
Folic Acid Homeostasis and Its Pathways Related to Hepatic Oxidation in Adolescent Rats Exposed to Binge Drinking

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Diet Used
2.3. Binge Drinking Treatment
2.4. Nutritional Control
2.5. Samples
2.6. Serum Measurements
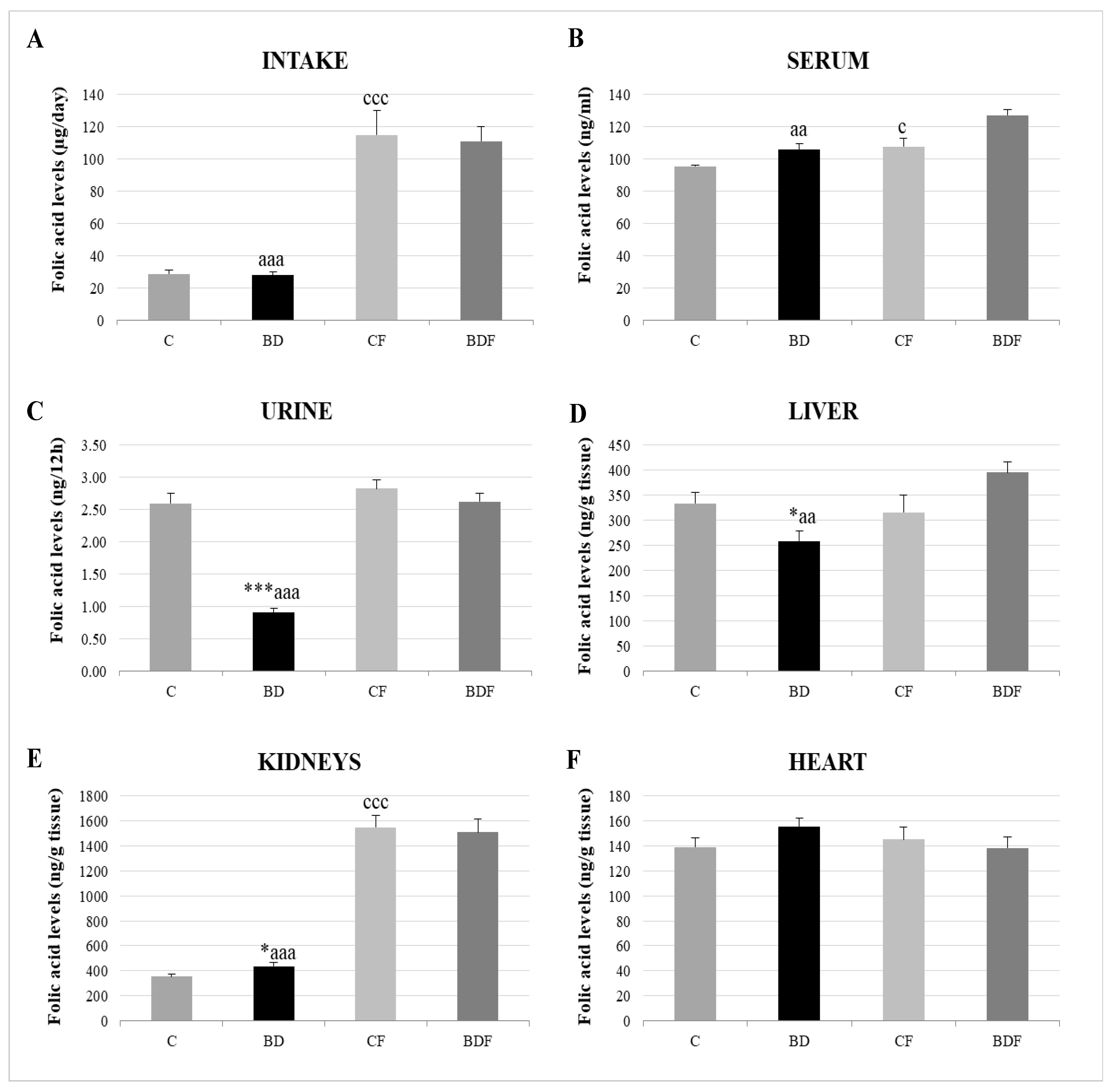
2.7. Folic Acid Homeostasis
2.8. Activity of Antioxidant Enzymes and Oxidative Stress Markers in the Liver
2.9. GSH Hepatic Levels
2.10. Nitric Oxide Hepatic Levels
2.11. Immunoblotting Analysis in the Liver
2.12. Ratios of Measurements
2.13. Statistical Analysis
3. Results
3.1. Nutritional and Hepatic Parameters
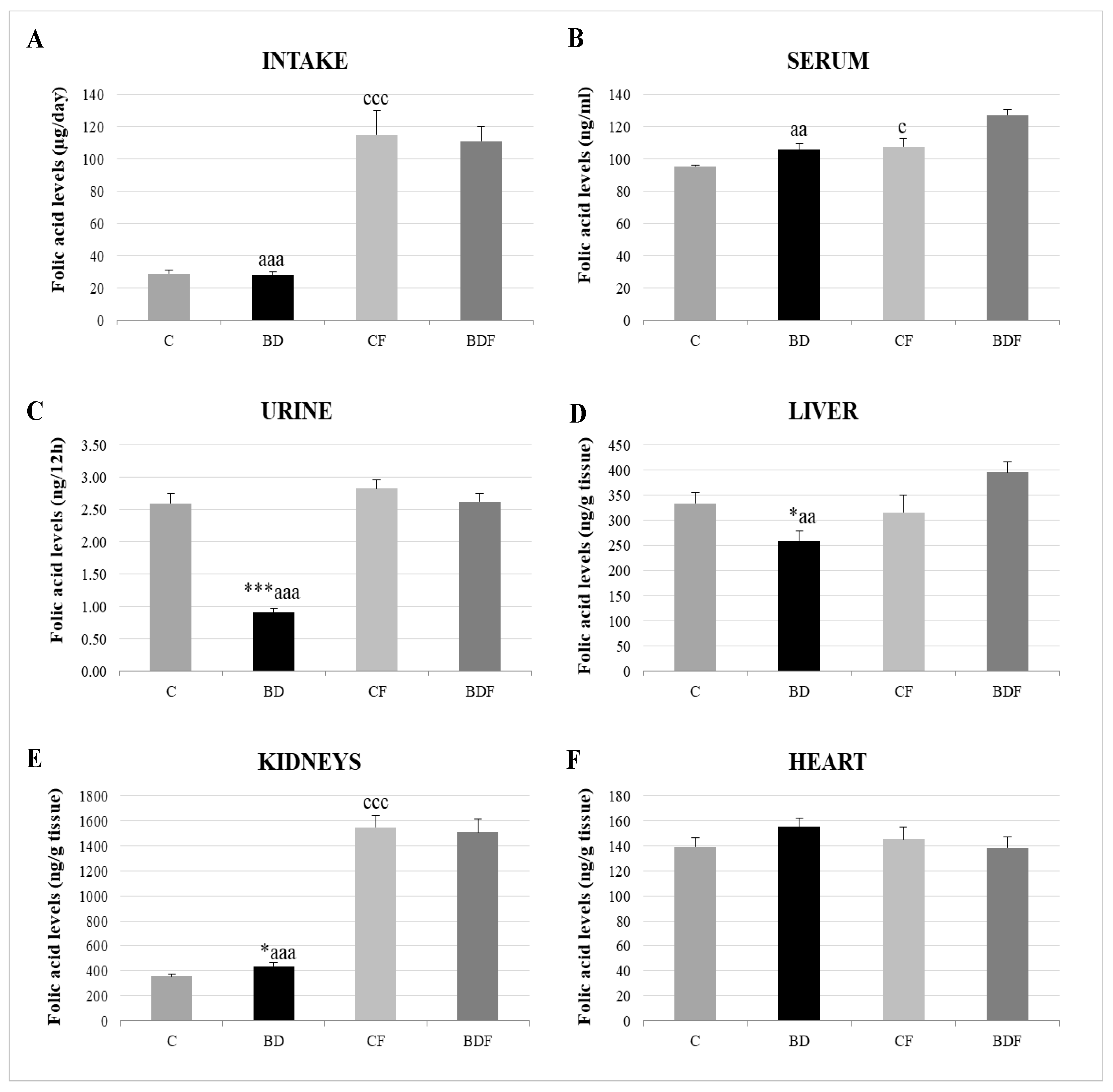
3.2. Folic Acid Homeostasis
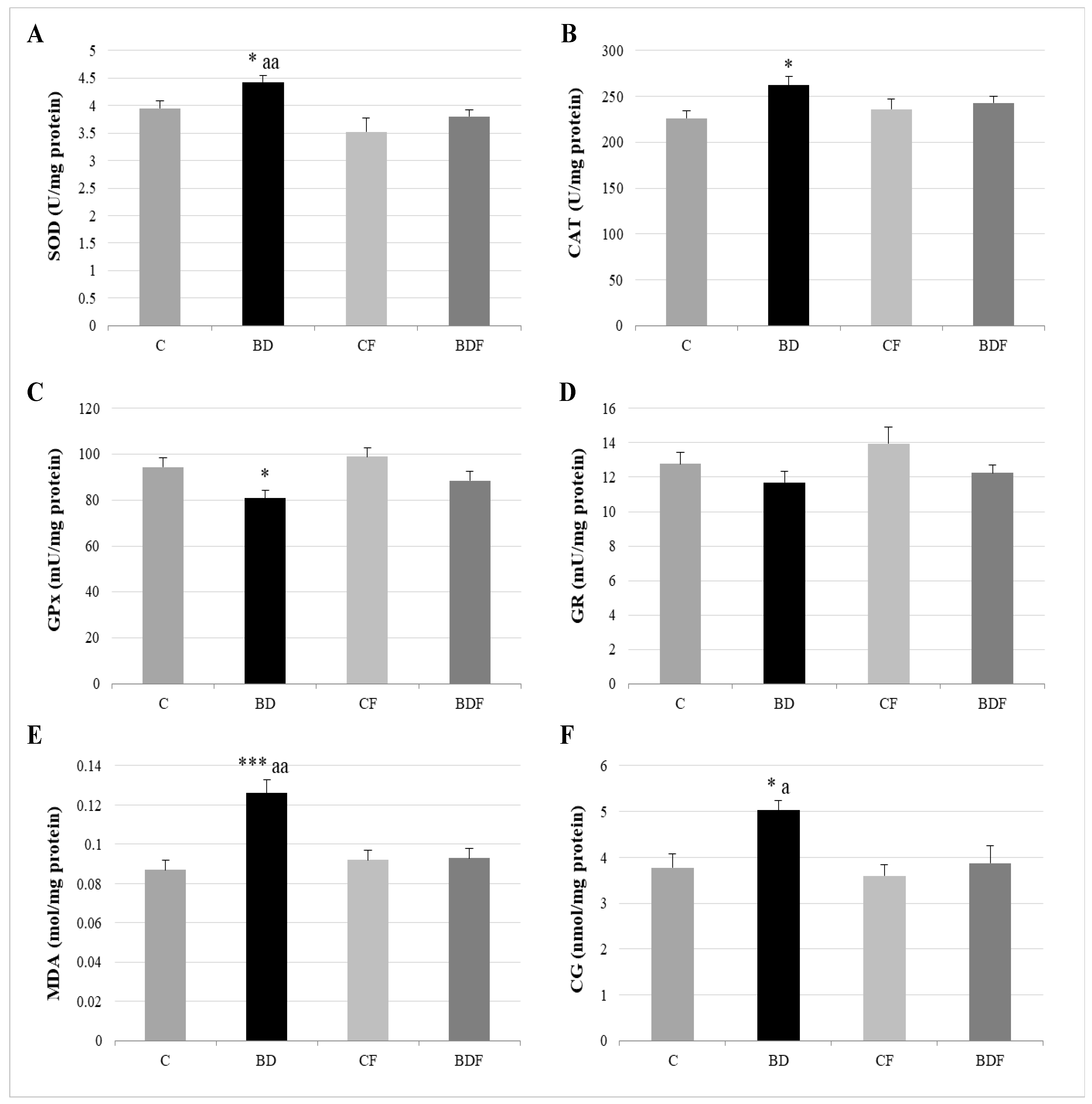
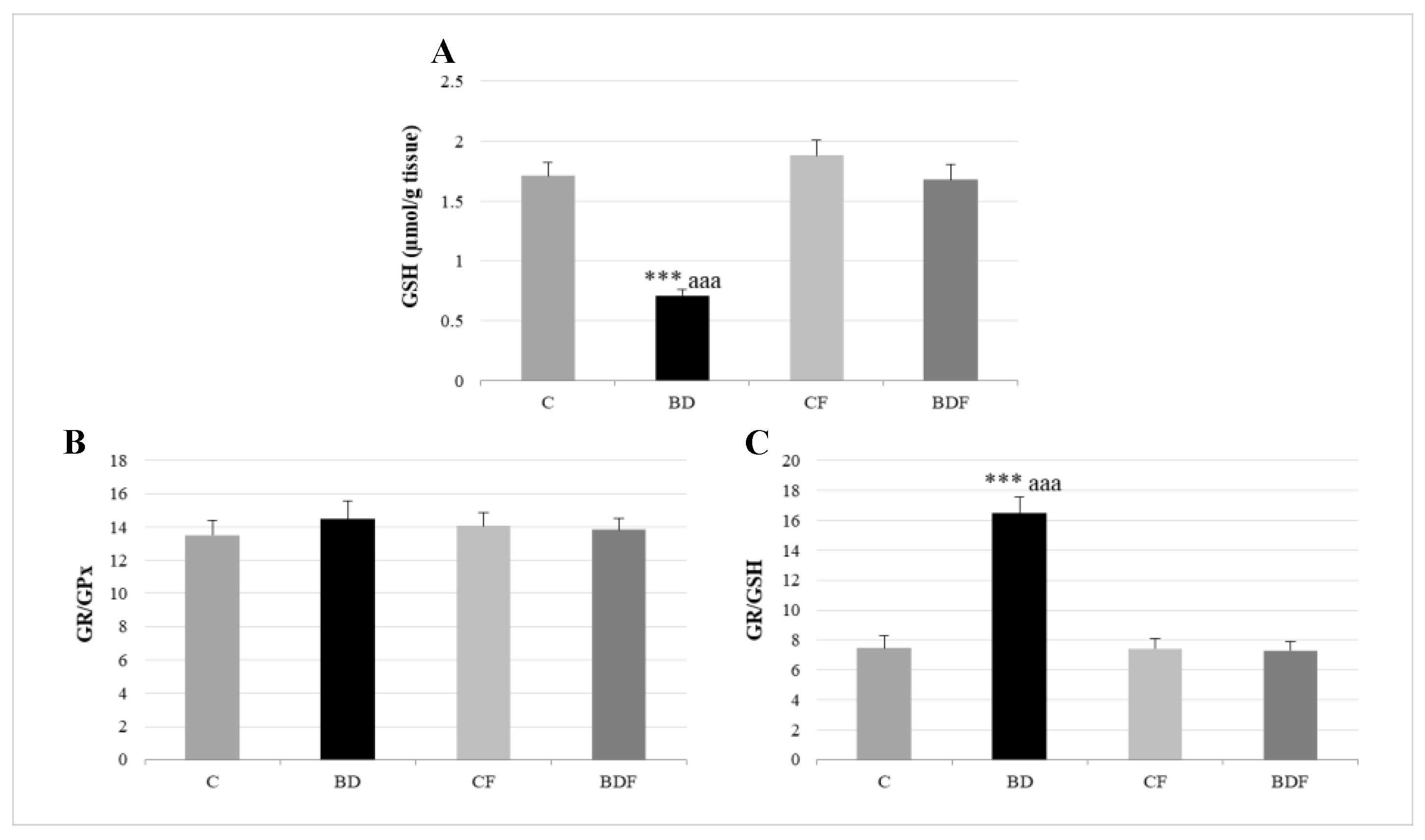
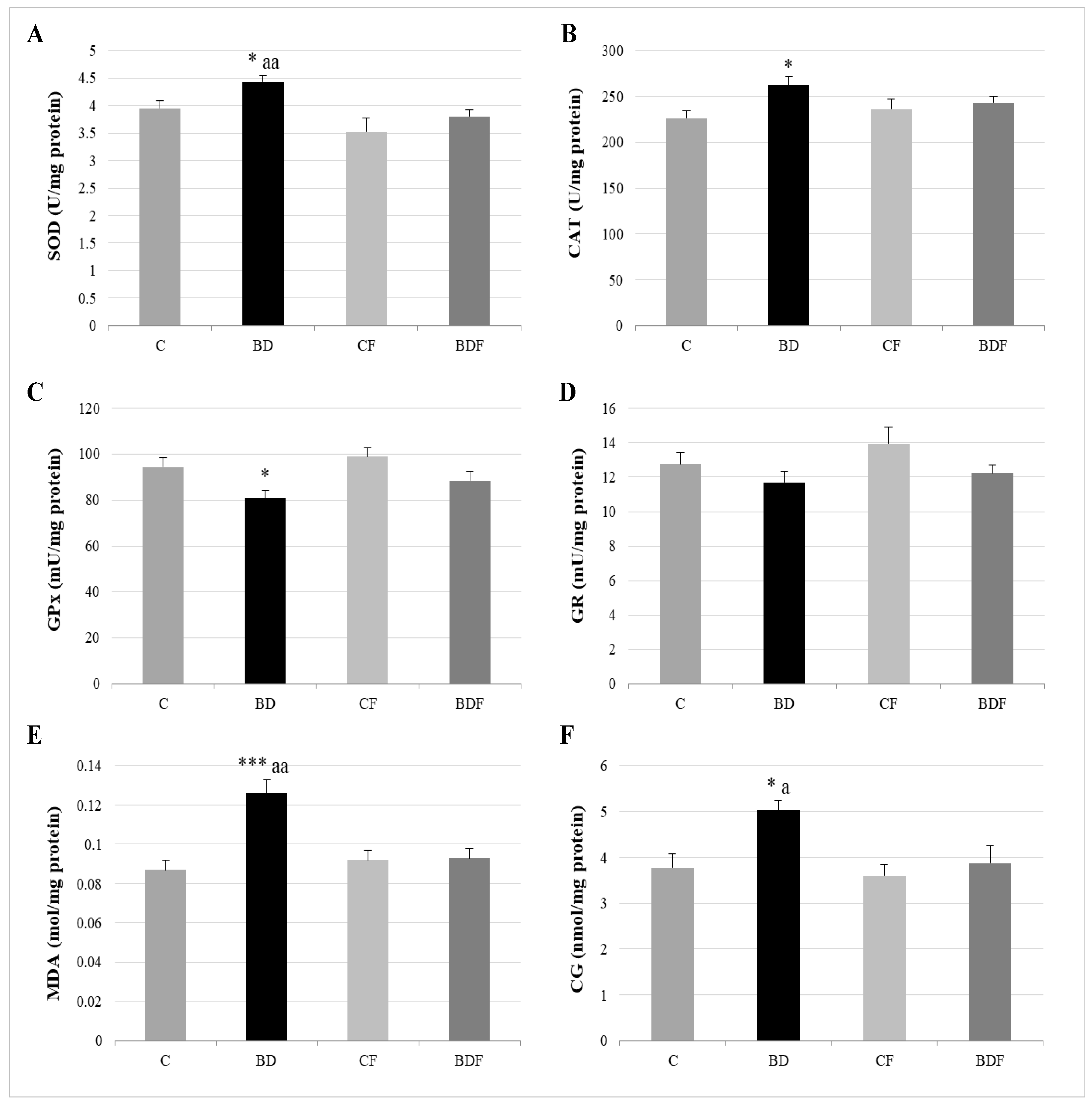
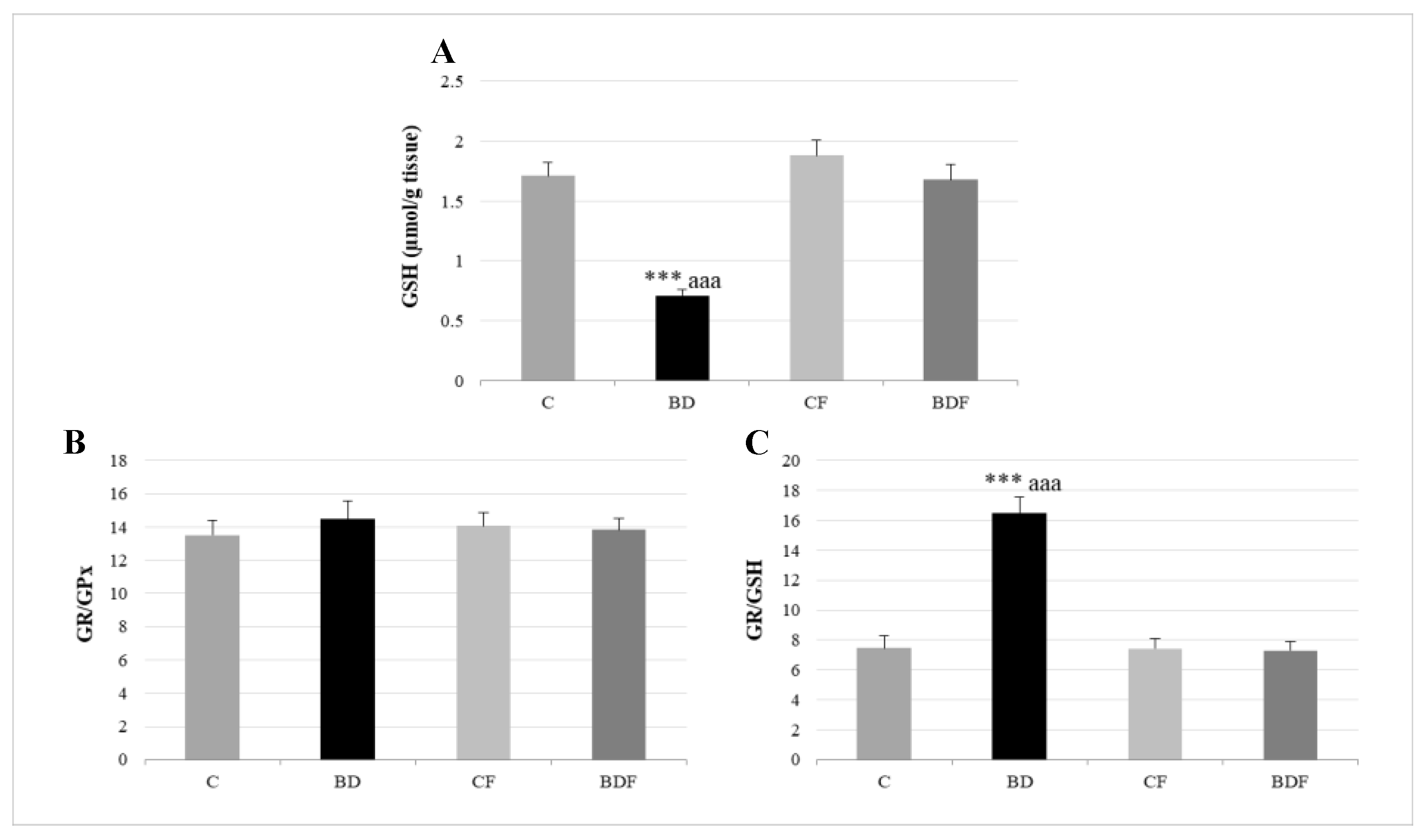
3.3. Oxidative Hepatic Balance
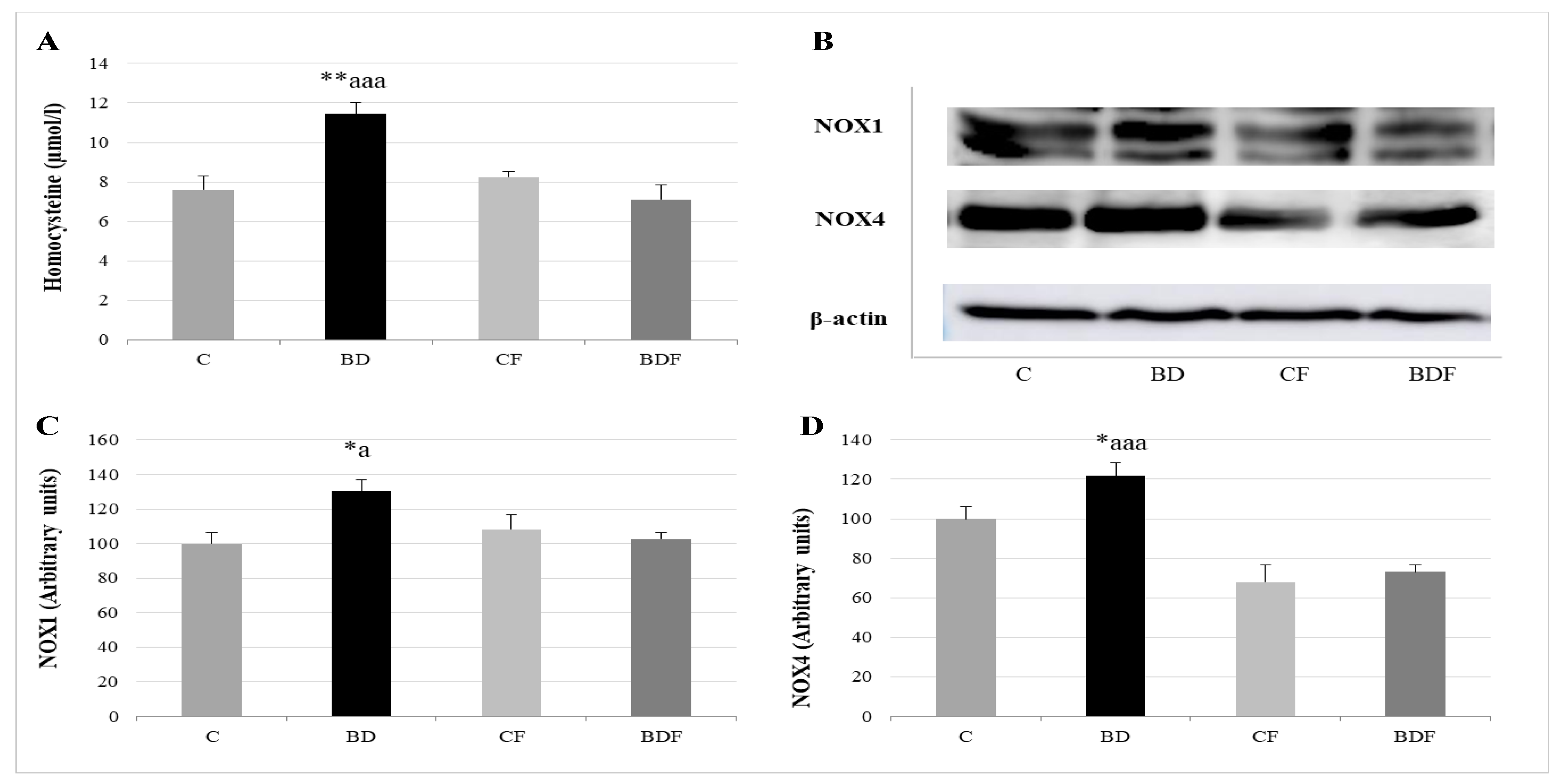
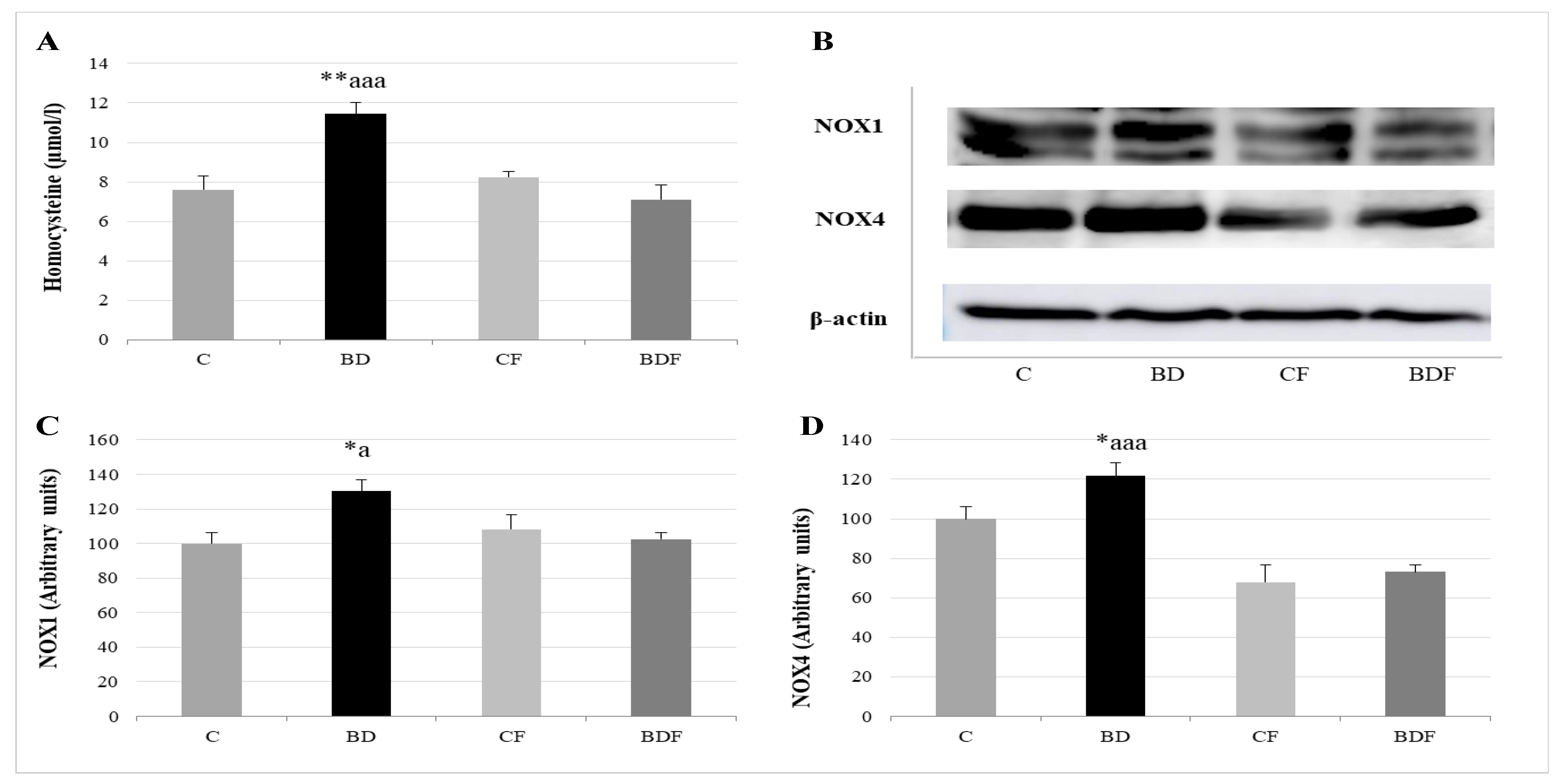
3.4. Serum Homocysteine and NOX Hepatic Expression
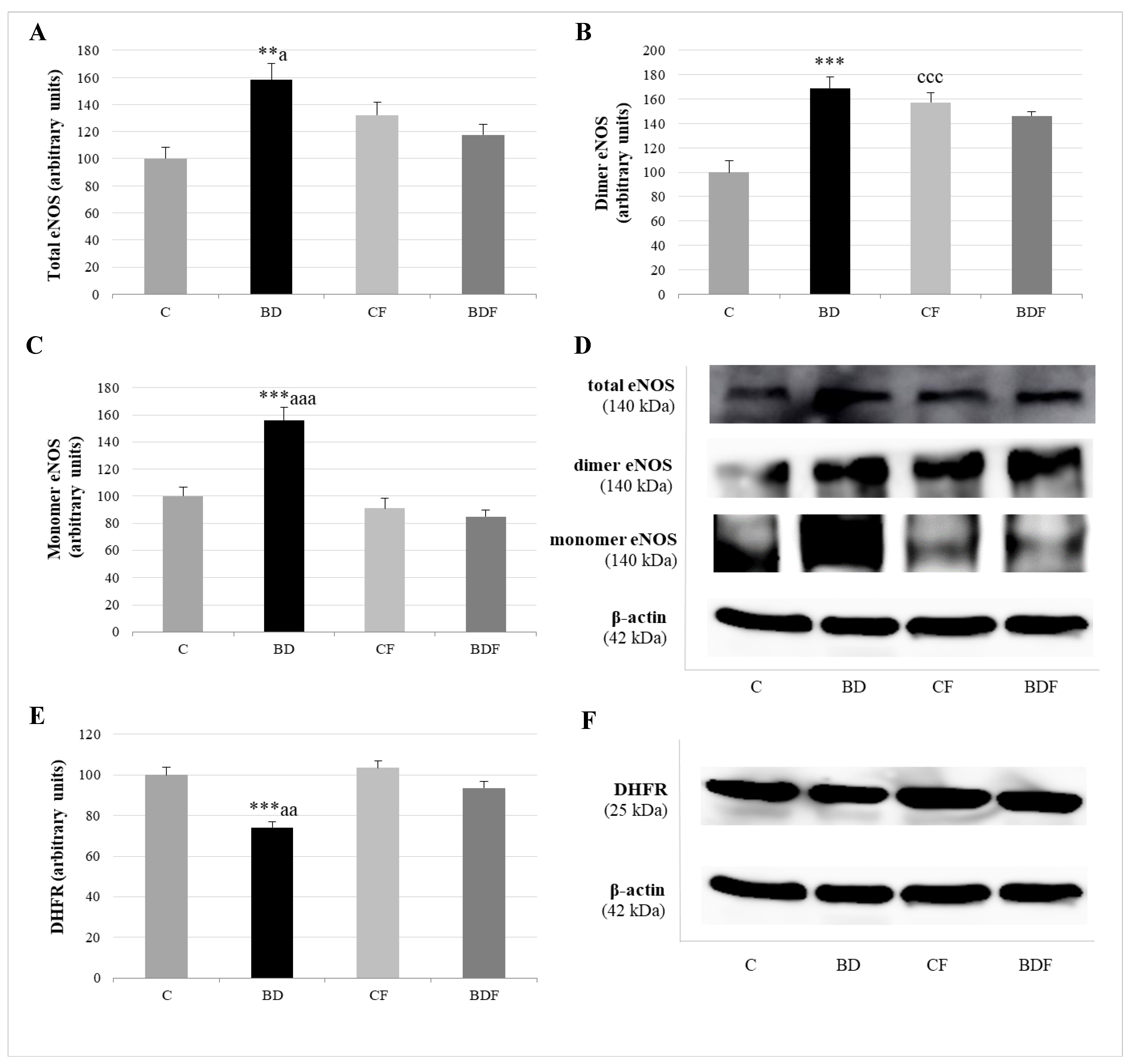
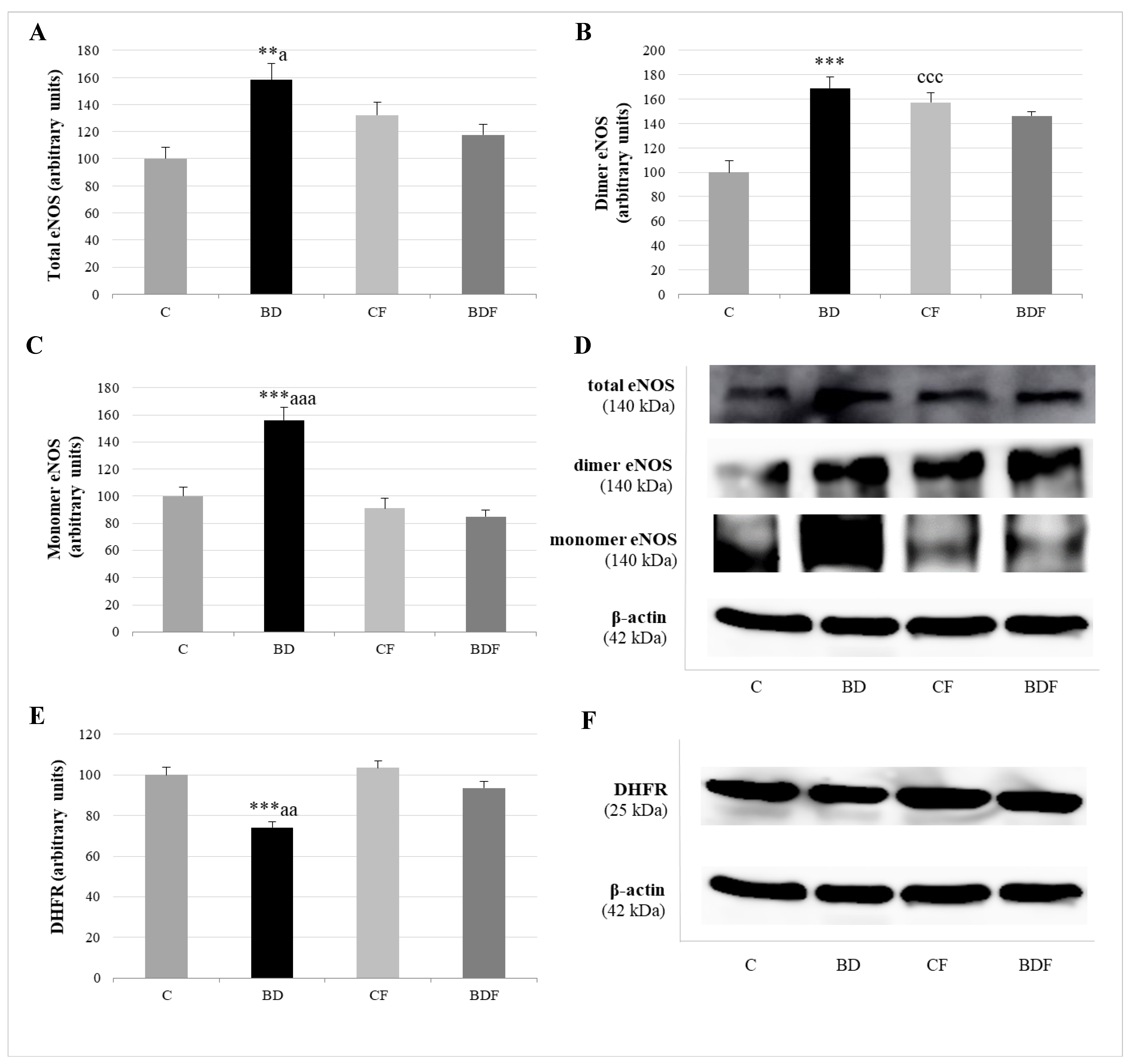
3.5. Hepatic eNOS Expression
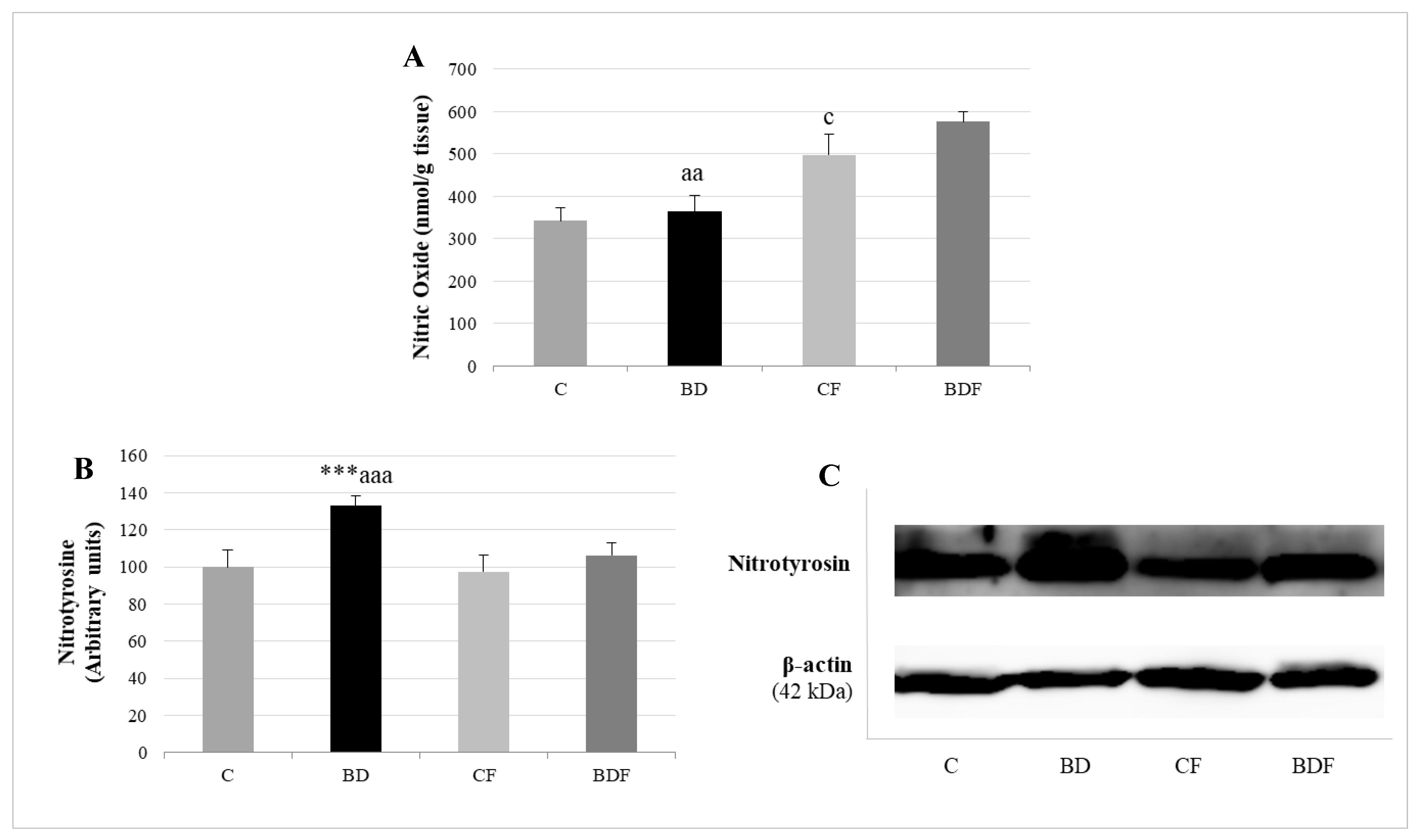
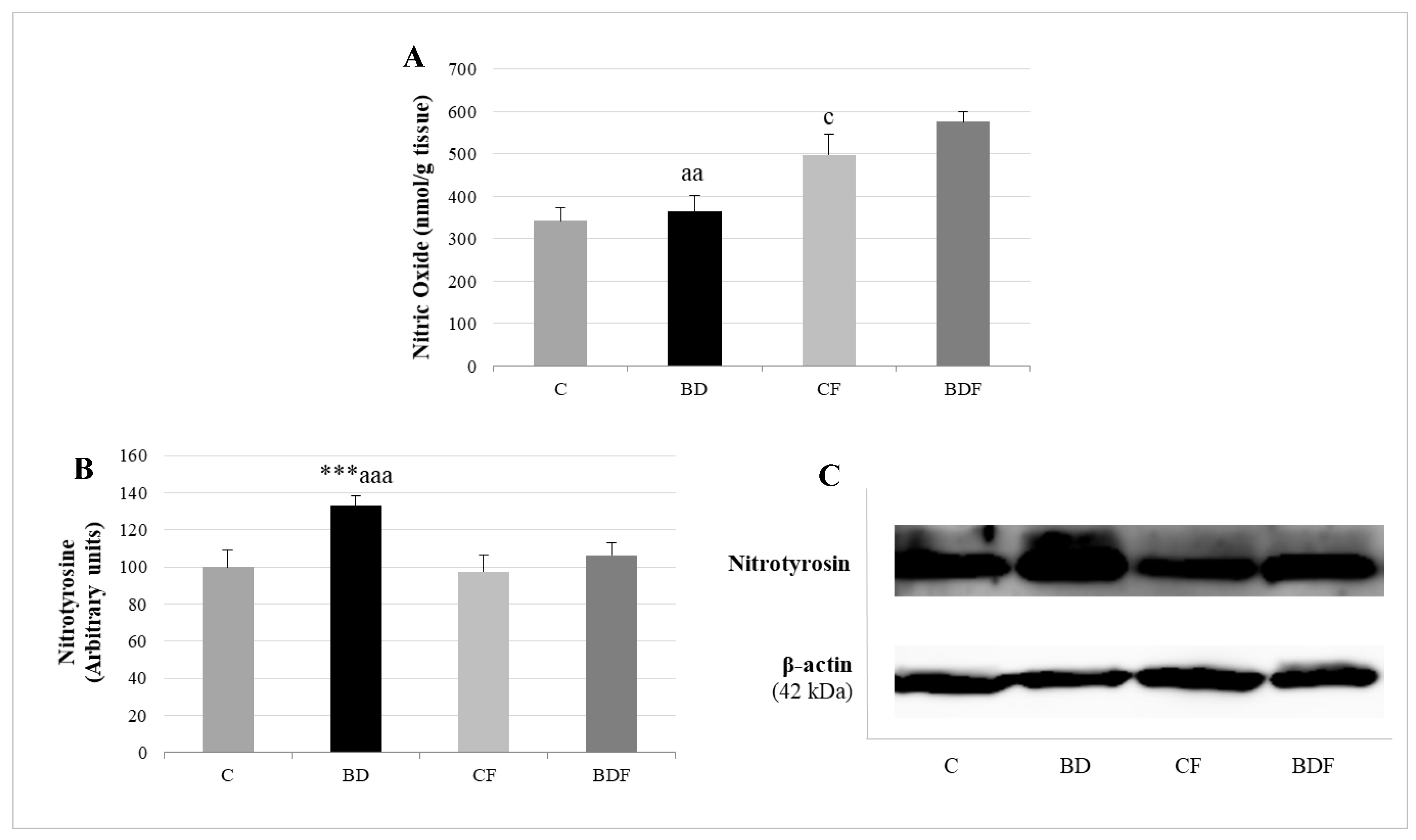
3.6. Hepatic Nitrosative Stress
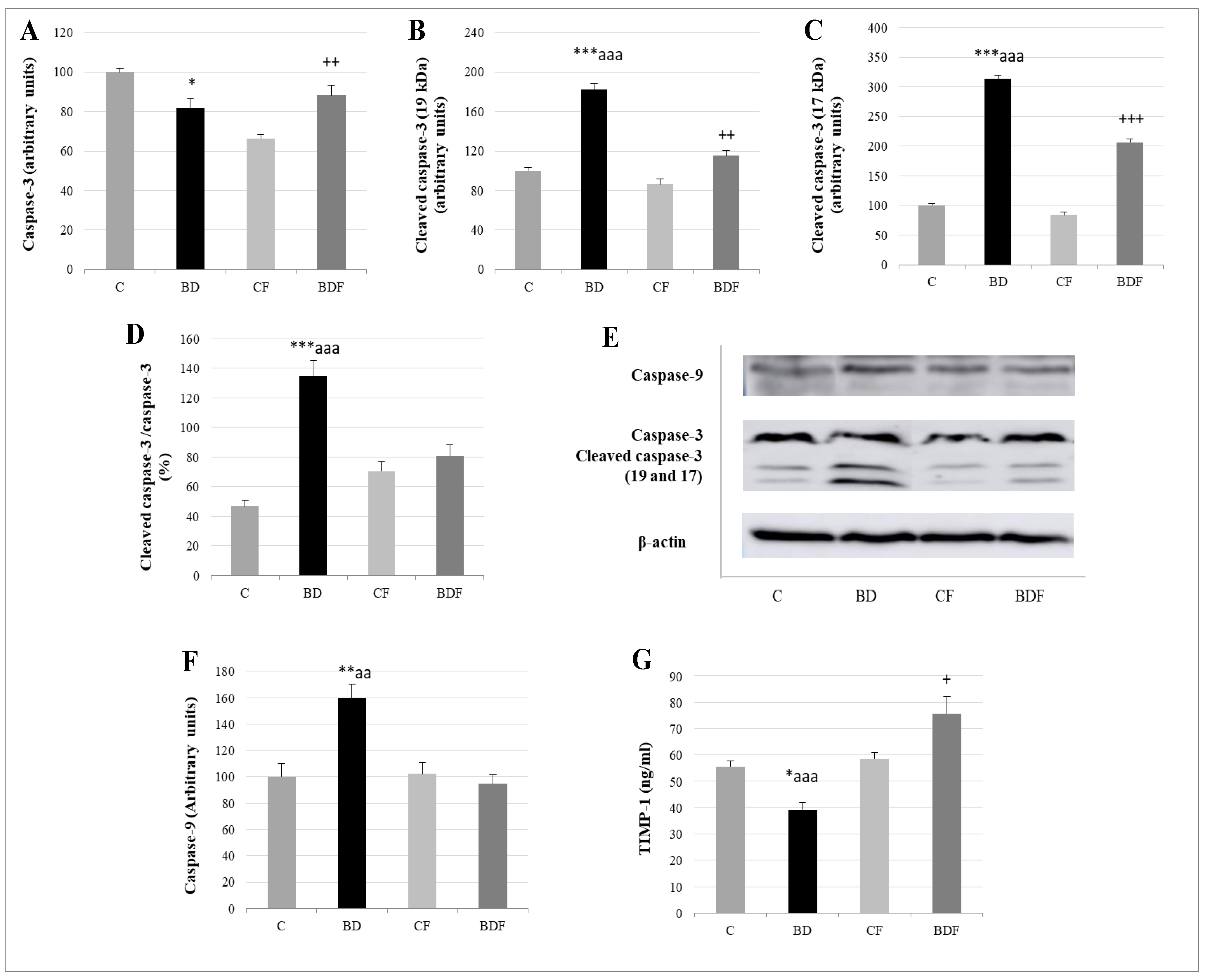
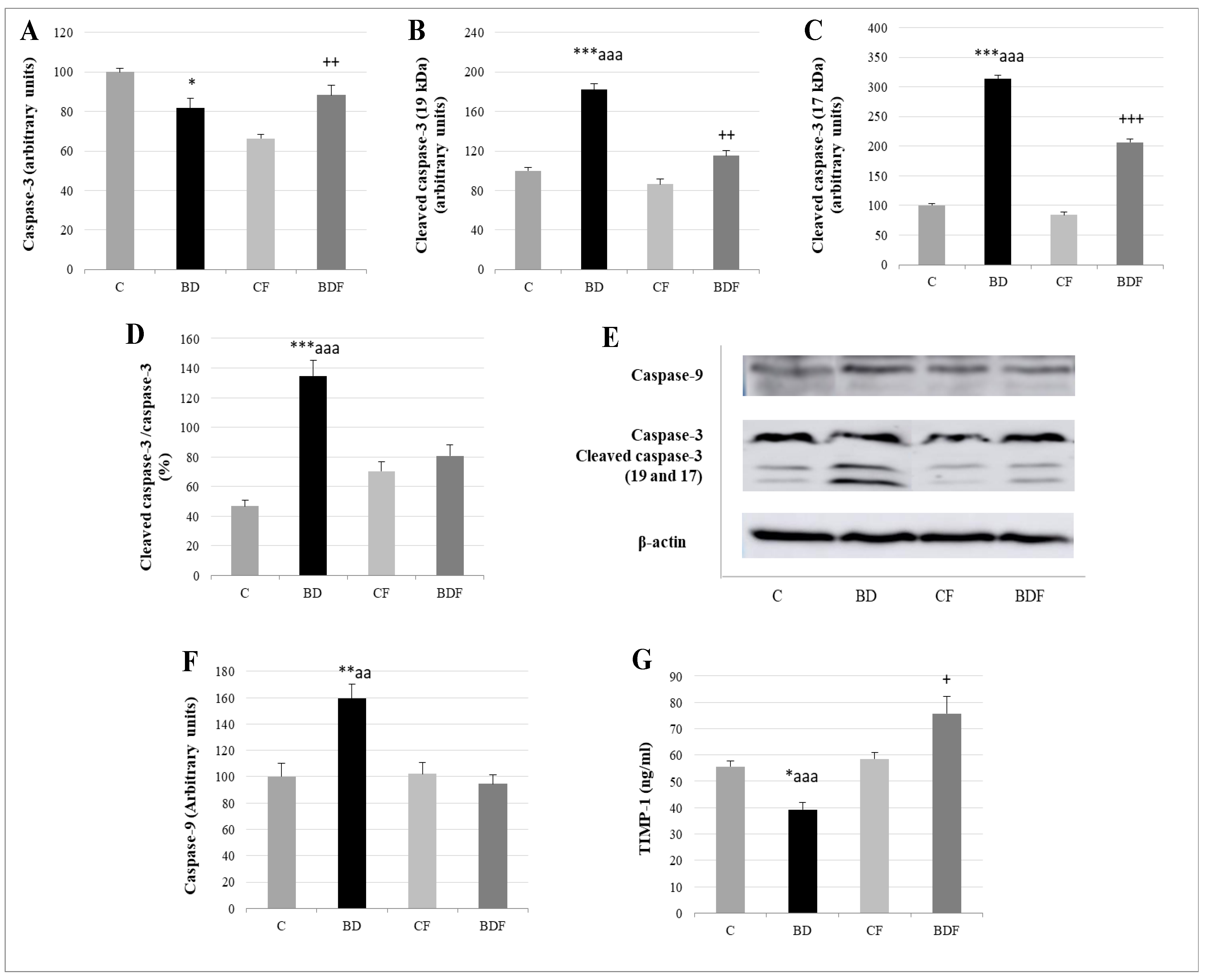
3.7. Liver Apoptosis
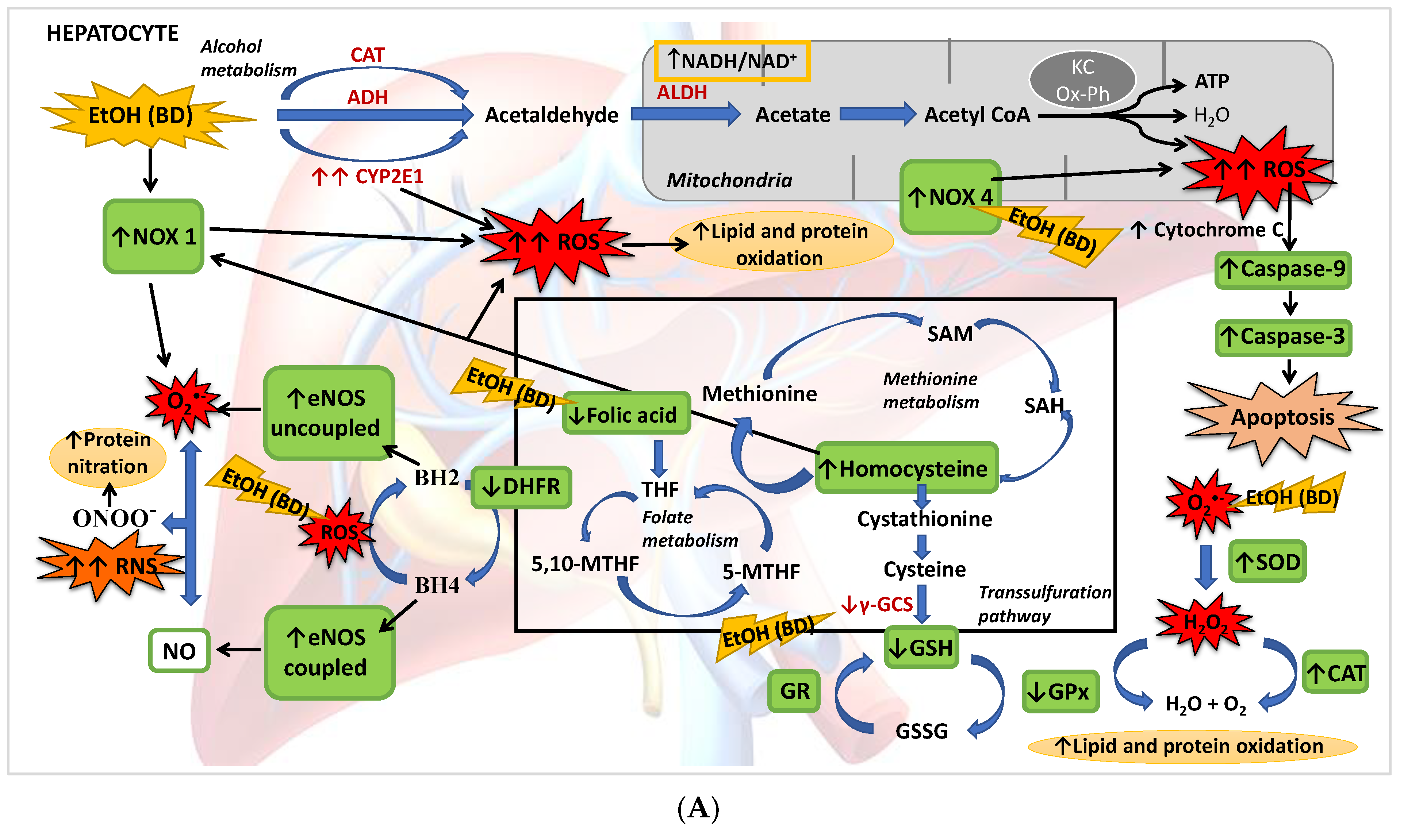
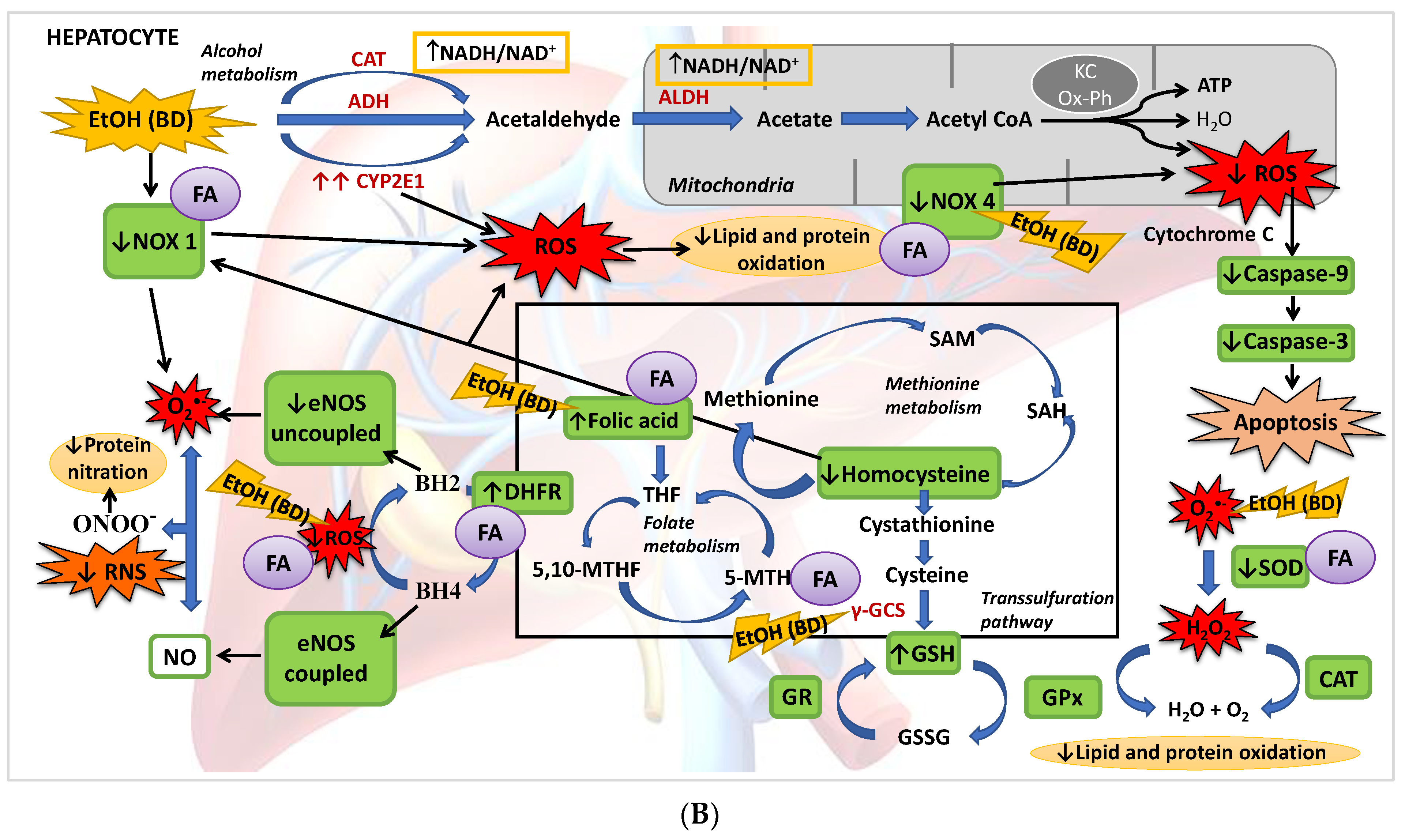
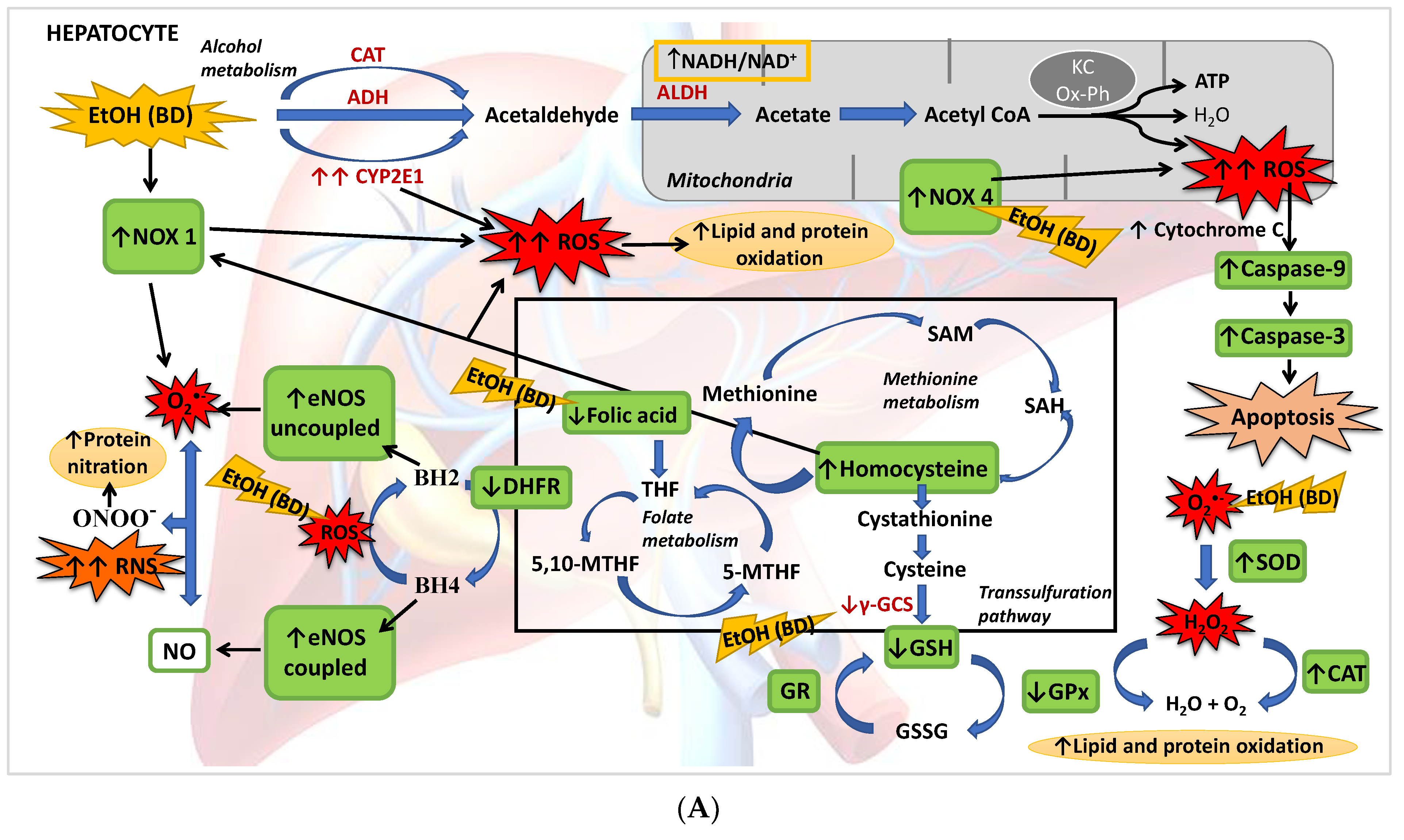
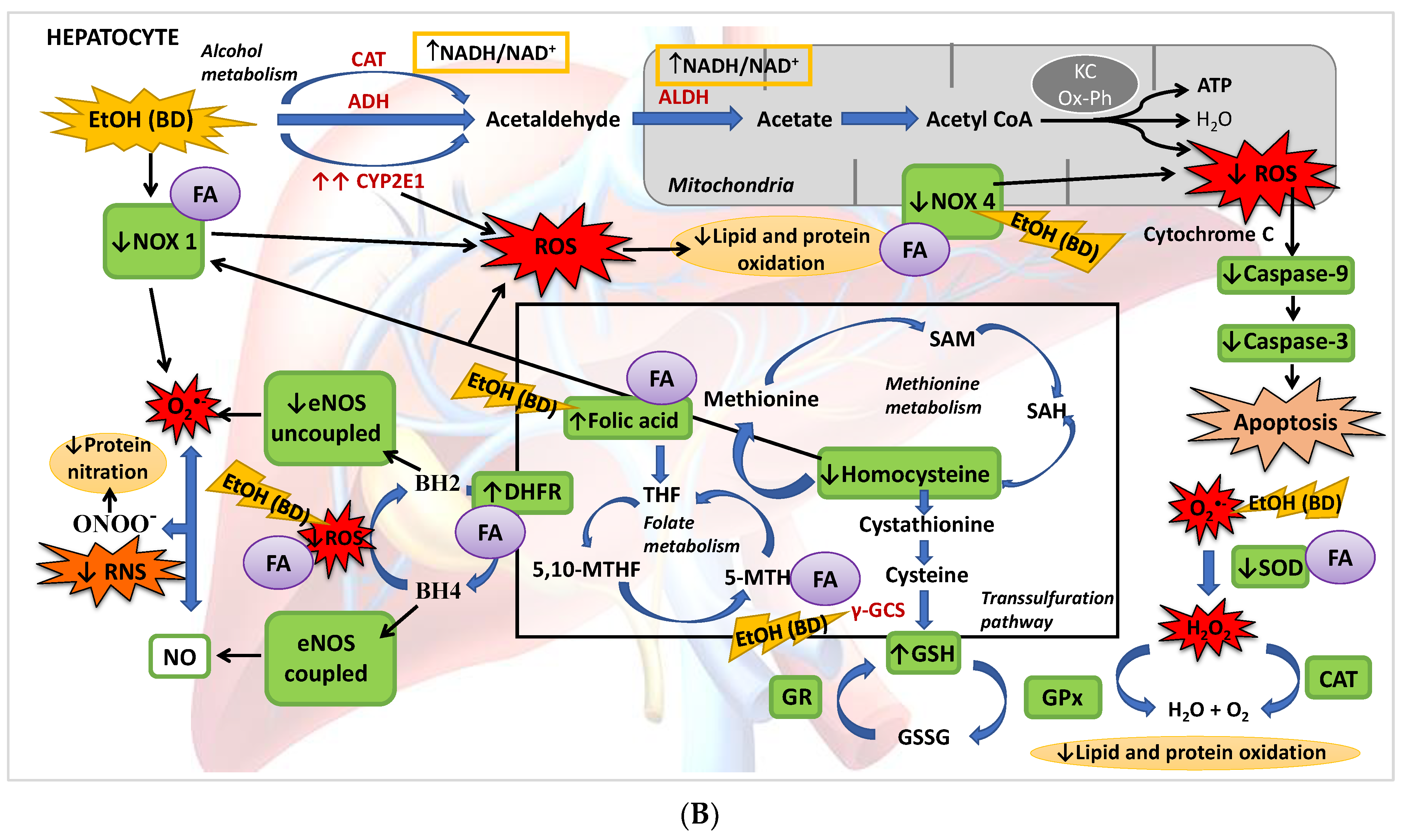
4. Discussion
4.1. Hepatic Function and FA Homeostasis
4.2. Hepatic Antioxidant Endogenous Enzymatic Defense System
4.3. Hepatic GSH Levels
4.4. Hcy Levels in Serum and Hepatic Pro-Oxidant Expression of NOX1 and NOX4
4.5. eNOS, NO and NS
4.6. Apoptosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Medici, V.; Halsted, C.H. Folate, Alcohol, and Liver Disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halsted, C.H. Alcohol and Folate Interactions: Clinical Implications. In Folate in Health and Disease; Marcel Dekker: New York, NY, USA, 1995; pp. 313–328. [Google Scholar]
- Hernández-Rodríguez, S.; Gutiérrez-Salinas, J.; García-Ortíz, L.; Mondragón-Terán, P.; Ramírez-García, S.; Núñez-Ramos, N.R. Estrésoxidativo y Nitrosativo Como Mecanismo de Daño al Hepatocito Producido Por El Metabolismo Del Etanol. Med. Interna Mex. 2014, 30, 295–308. [Google Scholar]
