM (mono-methyl arsenite) in medium, 24 h | [104] | |
| L6 rat skeletal muscle cells | 0.5 mM, sodium arsenite in medium, 30 min | [123] | |
| NIH3T3, HeLa cells | 2 μM, sodium arsenite in medium, 48 h | [145] | |
| P19 mouse embryonic stem cells | 1 μM, sodium arsenite in medium, 5 days until full differentiation | [91] | |
| PAEC, porcine aortic endothelial cells | 5 μM, sodium arsenite in medium, 2 h | [32] | |
| Pancreatic eyelets (ex vivo) of B6 mice | 0.1 μM (methylarsonite, dimethylarsinite) in medium, 48 h | [103] | |
| Primary SECs, Liver sinusoidal endothelial cells | 5 μM, sodium arsenite in medium, 8 h | [33] | |
| UROtsa, Non-tumorigenic human urothelial cell lines | 1 μM, sodium arsenite in medium, 12 weeks 50 nM, MMA (III) in medium, 24 weeks | [36] | |
| Animals | Chicken skeletal muscle (in vivo) | 2.5 mg/kg/day, until reach 30 mg/kg, H3AsO3 in diet | [85] |
| Chickens | 2.5 mg/kg, As2O3 in diet, 12 weeks | [140] | |
| Hamster LD50 | 112 μmol/kg and 29.3 μmol/kg (MMAIII) in diet | [42] | |
| Mice (C57BL/6 aka B6, male and female) | 10 mg/kg/d, sodium arsenite in gavage, 9 days | [79] | |
| Mice (B6, male) | 45 ppm, sodium arsenite in drinking water, 48 weeks | [34] | |
| Mice (B6, male) | 50 ppm, sodium arsenite in drinking water, 18 weeks | [73] | |
| Mice (B6, male) | 50 ppm, sodium arsenate in drinking water, 8 weeks | [101] | |
| Mice (B6, female) | 20 ppm, sodium arsenite and sodium arsenate dibasic heptahydrate (1:1 ratio) in drinking water, 17 weeks | [80] | |
| Mice (C57BL/6J, male) eye | 250 ppm for 1 month or 50 ppm for 6 months (sodium arsenite) in drinking water | [62] | |
| Mice (B6) skeletal muscle | 4 μM or 4 mg/L, arsenic trioxide in drinking water, 12 weeks | [141] | |
| Mice (C57BL/6Tac) | 100 μg/L, sodium arsenite in drinking water, 5 weeks | [78] | |
| Mice (C57BL/6Tac) | 100 μg/L, trivalent arsenite (AsIII) in drinking water, 5 weeks | [112] | |
| Mice (C57BL/6NTac male) | 100 μg/L, sodium metaarsenite (NaAsO2) in drinking water, 5 weeks | [113] | |
| Mice (C57BL/6Ai p47phox knockout) | 250 ppb, sodium arsenite in drinking water, 2 weeks | [33] | |
| Mice (CrL:Sk1-hrBD, weanling female hairless mice) skin | 5 mg/L, sodium arsenite in drinking water, 26 weeks | [46] | |
| Mice (sciatic nerve denervated) | 0.5 ppm, arsenic trioxide in drinking water, 4 weeks | [86] | |
| Mice (specific pathogen free female) | 1 ppm, sodium arsenite in drinking water, 2 weeks | [68] | |
| Rat (Winstar, Albino male) | 150 ppm (sodium arsenite), 200 ppm (sodium arsenate) in drinking water, 12 weeks | [67] | |
| Rat (Albino male) | 25 ppm, sodium arsenite in drinking water, 8 weeks | [69] | |
| Rat (Albino male) | 133 μg/mL (arsenic trioxide) in drinking water, 8 weeks | [71] | |
| Rat (Albino pregnant) | 13 ppm, sodium arsenite, from gestation, lactation through full adulthood in drinking water | [72] | |
| Rat liver tissue in test tube | 40 μM sodium arsenate uptake upto 30 min | [106] | |
| Rat liver mitochondria | 100 μM, sodium arsenite, 5 min | [107] | |
| Rat liver mitochondria | 25 ppm, sodium arsenite, 12 weeks | [109] | |
| Rat (Winstar, TR−) | 0.5 mg/kg, 1 mmol, 0.4 mmol (MMA), 1 mmol (DMA), injection through bile duct cannulation less than 5 s | [50] | |
| Zebrafish | 500 ppb, sodium arsenite in water, 7 days | [70] | |
| Human patients | Arsenic biomarkers in human | 1–2 mg/kg (LD50 of iAs), accumulated arsenic from food, water, air and soil | [29] |
| Hair, nails and skin scales of arsenic-exposed patients in West Bengal, India | Arsenic patients exposed to above 50 mg/L in drinking groundwater | [44] | |
| Human (2–14 years old adolescents) arsenicosis patients | Upto 31.6 μg/L total inorganic arsenic exposure, upto 14 years | [66] | |
| Human acute promyelocytic leukaemia (APL) patients | All patients were treated with intravenous infusion of 10 mg of As2O3 (10 mL, 0.1% solution) over 2–8 h daily for 28 d and the cycle was repeated at intervals of 2 weeks until complete remission (CR) was achieved. After CR, they were maintained with several cycles of As2O3 at intervals of 2–3 months. | [87] | |
| Human cardiovascular disease patients | 100 μg/L of arsenic above or below in meta-analysis | [95] | |
| Human patients (arsenic exposure) | Chronic exposure of arsenic in Bandladeshi (above 10 μg/L exposure) for 9 days | [98] | |
