
The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage


 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Murine Models of Cartilage PC
2.3. Murine Knee microCT-Scan
2.4. Murine Knee Histology
2.5. Murine Chondrocyte Isolation and Induction of Calcification
2.6. Quantification of Chondrocyte Calcification
2.7. Stimulation with Calcium Phosphate Crystals
2.8. Alkaline Phosphatase Activity
2.9. Real Time PCR Analysis
2.10. Human Cartilage Samples
2.11. Human Chondrocyte Isolation and Induction of Calcification
2.12. Human Cartilage Histology and Quantification of Calcification
2.13. Immunohistochemical Analysis
2.14. H2S Measurements
2.15. IL-6 Quantification
2.16. LDH Measurement
2.17. Statistical Analysis
3. Results
3.1. Cse Expression Regulates Pathologic Calcification and Cartilage Damage in Murine Knees
3.2. Cse Regulates Chondrocyte Calcification and IL-6 Secretion In Vitro
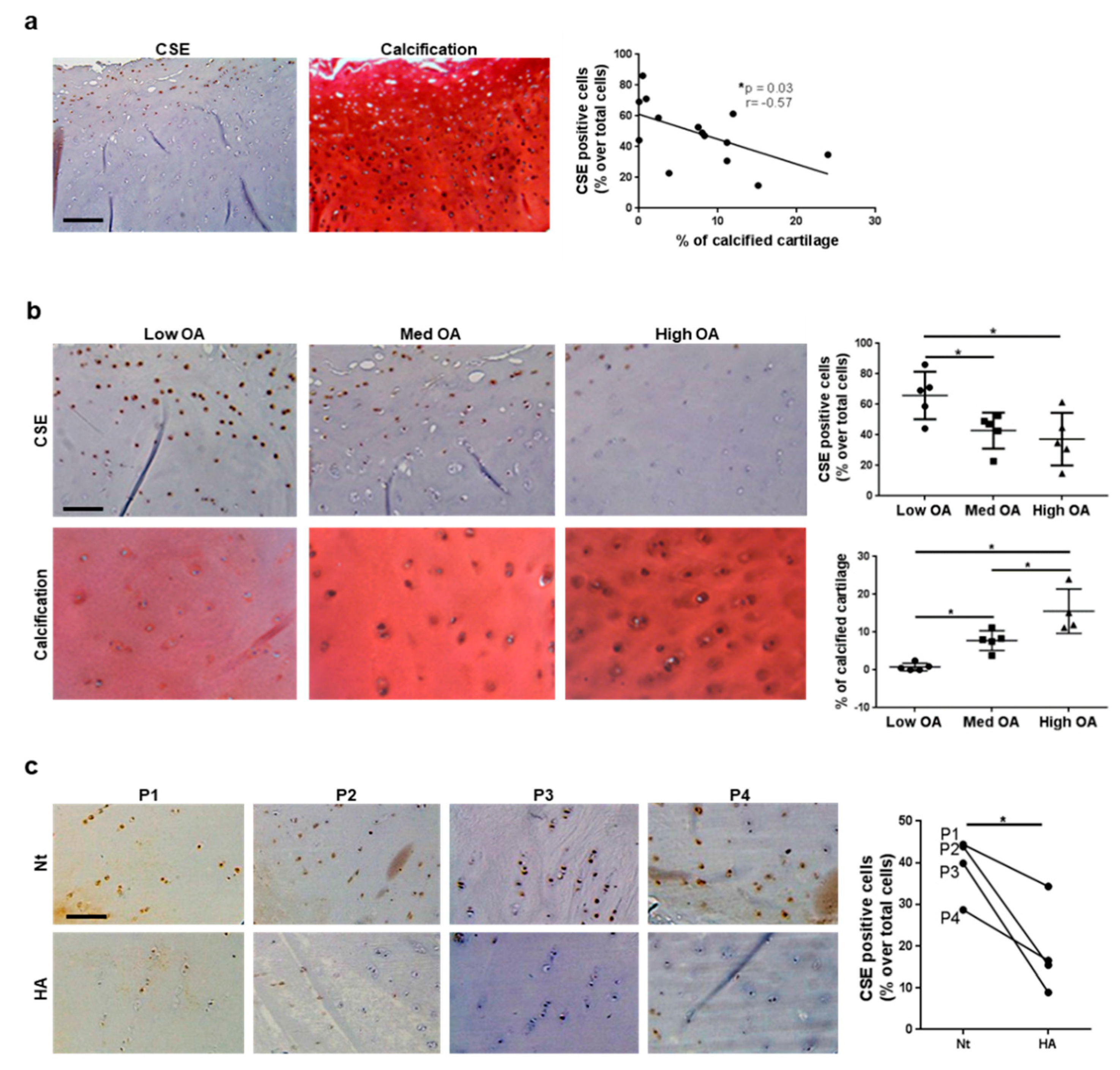
3.3. CSE Expression in Human Cartilage Negatively Correlates with Tissue Calcification and Disease Severity
3.4. A CSE Positive Allosteric Activator Reduces Chondrocyte Calcification and IL-6 Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3, e10172. [Google Scholar] [CrossRef]
- Ranganathan, K.; Loder, S.; Agarwal, S.; Wong, V.W.; A Forsberg, J.; Davis, T.A.; Wang, S.; James, A.W.; Levi, B. Heterotopic Ossification: Basic-Science Principles and Clinical Correlates. J. Bone Jt. Surg.-Am. Vol. 2015, 97, 1101–1111. [Google Scholar] [CrossRef]
- Foreman, S.; Gersing, A.; von Schacky, C.; Joseph, G.; Neumann, J.; Lane, N.; McCulloch, C.; Nevitt, M.; Link, T. Chondrocalcinosis is associated with increased knee joint degeneration over 4 years: Data from the Osteoarthritis Initiative. Osteoarthr. Cartil. 2020, 28, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; So, A.; Combes, C.; Daudon, M.; Busso, N. Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann. Rheum. Dis. 2015, 75, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.; McCarthy, G.M. Cartilage calcification and osteoarthritis: A pathological association? Osteoarthr. Cartil. 2020, 28, 1301–1302. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.F.; Qin, W.P.; Xiao, B.C.; Wan, Q.Q.; Tay, F.R.; Niu, L.N.; Jiao, K. Pathological calcification in osteoarthritis: An outcome or a disease initiator? Biol. Rev. Camb. Philos. Soc. 2020, 95, 960–985. [Google Scholar] [CrossRef]
- Fuerst, M.; Bertrand, J.; Lammers, L.; Dreier, R.; Echtermeyer, F.; Nitschke, Y.; Rutsch, F.; Schäfer, F.K.W.; Niggemeyer, O.; Steinhagen, J.; et al. Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum. 2009, 60, 2694–2703. [Google Scholar] [CrossRef]
- McCarthy, G.M.; Dunne, A. Calcium crystal deposition diseases — beyond gout. Nat. Rev. Rheumatol. 2018, 14, 592–602. [Google Scholar] [CrossRef]
- Liu, Y.; Jackson, A.; Cosgrove, S. Contribution of calcium-containing crystals to cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthr. Cartil. 2009, 17, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R.; Lioté, F. Monosodium urate and calcium pyrophosphate dihydrate (CPPD) crystals, inflammation, and cellular signaling. Jt. Bone Spine 2005, 72, 295–302. [Google Scholar] [CrossRef]