- Warzyszynska, J.E.; Kim, Y.-I.J. Folate in Human Health and Disease. In eLS; John Wiley & Sons: Hoboken, NJ, USA, 2014; ISBN 9780470015902. [Google Scholar]
- Sharma, J.; Krupenko, S.A. Folate Pathways Mediating the Effects of Ethanol in Tumorigenesis. Chem.-Biol. Interact. 2020, 324, 109091. [Google Scholar] [CrossRef] [PubMed]
- Asbaghi, O.; Ghanavati, M.; Ashtary-larky, D.; Bagheri, R.; Rezaei Kelishadi, M.; Nazarian, B.; Nordvall, M.; Wong, A.; Dutheil, F.; Suzuki, K.; et al. Effects of Folic Acid Supplementation on Oxidative Stress Markers: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Antioxidants 2021, 10, 871. [Google Scholar] [CrossRef]
- Lai, K.G.; Chen, C.F.; Ho, C.T.; Liu, J.J.; Liu, T.Z.; Chern, C.L. Novel Roles of Folic Acid as Redox Regulator: Modulation of Reactive Oxygen Species Sinker Protein Expression and Maintenance of Mitochondrial Redox Homeostasis on Hepatocellular Carcinoma. Tumor Biol. 2017, 39, 1010428317702649. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Adhikari, S.; Patro, B.S.; Chattopadhyay, S.; Mukherjee, T. Free Radical Scavenging Behavior of Folic Acid: Evidence for Possible Antioxidant Activity. Free. Radic. Biol. Med. 2001, 30, 1390–1399. [Google Scholar] [CrossRef]
- Stanger, O.; Wonisch, W. Enzymatic and Non-Enzymatic Antioxidative Effects of Folic Acid and Its Reduced Derivates. In Water Soluble Vitamins, Subcellular Biochemistry; O. Stanger: London, UK, 2012; Volume 56, pp. 131–161. ISBN 9789400721999. [Google Scholar]
- Cano, M.J.; Ayala, A.; Murillo, M.L.; Carreras, O. Protective Effect of Folic Acid against Oxidative Stress Produced in 21-Day Postpartum Rats by Maternal-Ethanol Chronic Consumption during Pregnancy and Lactation Period. Free. Radic. Res. 2001, 34, 1–8. [Google Scholar] [CrossRef]
- Woo, C.W.H.; Prathapasinghe, G.A.; Siow, Y.L.; Karmin, O. Hyperhomocysteinemia Induces Liver Injury in Rat: Protective Effect of Folic Acid Supplementation. Biochim. Biophys. Acta 2006, 1762, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.Y.; Siow, Y.L.; Au-Yeung, K.K.W.; House, J.; Karmin, O. Folic Acid Supplementation Inhibits NADPH Oxidase-Mediated Superoxide Anion Production in the Kidney. Am. J. Physiol. Ren. Physiol. 2011, 300, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.C.; Chang, W.M.; Wu, J.Y.; Huang, C.C.; Lu, F.J.; Chuang, Y.W.; Chang, P.J.; Chen, K.H.; Hong, C.Z.; Yeh, R.H.; et al. Folate Deficiency Triggered Apoptosis of Synoviocytes: Role of Overproduction of Reactive Oxygen Species Generated via NADPH Oxidase/Mitochondrial Complex II and Calcium Perturbation. PLoS ONE 2016, 11, e0146440. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Zuo, Z.X.; Liu, Z.X.; Wu, Q.N.; Mo, H.Y.; Wang, Z.X.; Wang, D.S.; Pu, H.Y.; et al. Modulation of Redox Homeostasis by Inhibition of MTHFD2 in Colorectal Cancer: Mechanisms and Therapeutic Implications. J. Natl. Cancer Inst. 2019, 111, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Lan, J.; Xia, W.; Tu, C.; Chen, B.; Li, S.; Pan, W. Folic Acid Attenuates Vascular Endothelial Cell Injury Caused by Hypoxia via the Inhibition of ERK1/2/NOX4/ROS Pathway. Cell Biochem. Biophys. 2016, 74, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Chalupsky, K.; Stefani, E.; Cai, H. Mechanistic Insights into Folic Acid-Dependent Vascular Protection: Dihydrofolate Reductase-Mediated Reduction in Oxidant Stress in Endothelial Cells and Angiotensin II-Infused Mice: A Novel HPLC-Based Fluorescent Assay for DHFR Activity. J. Mol. Cell. Cardiol. 2009, 47, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Chalupsky, K.; Kračun, D.; Kanchev, I.; Bertram, K.; Görlach, A. Folic Acid Promotes Recycling of Tetrahydrobiopterin and Protects Against Hypoxia-Induced Pulmonary Hypertension by Recoupling Endothelial Nitric Oxide Synthase. Antioxid. Redox Signal. 2015, 23, 1076–1091. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.P.; Schlaich, M.P.; Harazny, J.M.; Raff, U.; Ritt, M.; Ott, C.; Schmieder, R.E. Folic Acid Treatment Normalizes Nos-Dependence of Vascular Tone in the Metabolic Syndrome. Obesity 2011, 19, 960–967. [Google Scholar] [CrossRef]
- National Institute on Alcohol Abuse and Alcoholism What Is Binge Drinking? Available online: https://www.niaaa.nih.gov/publications/brochures-and-fact-sheets/binge-drinking (accessed on 23 March 2021).
- Bonar, E.E.; Parks, M.J.; Gunlicks-Stoessel, M.; Lyden, G.R.; Mehus, C.J.; Morrell, N.; Patrick, M.E. Binge Drinking before and after a COVID-19 Campus Closure among First-Year College Students. Addict. Behav. 2021, 118, 106879. [Google Scholar] [CrossRef]