| Human patients (Diabetes) | Geographical tracing (0.1 μg/L detection limit of arsenic) in Denmark for mean follow-up for 9.7 years | [105] | |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meliker, J.R.; Wahl, R.L.; Cameron, L.L.; Nriagu, J.O. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: A standardized mortality ratio analysis. Environ. Health 2007, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmaus, C.; Yuan, Y.; Liaw, J.; Smith, A.H. Low-level population exposure to inorganic arsenic in the United States and diabetes mellitus: A reanalysis. Epidemiology 2009, 20, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Silbergeld, E.K.; Pastor-Barriuso, R.; Guallar, E. Arsenic exposure and prevalence of type 2 diabetes in US adults. JAMA 2008, 300, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, D.C.; Klawer, L.M.; Joeckel, R.M.; Harvey, F.E.; Reade, A.R.; McVey, K. Arsenic in Groundwater and Rural Public Water Supplies in Nebraska, USA. Great Plains Res. 2006, 16, 137–148. [Google Scholar]
- Cohen, S. Agricultural Chemical News. Groundw. Monit. Remediat. 1989, 9, 57–64. [Google Scholar] [CrossRef]
- NDA. Nebraska Department of Agriculture Report; NDA: Lincoln, NE, USA, 2020. [Google Scholar]
- Parsons, J.G.; Martinez-Martinez, A.; Peralta-Videa, J.R.; Gardea-Torresdey, J.L. Speciation and uptake of arsenic accumulated by corn seedlings using XAS and DRC-ICP-MS. Chemosphere 2008, 70, 2076–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armendariz, A.L.; Talano, M.A.; Travaglia, C.; Reinoso, H.; Wevar Oller, A.L.; Agostini, E. Arsenic toxicity in soybean seedlings and their attenuation mechanisms. Plant Physiol. Biochem. 2016, 98, 119–127. [Google Scholar] [CrossRef]
- Palma-Lara, I.; Martinez-Castillo, M.; Quintana-Perez, J.C.; Arellano-Mendoza, M.G.; Tamay-Cach, F.; Valenzuela-Limon, O.L.; Garcia-Montalvo, E.A.; Hernandez-Zavala, A. Arsenic exposure: A public health problem leading to several cancers. Regul. Toxicol. Pharm. 2019, 110, 104539. [Google Scholar] [CrossRef]
- Karagas, M.R.; Gossai, A.; Pierce, B.; Ahsan, H. Drinking Water Arsenic Contamination, Skin Lesions, and Malignancies: A Systematic Review of the Global Evidence. Curr. Environ. Health Rep. 2015, 2, 52–68. [Google Scholar] [CrossRef]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Abdul, K.S.; Jayasinghe, S.S.; Chandana, E.P.; Jayasumana, C.; De Silva, P.M. Arsenic and human health effects: A review. Environ. Toxicol. Pharm. 2015, 40, 828–846. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Moon, K.A.; Wang, S.L.; Silbergeld, E.; Navas-Acien, A. The Association of Arsenic Metabolism with Cancer, Cardiovascular Disease, and Diabetes: A Systematic Review of the Epidemiological Evidence. Environ. Health Perspect. 2017, 125, 087001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castriota, F.; Acevedo, J.; Ferreccio, C.; Smith, A.H.; Liaw, J.; Smith, M.T.; Steinmaus, C. Obesity and increased susceptibility to arsenic-related type 2 diabetes in Northern Chile. Environ. Res. 2018, 167, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Maull, E.A.; Ahsan, H.; Edwards, J.; Longnecker, M.P.; Navas-Acien, A.; Pi, J.; Silbergeld, E.K.; Styblo, M.; Tseng, C.H.; Thayer, K.A.; et al. Evaluation of the association between arsenic and diabetes: A National Toxicology Program workshop review. Environ. Health Perspect. 2012, 120, 1658–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkhondeh, T.; Samarghandian, S.; Azimi-Nezhad, M. The role of arsenic in obesity and diabetes. J. Cell Physiol. 2019, 234, 12516–12529. [Google Scholar] [CrossRef] [PubMed]
- Spratlen, M.J.; Grau-Perez, M.; Best, L.G.; Yracheta, J.; Lazo, M.; Vaidya, D.; Balakrishnan, P.; Gamble, M.V.; Francesconi, K.A.; Goessler, W.; et al. The Association of Arsenic Exposure and Arsenic Metabolism With the Metabolic Syndrome and Its Individual Components: Prospective Evidence From the Strong Heart Family Study. Am. J. Epidemiol. 2018, 187, 1598–1612. [Google Scholar] [CrossRef] [PubMed]
- Ayotte, J.D.; Medalie, L.; Qi, S.L.; Backer, L.C.; Nolan, B.T. Estimating the High-Arsenic Domestic-Well Population in the Conterminous United States. Environ. Sci. Technol. 2017, 51, 12443–12454. [Google Scholar] [CrossRef]
- Preston, S.H.; Vierboom, Y.C.; Stokes, A. The role of obesity in exceptionally slow US mortality improvement. Proc. Natl. Acad. Sci. USA 2018, 115, 957–961. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; McPherson, K.; Marsh, T.; Gortmaker, S.L.; Brown, M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 2011, 378, 815–825. [Google Scholar] [CrossRef]
- Kim, D.D.; Basu, A. Estimating the Medical Care Costs of Obesity in the United States: Systematic Review, Meta-Analysis, and Empirical Analysis. Value Health 2016, 19, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, S.H.; Jang, Y.; Bae, J.; Kim, I.M.; Schaecher, C.; Shomo, Z.D. Autophagy in Adipocyte Browning: Emerging Drug Target for Intervention in Obesity. Front. Physiol. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.D.; Noseworthy, M.D.; Hamilton, J.; Tarnopolski, M.; Tein, I. Skeletal muscle metabolic dysfunction in obesity and metabolic syndrome. Can. J. Neurol. Sci. 2008, 35, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marette, A.; Liu, Y.; Sweeney, G. Skeletal muscle glucose metabolism and inflammation in the development of the metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 299–305. [Google Scholar] [CrossRef]
- Kuivenhoven, M.; Mason, K. Arsenic Toxicity. In StatPearls; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Arsenic in Drinking Water; The National Academies Press: Washington, DC, USA, 1999.
- Ravenscroft, B.H.; Richards, K. Arsenic Pollution: A Global Synthesis; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-1-405-18601-8. [Google Scholar]
- Hughes, M.F. Biomarkers of exposure: A case study with inorganic arsenic. Environ. Health Perspect 2006, 114, 1790–1796. [Google Scholar] [CrossRef]
- Klein, C.B.; Leszczynska, J.; Hickey, C.; Rossman, T.G. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol. Appl. Pharm. 2007, 222, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad Med. J. 2003, 79, 391–396. [Google Scholar] [CrossRef]
- Smith, K.R.; Klei, L.R.; Barchowsky, A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L442–L449. [Google Scholar] [CrossRef]
- Straub, A.C.; Clark, K.A.; Ross, M.A.; Chandra, A.G.; Li, S.; Gao, X.; Pagano, P.J.; Stolz, D.B.; Barchowsky, A. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J. Clin. Investig. 2008, 118, 3980–3989. [Google Scholar] [CrossRef]
- Chen, H.; Li, S.; Liu, J.; Diwan, B.A.; Barrett, J.C.; Waalkes, M.P. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: Implications for arsenic hepatocarcinogenesis. Carcinogenesis 2004, 25, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Reichard, J.F.; Schnekenburger, M.; Puga, A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 2007, 352, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.J.; Novak, P.; Wnek, S.M.; Gandolfi, A.J.; Futscher, B.W. Arsenicals produce stable progressive changes in DNA methylation patterns that are linked to malignant transformation of immortalized urothelial cells. Toxicol. Appl. Pharm. 2009, 241, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajees, A.A.; Rosen, B.P. As(III) S-adenosylmethionine methyltransferases and other arsenic binding proteins. Geomicrobiol. J. 2015, 32, 570–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.K.; Murphy, A.; Costa, M. Arsenic Methyltransferase and Methylation of Inorganic Arsenic. Biomolecules 2020, 10, 1351. [Google Scholar] [CrossRef] [PubMed]
- Chen Jian, R.B.P. The Arsenic Methylation Cycle: How Microbial Communities Adapted Methylarsenicals for Use as Weapons in the Continuing War for Dominance. Front. Environ. Sci. 2020, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Ajees, A.A.; Marapakala, K.; Packianathan, C.; Sankaran, B.; Rosen, B.P. Structure of an As(III) S-adenosylmethionine methyltransferase: Insights into the mechanism of arsenic biotransformation. Biochemistry 2012, 51, 5476–5485. [Google Scholar] [CrossRef] [Green Version]
- Kitchin, K.T. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharm. 2001, 172, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Petrick, J.S.; Jagadish, B.; Mash, E.A.; Aposhian, H.V. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001, 14, 651–656. [Google Scholar] [CrossRef]
- Lansdown, A.B. Physiological and toxicological changes in the skin resulting from the action and interaction of metal ions. Crit. Rev. Toxicol. 1995, 25, 397–462. [Google Scholar] [CrossRef]
- Samanta, G.; Sharma, R.; Roychowdhury, T.; Chakraborti, D. Arsenic and other elements in hair, nails, and skin-scales of arsenic victims in West Bengal, India. Sci. Total Environ. 2004, 326, 33–47. [Google Scholar] [CrossRef]
- Scott, N.; Hatlelid, K.M.; MacKenzie, N.E.; Carter, D.E. Reactions of arsenic(III) and arsenic(V) species with glutathione. Chem. Res. Toxicol. 1993, 6, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Uddin, A.N.; Burns, F.J.; Rossman, T.G. Vitamin E and organoselenium prevent the cocarcinogenic activity of arsenite with solar UVR in mouse skin. Carcinogenesis 2005, 26, 2179–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Styblo, M.; Rosen, B.P. Methylarsonous acid transport by aquaglyceroporins. Environ. Health Perspect 2006, 114, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latowski, A.K.a.D. Biochemical pathways of arsenic uptake from the environment to human cells. Mol. Biophys. Biochem. 2016, 1, 9–16. [Google Scholar] [CrossRef]