- Muzi, S.; Sansò, A.; Serena Pace, C. What’s Happened to Italian Adolescents During the COVID-19 Pandemic? A Preliminary Study on Symptoms, Problematic Social Media Usage, and Attachment: Relationships and Differences with Pre-Pandemic Peers. Front. Psychiatry 2021, 12, 590543. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and Oxidative Liver Injury by Alcohol. Free. Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Eiciner, E.R.; Haliman, R.S. Effect of Alcohol on Serum Folate Level. J. Clin. Investig. 1973, 52, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Ojeda, M.L.; Rua, R.M.; Nogales, F.; Díaz-Castro, J.; Murillo, M.L.; Carreras, O. The Benefits of Administering Folic Acid in Order to Combat the Oxidative Damage Caused by Binge Drinking in Adolescent Rats. Alcohol Alcohol. 2016, 51, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, T.M.; Reed, M.C.; Frederik Nijhou, H. A Population Model of Folate-Mediated One-Carbon Metabolism. Nutrients 2013, 5, 2457–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, L. Modeling Adolescent Development and Alcohol Use in Animals. Alcohol. Res. Health 2000, 24, 115. [Google Scholar]
- Callaci, J.J.; Himes, R.; Lauing, K.; Roper, P. Long-Term Modulations in the Vertebral Transcriptome of Adolescent-Stage Rats Exposed to Binge Alcohol. Alcohol Alcohol. 2010, 45, 332–346. [Google Scholar] [CrossRef]
- Fridovich, I. Cytochrome c. In CRC Handbook of Methods for Oxygen Radical Research; CRC Press: Boca Raton, FL, USA, 1985; pp. 213–215. [Google Scholar]
- Beers, R.F.; Sizer, I.W. A Spectrophotometric Method for Measuring the Breakdown of Hydrogen Peroxide by Catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Lawrence, R.A.; Burk, R.F. Glutathione Peroxidase Activity in Selenium-Deficient Rat Liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef]
- Worthington, D.J.; Rosemeyer, M.A. Human Glutathione Reductase: Purification of the Crystalline Enzyme from Erythrocytes. Eur. J. Biochem. 1974, 48, 167–177. [Google Scholar] [CrossRef]
- Draper, H.H.; Hadley, M. Malondialdehyde Determination as Index of Lipid Peroxidation. Methods Enzymol. 1990, 186, 421–431. [Google Scholar] [CrossRef]
- Reznick, A.Z.; Packer, L. Oxidative Damage to Proteins: Spectrophotometric Method for Carbonyl Assay. Methods Enzymol. 1994, 233, 357–363. [Google Scholar] [CrossRef]
- Mathews, S.; Xu, M.; Wang, H.; Bertola, A.; Gao, B. Animals Models of Gastrointestinal and Liver Diseases. Animal Models of Alcohol-Induced Liver Disease: Pathophysiology, Translational Relevance, and Challenges. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 306, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.; Knibbe, K.; Kreissl, A.; Repa, A.; Thanhaeuser, M.; Greber-Platzer, S.; Berger, A.; Jilma, B.; Haiden, N. Does Acute Alcohol Intoxication Cause Transaminase Elevations in Children and Adolescents? Alcohol 2016, 51, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Fernandes, L.M.; Lopes, K.S.; Santana, L.N.S.; Fontes-Júnior, E.A.; Ribeiro, C.H.M.A.; Silva, M.C.F.; de Oliveira Paraense, R.S.; Crespo-López, M.E.; Gomes, A.R.Q.; Lima, R.R.; et al. Repeated Cycles of Binge-like Ethanol Intake in Adolescent Female Rats Induce Motor Function Impairment and Oxidative Damage in Motor Cortex and Liver, but Not in Blood. Oxid. Med. Cell. Longev. 2018, 2018, 3467531. [Google Scholar] [CrossRef] [Green Version]
- Ojeda, M.L.; Rua, R.M.; Murillo, M.L.; Carreras, O.; Nogales, F. Binge Drinking during Adolescence Disrupts Se Homeostasis and Its Main Hepatic Selenoprotein Expression. Alcohol. Clin. Exp. Res. 2015, 39, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Nyblom, H.; Berggren, U.; Balldin, J.; Olsson, R. High AST/ALT Ratio May Indicate Advanced Alcoholic Liver Disease Rather than Heavy Drinking. Alcohol Alcohol. 2004, 39, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, M.L.; Carreras, O.; Sobrino, P.; Murillo, M.L.; Nogales, F. Biological Implications of Selenium in Adolescent Rats Exposed to Binge Drinking: Oxidative, Immunologic and Apoptotic Balance. Toxicol. Appl. Pharmacol. 2017, 329, 165–172. [Google Scholar] [CrossRef]
- Shaw, S.; Jayatilleke, E.; Herbert, V.; Colman, N. Cleavage of Folates during Ethanol Metabolism. Role of Acetaldehyde/Xanthine Oxidase-Generated Superoxide. Biochem. J. 1989, 257, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed Mitochondrial Biogenesis in Folic Acid-Induced Acute Kidney Injury and Early Fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Nogales, F.; Rua, R.M.; Ojeda, M.L.; Murillo, M.L.; Carreras, O. Oral or Intraperitoneal Binge Drinking and Oxidative Balance in Adolescent Rats. Chem. Res. Toxicol. 2014, 27, 1926–1933. [Google Scholar] [CrossRef]
- Vatsalya, V.; Gala, K.S.; Hassan, A.Z.; Frimodig, J.; Kong, M.; Sinha, N.; Schwandt, M.L. Characterization of Early-Stage Alcoholic Liver Disease with Hyperhomocysteinemia and Gut Dysfunction and Associated Immune Response in Alcohol Use Disorder Patients. Biomedicines 2021, 9, 7. [Google Scholar] [CrossRef]