- Drobna, Z.; Walton, F.S.; Paul, D.S.; Xing, W.; Thomas, D.J.; Styblo, M. Metabolism of arsenic in human liver: The role of membrane transporters. Arch. Toxicol. 2010, 84, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kala, S.V.; Neely, M.W.; Kala, G.; Prater, C.I.; Atwood, D.W.; Rice, J.S.; Lieberman, M.W. The MRP2/cMOAT transporter and arsenic-glutathione complex formation are required for biliary excretion of arsenic. J. Biol. Chem. 2000, 275, 33404–33408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herath, I.; Vithanage, M.; Seneweera, S.; Bundschuh, J. Thiolated arsenic in natural systems: What is current, what is new and what needs to be known. Environ. Int. 2018, 115, 370–386. [Google Scholar] [CrossRef]
- Planer-Friedrich, B.; London, J.; McCleskey, R.B.; Nordstrom, D.K.; Wallschlager, D. Thioarsenates in geothermal waters of Yellowstone National Park: Determination, preservation, and geochemical importance. Environ. Sci. Technol. 2007, 41, 5245–5251. [Google Scholar] [CrossRef]
- Colina Blanco, A.E.; Kerl, C.F.; Planer-Friedrich, B. Detection of Thioarsenates in Rice Grains and Rice Products. J. Agric. Food Chem. 2021, 69, 2287–2294. [Google Scholar] [CrossRef]
- Wang, J.; Kerl, C.F.; Hu, P.; Martin, M.; Mu, T.; Brüggenwirth, L.; Wu, G.; Said-Pullicino, D.; Romani, M.; Wu, L.; et al. Thiolated arsenic species observed in rice paddy pore waters. Nat. Geosci. 2020, 13, 282–287. [Google Scholar] [CrossRef]
- SS, D.C.R.; Alava, P.; Zekker, I.; Du Laing, G.; Van de Wiele, T. Arsenic thiolation and the role of sulfate-reducing bacteria from the human intestinal tract. Environ. Health Perspect. 2014, 122, 817–822. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, G.; Cai, Y. Thiolated arsenicals in arsenic metabolism: Occurrence, formation, and biological implications. J. Environ. Sci. 2016, 49, 59–73. [Google Scholar] [CrossRef]
- Fricke, M.; Zeller, M.; Cullen, W.; Witkowski, M.; Creed, J. Dimethylthioarsinic anhydride: A standard for arsenic speciation. Anal. Chim. Acta 2007, 583, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Naranmandura, H.; Carew, M.W.; Xu, S.; Lee, J.; Leslie, E.M.; Weinfeld, M.; Le, X.C. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells. Chem. Res. Toxicol. 2011, 24, 1586–1596. [Google Scholar] [CrossRef]
- Guha Mazumder, D.N.; De, B.K.; Santra, A.; Ghosh, N.; Das, S.; Lahiri, S.; Das, T. Randomized placebo-controlled trial of 2,3-dimercapto-1-propanesulfonate (DMPS) in therapy of chronic arsenicosis due to drinking arsenic-contaminated water. J. Toxicol. Clin. Toxicol. 2001, 39, 665–674. [Google Scholar] [CrossRef]
- Bjorklund, G.; Oliinyk, P.; Lysiuk, R.; Rahaman, M.S.; Antonyak, H.; Lozynska, I.; Lenchyk, L.; Peana, M. Arsenic intoxication: General aspects and chelating agents. Arch. Toxicol. 2020, 94, 1879–1897. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Djordjevic, A.B.; Crisponi, G.; Alexander, J.; Bjorklund, G.; Aaseth, J. Arsenic Toxicity: Molecular Targets and Therapeutic Agents. Biomolecules 2020, 10, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiman, N.J.; Quinn, A.M.; Fields, K.G.; Slavkovich, V.; Graziano, J.H. Arsenite accumulation in the mouse eye. J. Toxicol. Environ. Health A 2016, 79, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athyros, V.G.; Doumas, M.; Imprialos, K.P.; Stavropoulos, K.; Georgianou, E.; Katsimardou, A.; Karagiannis, A. Diabetes and lipid metabolism. Hormones 2018, 17, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloska, A.; Węsierska, M.; Malinowska, M.; Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J. Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. Int. J. Mol. Sci. 2020, 21, 6113. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Su, P.H.; Sun, C.W.; Liu, H.J.; Chang, C.L.; Wang, S.L. Early-life arsenic exposure promotes atherogenic lipid metabolism in adolescence: A 15-year birth cohort follow-up study in central Taiwan. Environ. Int. 2018, 118, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, O.K.; Wusu, A.D.; Ogunrinola, O.O.; Abam, E.O.; Babayemi, D.O.; Dosumu, O.A.; Onunkwor, O.B.; Balogun, E.A.; Odukoya, O.O.; Ademuyiwa, O. Arsenic-induced dyslipidemia in male albino rats: Comparison between trivalent and pentavalent inorganic arsenic in drinking water. BMC Pharm. Toxicol. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, L.; Lai, Y.; Tu, P.; Liu, C.W.; Xue, J.; Ru, H.; Lu, K. Lipid and Cholesterol Homeostasis after Arsenic Exposure and Antibiotic Treatment in Mice: Potential Role of the Microbiota. Environ. Health Perspect 2019, 127, 97002. [Google Scholar] [CrossRef] [PubMed]