- Murray, T.V.A.; Dong, X.; Sawyer, G.J.; Caldwell, A.; Halket, J.; Sherwood, R.; Quaglia, A.; Dew, T.; Anilkumar, N.; Burr, S.; et al. NADPH Oxidase 4 Regulates Homocysteine Metabolism and Protects against Acetaminophen-Induced Liver Damage in Mice. Free. Radic. Biol. Med. 2015, 89, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Berasain, C.; Prieto, J.; Mato, J.M.; García-Trevijano, E.R.; Corrales, F.J. Influence of Impaired Liver Methionine Metabolism on the Development of Vascular Disease and Inflammation. Current medicinal chemistry. Cardiovasc. Hematol. Agents 2005, 3, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K. Alcoholic Liver Disease and Methionine Metabolism. Semin. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; de Santo, N.G. Possible Mechanisms of Homocysteine Toxicity. Kidney Int. 2003, 63, S137–S140. [Google Scholar] [CrossRef] [Green Version]
- Karolczak, K.; Watala, C. Melatonin as a Reducer of Neuro- and Vasculotoxic Oxidative Stress Induced by Homocysteine. Antioxidants 2021, 10, 1178. [Google Scholar] [CrossRef]
- Dumitrescu, R.G. Alcohol-Induced Epigenetic Changes in Cancer. Methods Mol. Biol. 2018, 1856, 157–172. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Ishida, K.; Kaji, K.; Sato, S.; Ogawa, H.; Takagi, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane Ameliorates Ethanol plus Carbon Tetrachloride-Induced Liver Fibrosis in Mice through the Nrf2-Mediated Antioxidant Response and Acetaldehyde Metabolization with Inhibition of the LPS/TLR4 Signaling Pathway. J. Nutr. Biochem. 2021, 89, 108573. [Google Scholar] [CrossRef]
- Andueza, A.; Garde, N.; García-Garzón, A.; Ansorena, E.; López-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martínez-Irujo, J.J. NADPH Oxidase 5 Promotes Proliferation and Fibrosis in Human Hepatic Stellate Cells. Free. Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, W.; Zhong, W.; Sun, X.; Zhou, Z. Pharmacological Inhibition of NOX4 Ameliorates Alcohol-Induced Liver Injury in Mice through Improving Oxidative Stress and Mitochondrial Function. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 2912–2921. [Google Scholar] [CrossRef] [Green Version]
- Sarna, L.K.; Wu, N.; Wang, P.; Hwang, S.Y.; Siow, Y.L.; Karmin, O. Folic Acid Supplementation Attenuates High Fat Diet Induced Hepatic Oxidative Stress via Regulation of NADPH Oxidase. Can. J. Physiol. Pharmacol. 2012, 90, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wang, Y.; Siu, K.L.; Zhang, Y.; Cai, H. Targeting Feed-Forward Signaling of TGFβ/NOX4/DHFR/ENOS Uncoupling/TGFβ Axis with Anti-TGFβ and Folic Acid Attenuates Formation of Aortic Aneurysms: Novel Mechanisms and Therapeutics. Redox Biol. 2021, 38, 101757. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Siu, K.L.; Chalupsky, K.; Nguyen, A.; Chen, P.; Weintraub, N.L.; Galis, Z.; Cai, H. Role of Uncoupled Endothelial Nitric Oxide Synthase in Abdominal Aortic Aneurysm Formation: Treatment with Folic Acid. Hypertension 2012, 59, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Aircart-Ramos, C. Papel Del Módulo Carboxi-Terminal De Las Óxido Nítrico Sintasas En La Regulación De La Síntesis De No: Caracterización Del Receptor Nuclear E75; Un Sensor De Óxido Nítrico: Madrid, Spain, 2013. [Google Scholar]
- Hwang, C.-L.; Bian, J.-T.; Thur, L.A.; Peters, T.A.; Piano, M.R.; Phillips, S.A. Tetrahydrobiopterin Restores Microvascular Dysfunction in Young Adult Binge Drinkers. Alcohol. Clin. Exp. Res. 2020, 44, 407–414. [Google Scholar] [CrossRef]
- Fleming, I. NO Signaling Defects in Hypertension. In Nitric Oxide: Biology and Pathobiology; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 301–311. ISBN 9780128042731. [Google Scholar]
- Htet Hlaing, K.; Clément, M.v. Formation of Protein S-Nitrosylation by Reactive Oxygen Species. Free Radic. Res. 2014, 48, 996–1010. [Google Scholar] [CrossRef]
- Pignatelli, P.; Menichelli, D.; Pastori, D.; Violi, F. Oxidative Stress and Cardiovascular Disease: New Insights. Kardiol. Pol. 2018, 76, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.; Zhao, C.; Zhang, L.; Tang, S.; Jia, X.; Tong, H.; Liu, R.; Tang, C.; Gao, J. Celecoxib Reduces Hepatic Vascular Resistance in Portal Hypertension by Amelioration of Endothelial Oxidative Stress. J. Cell. Mol. Med. 2021, 25, 10389–10402. [Google Scholar] [CrossRef]
- Stanhewicz, A.E.; Kenney, W.L. Role of Folic Acid in Nitric Oxide Bioavailability and Vascular Endothelial Function. Nutr. Rev. 2017, 75, 61–70. [Google Scholar] [CrossRef]
- Ting-Jun, F.; Li-Hui, H.; Ri-Shan, C.; Jin, L. Caspase Family Proteases and Apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Ries, C. Cytokine Functions of TIMP-1. Cell. Mol. Life Sci. 2014, 71, 659–672. [Google Scholar] [CrossRef]