- Soni, M.; Prakash, C.; Dabur, R.; Kumar, V. Protective Effect of Hydroxytyrosol Against Oxidative Stress Mediated by Arsenic-Induced Neurotoxicity in Rats. Appl Biochem. Biotechnol. 2018, 186, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.; Van Beneden, R.J. Arsenic exposure alters expression of cell cycle and lipid metabolism genes in the liver of adult zebrafish (Danio rerio). Aquat. Toxicol. 2014, 153, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.J.; Chuu, J.J.; Chang, C.Y.; Tsai, W.C.; Chen, K.J.; Guo, H.R. Atherosclerosis induced by arsenic in drinking water in rats through altering lipid metabolism. Toxicol. Appl. Pharm. 2011, 256, 146–153. [Google Scholar] [CrossRef]
- Rivas-Santiago, C.; Gonzalez-Curiel, I.; Zarazua, S.; Murgu, M.; Ruiz Cardona, A.; Lazalde, B.; Lara-Ramirez, E.E.; Vazquez, E.; Castaneda-Delgado, J.E.; Rivas-Santiago, B.; et al. Lipid Metabolism Alterations in a Rat Model of Chronic and Intergenerational Exposure to Arsenic. Biomed. Res. Int. 2019, 2019, 4978018. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, Y.; Liu, J.; Ji, X.; Zhao, L.; Wei, Y. Changes in Serum Adiponectin in Mice Chronically Exposed to Inorganic Arsenic in Drinking Water. Biol. Trace Elem. Res. 2017, 179, 140–147. [Google Scholar] [CrossRef]
- Renu, K.; Madhyastha, H.; Madhyastha, R.; Maruyama, M.; Arunachlam, S.; V G, A. Role of arsenic exposure in adipose tissue dysfunction and its possible implication in diabetes pathophysiology. Toxicol. Lett. 2018, 284, 86–95. [Google Scholar] [CrossRef]
- Hou, Y.; Xue, P.; Woods, C.G.; Wang, X.; Fu, J.; Yarborough, K.; Qu, W.; Zhang, Q.; Andersen, M.E.; Pi, J. Association between arsenic suppression of adipogenesis and induction of CHOP10 via the endoplasmic reticulum stress response. Environ. Health Perspect 2013, 121, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmaja Divya, S.; Pratheeshkumar, P.; Son, Y.O.; Vinod Roy, R.; Andrew Hitron, J.; Kim, D.; Dai, J.; Wang, L.; Asha, P.; Huang, B.; et al. Arsenic Induces Insulin Resistance in Mouse Adipocytes and Myotubes Via Oxidative Stress-Regulated Mitochondrial Sirt3-FOXO3a Signaling Pathway. Toxicol. Sci. 2015, 146, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Trouba, K.J.; Wauson, E.M.; Vorce, R.L. Sodium arsenite inhibits terminal differentiation of murine C3H 10T1/2 preadipocytes. Toxicol. Appl. Pharm. 2000, 168, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Garciafigueroa, D.Y.; Klei, L.R.; Ambrosio, F.; Barchowsky, A. Arsenic-stimulated lipolysis and adipose remodeling is mediated by G-protein-coupled receptors. Toxicol. Sci. 2013, 134, 335–344. [Google Scholar] [CrossRef]
- Bae, J.; Jang, Y.; Kim, H.; Mahato, K.; Schaecher, C.; Kim, I.M.; Kim, E.; Ro, S.H. Arsenite exposure suppresses adipogenesis, mitochondrial biogenesis and thermogenesis via autophagy inhibition in brown adipose tissue. Sci. Rep. 2019, 9, 14464. [Google Scholar] [CrossRef]
- Zuo, Z.; Liu, Z.; Gao, T.; Yin, Y.; Wang, Z.; Hou, Y.; Fu, J.; Liu, S.; Wang, H.; Xu, Y.; et al. Prolonged inorganic arsenic exposure via drinking water impairs brown adipose tissue function in mice. Sci. Total Environ. 2019, 668, 310–317. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Ceriani, F.; Procopio, A.; Bonacci, S.; Malandra, R.; Panseri, S.; Arioli, F. Exposure to metals and arsenic from yellow and red tuna consumption. Food Addit. Contam. Part A Chem Anal. Control. Expo. Risk Assess. 2019, 36, 1228–1235. [Google Scholar] [CrossRef]
- Farzad, R.; Kuhn, D.D.; Smith, S.A.; O’Keefe, S.F.; Ralston, N.V.C.; Neilson, A.P.; Gatlin, D.M. Trace minerals in tilapia fillets: Status in the United States marketplace and selenium supplementation strategy for improving consumer’s health. PLoS ONE 2019, 14, e0217043. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Zuo, J.; Wang, X.; Yin, J.; Liu, L.; Fan, W. The Bioaccumulation and Tissue Distribution of Arsenic Species in Tilapia. Int J. Environ. Res. Public Health 2019, 16, 757. [Google Scholar] [CrossRef] [Green Version]