- Li, W.; Ma, Y.; Li, Z.; Lv, X.; Wang, X.; Zhou, D.; Luo, S.; Wilson, J.X.; Huang, G. Folic Acid Decreases Astrocyte Apoptosis by Preventing Oxidative Stress-Induced Telomere Attrition. Int. J. Mol. Sci. 2020, 21, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Z.; Zhou, D.; Zhang, X.; Yan, J.; Huang, G. Maternal Folic Acid Deficiency Stimulates Neural Cell Apoptosis via MiR-34a Associated with Bcl-2 in the Rat Foetal Brain. Int. J. Dev. Neurosci. 2019, 72, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Li, W.; Wang, P.; Lv, X.; Gao, Y.; Huang, G. Folic Acid Inhibits Homocysteine-Induced Cell Apoptosis in Human Umbilical Vein Endothelial Cells. Mol. Cell. Biochem. 2018, 444, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Chen, D.; Wang, A.; Ding, X.; Liu, Z.; Ling, L.; He, Q.; Zhao, T. Folic Acid Rescue of ATRA-Induced Cleft Palate by Restoring the TGF-β Signal and Inhibiting Apoptosis. J. Oral Pathol. Med. 2011, 40, 433–439. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | C | BD | CF | BDF |
|---|---|---|---|---|
| Body weight increase (g/day) | 5.72 ± 0.17 | 5.29 ± 0.14 | 5.80 ± 0.21 | 5.20 ± 0.53 |
| Food intake (g/day) | 14.29 ± 1.31 | 13.84 ± 1.09 | 14.35 ± 1.85 | 13.95 ± 1.04 |
| LSI (%) | 3.48 ± 0.17 | 3.43 ± 0.067 | 3.65 ± 0.054 | 3.65 ± 0.12 |
| Liver protein content (mg/g liver wet tissue) | 8.95 ± 0.33 | 9.53 ± 0.50 | 9.37 ± 0.50 | 9.19 ± 0.89 |
| Serum AST (U/L) | 118 ± 13 | 210 ± 8 *** a | 112 ± 13 | 168 ± 13 + |
| Serum ALT (U/L) | 29.4 ± 1.4 | 38.8 ± 2.2 * aa | 27.6 ± 1.7 | 29.12 ± 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Lopez, M.d.C.; Ojeda, M.L.; Romero-Herrera, I.; Nogales, F.; Carreras, O. Folic Acid Homeostasis and Its Pathways Related to Hepatic Oxidation in Adolescent Rats Exposed to Binge Drinking. Antioxidants 2022, 11, 362. https://doi.org/10.3390/antiox11020362
Gallego-Lopez MdC, Ojeda ML, Romero-Herrera I, Nogales F, Carreras O. Folic Acid Homeostasis and Its Pathways Related to Hepatic Oxidation in Adolescent Rats Exposed to Binge Drinking. Antioxidants. 2022; 11(2):362. https://doi.org/10.3390/antiox11020362
Chicago/Turabian StyleGallego-Lopez, María del Carmen, María Luisa Ojeda, Inés Romero-Herrera, Fátima Nogales, and Olimpia Carreras. 2022. "Folic Acid Homeostasis and Its Pathways Related to Hepatic Oxidation in Adolescent Rats Exposed to Binge Drinking" Antioxidants 11, no. 2: 362. https://doi.org/10.3390/antiox11020362
APA StyleGallego-Lopez, M. d. C., Ojeda, M. L., Romero-Herrera, I., Nogales, F., & Carreras, O. (2022). Folic Acid Homeostasis and Its Pathways Related to Hepatic Oxidation in Adolescent Rats Exposed to Binge Drinking. Antioxidants, 11(2), 362. https://doi.org/10.3390/antiox11020362