- Szymkowicz, D.B.; Schwendinger, K.L.; Tatnall, C.M.; Swetenburg, J.R.; Bain, L.J. Embryonic-only arsenic exposure alters skeletal muscle satellite cell function in killifish (Fundulus heteroclitus). Aquat. Toxicol. 2018, 198, 276–286. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Shao, Y.; Liu, J.; Li, J.; Luo, L.; Xing, M. Copper (II) and/or arsenite-induced oxidative stress cascades apoptosis and autophagy in the skeletal muscles of chicken. Chemosphere 2018, 206, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Chung, M.N.; Chiu, C.Y.; Liu, S.H.; Lan, K.C. Inorganic Arsenic Exposure Decreases Muscle Mass and Enhances Denervation-Induced Muscle Atrophy in Mice. Molecules 2020, 25, 3057. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Chang, C.S.; Tang, J.L.; Tien, H.F.; Kuo, T.L.; Huang, S.F.; Yao, Y.T.; Chou, W.C.; Chung, C.Y.; Wang, C.H.; et al. Acute and chronic arsenic poisoning associated with treatment of acute promyelocytic leukaemia. Br. J. Haematol. 1998, 103, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.K.; Tony, S.R.; Siddique, A.E.; Karim, M.R.; Haque, N.; Islam, Z.; Islam, M.S.; Khatun, M.; Islam, J.; Hossain, S.; et al. Arsenic Secondary Methylation Capacity Is Inversely Associated with Arsenic Exposure-Related Muscle Mass Reduction. Int. J. Environ. Res. Public Health 2021, 18, 9730. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.Y.A.; Briede, J.J.; van Herwijnen, M.; Krauskopf, J.; Jennen, D.G.J.; Malik, R.N.; Kleinjans, J.C.S. Integrating SNPs-based genetic risk factor with blood epigenomic response of differentially arsenic-exposed rural subjects reveals disease-associated signaling pathways. Environ. Pollut. 2022, 292, 118279. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Chung, M.N.; Lan, K.C.; Yang, R.S.; Liu, S.H. Exposure of low-concentration arsenic induces myotube atrophy by inhibiting an Akt signaling pathway. Toxicol. Vitr. 2020, 65, 104829. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.M.; Bain, L.J. Arsenic exposure inhibits myogenesis and neurogenesis in P19 stem cells through repression of the beta-catenin signaling pathway. Toxicol. Sci. 2012, 129, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Villasenor, A.; Burns, A.L.; Hiriart, M.; Cebrian, M.E.; Ostrosky-Wegman, P. Arsenic-induced alteration in the expression of genes related to type 2 diabetes mellitus. Toxicol. Appl. Pharm. 2007, 225, 123–133. [Google Scholar] [CrossRef]
- Drobna, Z.; Styblo, M.; Thomas, D.J. An Overview of Arsenic Metabolism and Toxicity. Curr. Protoc. Toxicol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.F. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 2002, 133, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Moon, K.A.; Oberoi, S.; Barchowsky, A.; Chen, Y.; Guallar, E.; Nachman, K.E.; Rahman, M.; Sohel, N.; D’Ippoliti, D.; Wade, T.J.; et al. A dose-response meta-analysis of chronic arsenic exposure and incident cardiovascular disease. Int. J. Epidemiol. 2017, 46, 1924–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.T.; López-Carrillo, L.; Gamboa-Loira, B.; Cebrián, M.E. Standards for arsenic in drinking water: Implications for policy in Mexico. J. Public Health Policy 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, D.J.; Naujokas, M.F.; Bradham, K.D.; Cowden, J.; Heacock, M.; Henry, H.F.; Lee, J.S.; Thomas, D.J.; Thompson, C.; Tokar, E.J.; et al. Arsenic and Environmental Health: State of the Science and Future Research Opportunities. Environ. Health Perspect. 2016, 124, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Argos, M.; Kalra, T.; Rathouz, P.J.; Chen, Y.; Pierce, B.; Parvez, F.; Islam, T.; Ahmed, A.; Rakibuz-Zaman, M.; Hasan, R.; et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): A prospective cohort study. Lancet 2010, 376, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Shao, K.; Zhou, Z.; Xun, P.; Cohen, S.M. Bayesian benchmark dose analysis for inorganic arsenic in drinking water associated with bladder and lung cancer using epidemiological data. Toxicology 2021, 455, 152752. [Google Scholar] [CrossRef]
- Sidhu, M.S.; Desai, K.P.; Lynch, H.N.; Rhomberg, L.R.; Beck, B.D.; Venditti, F.J. Mechanisms of action for arsenic in cardiovascular toxicity and implications for risk assessment. Toxicology 2015, 331, 78–99. [Google Scholar] [CrossRef]
- Paul, D.S.; Hernandez-Zavala, A.; Walton, F.S.; Adair, B.M.; Dedina, J.; Matousek, T.; Styblo, M. Examination of the effects of arsenic on glucose homeostasis in cell culture and animal studies: Development of a mouse model for arsenic-induced diabetes. Toxicol. Appl. Pharm. 2007, 222, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Walton, F.S.; Harmon, A.W.; Paul, D.S.; Drobna, Z.; Patel, Y.M.; Styblo, M. Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: Possible mechanism of arsenic-induced diabetes. Toxicol. Appl. Pharm. 2004, 198, 424–433. [Google Scholar] [CrossRef]
- Douillet, C.; Currier, J.; Saunders, J.; Bodnar, W.M.; Matousek, T.; Styblo, M. Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol. Appl. Pharm. 2013, 267, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Dover, E.N.; Beck, R.; Huang, M.C.; Douillet, C.; Wang, Z.; Klett, E.L.; Styblo, M. Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 beta cells. Arch. Toxicol. 2018, 92, 693–704. [Google Scholar] [CrossRef]
- Brauner, E.V.; Nordsborg, R.B.; Andersen, Z.J.; Tjonneland, A.; Loft, S.; Raaschou-Nielsen, O. Long-term exposure to low-level arsenic in drinking water and diabetes incidence: A prospective study of the diet, cancer and health cohort. Environ. Health Perspect. 2014, 122, 1059–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, R.K.; Lipmann, F. The effect of arsenate on aerobic phosphorylation. J. Biol. Chem. 1953, 201, 235–243. [Google Scholar] [CrossRef]
- Hosseini, M.J.; Shaki, F.; Ghazi-Khansari, M.; Pourahmad, J. Toxicity of Arsenic (III) on Isolated Liver Mitochondria: A New Mechanistic Approach. Iran. J. Pharm Res. 2013, 12, 121–138. [Google Scholar] [PubMed]
- Ramanathan, K.; Shila, S.; Kumaran, S.; Panneerselvam, C. Ascorbic acid and α-tocopherol as potent modulators on arsenic induced toxicity in mitochondria. J. Nutr. Biochem. 2003, 14, 416–420. [Google Scholar] [CrossRef]
- Prakash, C.; Kumar, V. Chronic Arsenic Exposure-Induced Oxidative Stress is Mediated by Decreased Mitochondrial Biogenesis in Rat Liver. Biol. Trace Elem. Res. 2016, 173, 87–95. [Google Scholar] [CrossRef]
- Partridge, M.A.; Huang, S.X.; Hernandez-Rosa, E.; Davidson, M.M.; Hei, T.K. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: Implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007, 67, 5239–5247. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic Exposure and the Induction of Human Cancers. J. Toxicol. 2011, 2011, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, F.; Brown, E.; Stolz, D.; Ferrari, R.; Goodpaster, B.; Deasy, B.; Distefano, G.; Roperti, A.; Cheikhi, A.; Garciafigueroa, Y.; et al. Arsenic induces sustained impairment of skeletal muscle and muscle progenitor cell ultrastructure and bioenergetics. Free Radic. Biol. Med. 2014, 74, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ferrari, R.; Beezhold, K.; Stearns-Reider, K.; D’Amore, A.; Haschak, M.; Stolz, D.; Robbins, P.D.; Barchowsky, A.; Ambrosio, F. Arsenic Promotes NF-Kappab-Mediated Fibroblast Dysfunction and Matrix Remodeling to Impair Muscle Stem Cell Function. Stem. Cells 2016, 34, 732–742. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.; Gjerstad, M.D.; Jakobsen, J. Atrophy of Foot Muscles: A measure of diabetic neuropathy. Diabetes Care 2004, 27, 2382–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.; Schmitz, O.; Nielsen, S. Decreased isometric muscle strength after acute hyperglycaemia in Type 1 diabetic patients. Diabet Med. 2005, 22, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive Adipose Tissue Infiltration in Skeletal Muscle in Individuals With Obesity, Diabetes Mellitus, and Peripheral Neuropathy: Association With Performance and Function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas-Acien, A.; Silbergeld, E.K.; Streeter, R.A.; Clark, J.M.; Burke, T.A.; Guallar, E. Arsenic exposure and type 2 diabetes: A systematic review of the experimental and epidemiological evidence. Environ. Health Perspect. 2006, 114, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.X.; Jiang, C.S.; Liu, L.; Wang, X.H.; Jin, H.J.; Wu, Q.; Chen, Q. The role of Akt on arsenic trioxide suppression of 3T3-L1 preadipocyte differentiation. Cell Res. 2005, 15, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Salazard, B.; Bellon, L.; Jean, S.; Maraninchi, M.; El-Yazidi, C.; Orsière, T.; Margotat, A.; Botta, A.; Bergé-Lefranc, J.L. Low-level arsenite activates the transcription of genes involved in adipose differentiation. Cell Biol. Toxicol. 2004, 20, 375–385. [Google Scholar] [CrossRef]
- Wauson, E.M.; Langan, A.S.; Vorce, R.L. Sodium arsenite inhibits and reverses expression of adipogenic and fat cell-specific genes during in vitro adipogenesis. Toxicol. Sci. 2002, 65, 211–219. [Google Scholar] [CrossRef] [Green Version]
- McDowell, H.E.; Walker, T.; Hajduch, E.; Christie, G.; Batty, I.H.; Downes, C.P.; Hundal, H.S. Inositol phospholipid 3-kinase is activated by cellular stress but is not required for the stress-induced activation of glucose transport in L6 rat skeletal muscle cells. Eur. J. Biochem. 1997, 247, 306–313. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Pawlowska, E.; Chojnacki, J.; Szczepanska, J.; Chojnacki, C.; Kaarniranta, K. Zinc and Autophagy in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4994. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Rimbach, G. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev. 2013, 12, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Nguyen, H.Q.; Hwang, J.S.; Zada, S.; Lai, T.H.; Kang, S.S.; Kim, D.R. Systematic characterization of autophagy-related genes during the adipocyte differentiation using public-access data. Oncotarget 2018, 9, 15526–15541. [Google Scholar] [CrossRef] [Green Version]
- Kovsan, J.; Bluher, M.; Tarnovscki, T.; Kloting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schon, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Kloting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Soussi, H.; Reggio, S.; Alili, R.; Prado, C.; Mutel, S.; Pini, M.; Rouault, C.; Clement, K.; Dugail, I. DAPK2 Downregulation Associates With Attenuated Adipocyte Autophagic Clearance in Human Obesity. Diabetes 2015, 64, 3452–3463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharm. 2017, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, X.; Jin, S. Autophagy in adipose tissue biology. Pharm. Res 2012, 66, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Baerga, R.; Zhang, Y.; Chen, P.H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, H.; Shao, Y.; Liu, J.; Li, J.; Luo, L.; Xing, M. Copper or/and arsenic induces autophagy by oxidative stress-related PI3K/AKT/mTOR pathways and cascaded mitochondrial fission in chicken skeletal muscle. J. Inorg. Biochem. 2018, 188, 1–8. [Google Scholar] [CrossRef]
- Yang, L.; Qiu, T.; Yao, X.; Jiang, L.; Wei, S.; Pei, P.; Wang, Z.; Bai, J.; Liu, X.; Yang, G.; et al. Taurine protects against arsenic trioxide-induced insulin resistance via ROS-Autophagy pathway in skeletal muscle. Int. J. Biochem. Cell Biol. 2019, 112, 50–60. [Google Scholar] [CrossRef]
- Bessho, M.; Aki, T.; Funakoshi, T.; Unuma, K.; Noritake, K.; Kato, C.; Uemura, K. Rho-kinase inhibitor Y-27632 attenuates arsenic trioxide toxicity in H9c2 cardiomyoblastoma cells. Cardiovasc. Toxicol. 2013, 13, 267–277. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox. Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Liu, P.; Jiang, T.; Ambrose, A.J.; Luo, G.; Rojo de la Vega, M.; Cholanians, A.B.; Wong, P.K.; Chapman, E.; Zhang, D.D. Increased O-GlcNAcylation of SNAP29 Drives Arsenic-Induced Autophagic Dysfunction. Mol. Cell Biol. 2018, 38, e00595-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahidnia, A.; van der Voet, G.B.; de Wolff, F.A. Arsenic neurotoxicity—A review. Hum. Exp. Toxicol. 2007, 26, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y.; Wu, Q.; Goyer, R.A.; Waalkes, M.P. Mineral arsenicals in traditional medicines: Orpiment, realgar, and arsenolite. J. Pharm. Exp. 2008, 326, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Au, W.Y. A biography of arsenic and medicine in Hong Kong and China. Hong Kong Med. J. 2011, 17, 507–513. [Google Scholar] [PubMed]
- Waxman, S.; Anderson, K.C. History of the development of arsenic derivatives in cancer therapy. Oncologist 2001, 6 (Suppl. 2), 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ro, S.-H.; Bae, J.; Jang, Y.; Myers, J.F.; Chung, S.; Yu, J.; Natarajan, S.K.; Franco, R.; Song, H.-S. Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues. Antioxidants 2022, 11, 689. https://doi.org/10.3390/antiox11040689
Ro S-H, Bae J, Jang Y, Myers JF, Chung S, Yu J, Natarajan SK, Franco R, Song H-S. Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues. Antioxidants. 2022; 11(4):689. https://doi.org/10.3390/antiox11040689
Chicago/Turabian StyleRo, Seung-Hyun, Jiyoung Bae, Yura Jang, Jacob F. Myers, Soonkyu Chung, Jiujiu Yu, Sathish Kumar Natarajan, Rodrigo Franco, and Hyun-Seob Song. 2022. "Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues" Antioxidants 11, no. 4: 689. https://doi.org/10.3390/antiox11040689
APA StyleRo, S.-H., Bae, J., Jang, Y., Myers, J. F., Chung, S., Yu, J., Natarajan, S. K., Franco, R., & Song, H.-S. (2022). Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues. Antioxidants, 11(4), 689. https://doi.org/10.3390/antiox11040689