- Campillo-Gimenez, L.; Renaudin, F.; Jalabert, M.; Gras, P.; Gosset, M.; Rey, C.; Sarda, S.; Collet, C.; Cohen-Solal, M.; Ea, H.K.; et al. Inflammatory Potential of Four Different Phases of Calcium Pyrophosphate Relies on NF-kappaB Activation and MAPK Pathways. Front. Immunol. 2018, 9, 2248. [Google Scholar] [CrossRef]
- Ea, H.K.; Chobaz, V.; Nguyen, C.; Nasi, S.; Van Lent, P.; Daudon, M.; Dessombz, A.; Bazin, D.; McCarthy, G.; Jolles-Haeberli, B.; et al. Pathogenic Role of Basic Calcium Phosphate Crystals in Destructive Arthropathies. PLoS ONE 2013, 8, e57352. [Google Scholar] [CrossRef]
- Bai, G.; Howell, D.; Howard, G.; Roos, B.; Cheung, H. Basic calcium phosphate crystals up-regulate metalloproteinases but down-regulate tissue inhibitor of metalloproteinase-1 and -2 in human fibroblasts. Osteoarthr. Cartil. 2001, 9, 416–422. [Google Scholar] [CrossRef][Green Version]
- McCarthy, G.M.; Westfall, P.R.; Masuda, I.A.; Christopherson, P.; Cheung, H.S.; Mitchell, P.G. Basic calcium phosphate crystals activate human osteoarthritic synovial fibroblasts and induce matrix metalloproteinase-13 (collagenase-3) in adult porcine articular chondrocytes. Ann. Rheum. Dis. 2001, 60, 399–406. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Yang, S.; Shin, Y.; Rhee, J.; Chun, C.-H.; Chun, J.-S. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011, 63, 2732–2743. [Google Scholar] [CrossRef]
- Desai, J.; Foresto-Neto, O.; Honarpisheh, M.; Steiger, S.; Nakazawa, D.; Popper, B.; Buhl, E.M.; Boor, P.; Mulay, S.R.; Anders, H.-J. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Sci. Rep. 2017, 7, 15003. [Google Scholar] [CrossRef]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun. 2016, 7, 10274. [Google Scholar] [CrossRef] [PubMed]
- Ea, H.K.; Monceau, V.; Camors, E.; Cohen-Solal, M.; Charlemagne, D.; Lioté, F. Annexin 5 overexpression increased articular chondrocyte apoptosis induced by basic calcium phosphate crystals. Ann. Rheum. Dis. 2008, 67, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Gimenez, L.; Renaudin, F.; Bobé, P.; Gosset, M.; Combes, C.; Cohen-Solal, M.; Lioté, F.; Ea, H.-K. OP0287 Calcium Pyrophosphate Dihydrate Crystals Induce IL-1β Production by Monocytes through A Potassium Efflux-Dependent Pathway. Ann. Rheum. Dis. 2016, 75, 166.3–167. [Google Scholar] [CrossRef]
- Nasi, S.; Ea, H.-K.; Lioté, F.; So, A.; Busso, N. Sodium Thiosulfate Prevents Chondrocyte Mineralization and Reduces the Severity of Murine Osteoarthritis. PLoS ONE 2016, 11, e0158196. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, M.; Nasi, S.; Pasch, A.; So, A.; Busso, N. The role of the gasotransmitter hydrogen sulfide in pathological calcification. Br. J. Pharmacol. 2019, 177, 778–792. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Miyamoto, T.; Fujita, N.; Kubota, Y.; Ito, K.; Takubo, K.; Miyamoto, K.; Ninomiya, K.; Suzuki, T.; Iwasaki, R.; et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 2007, 204, 1613–1623. [Google Scholar] [CrossRef]
- Nasi, S.; Ehirchiou, D.; Chatzianastasiou, A.; Nagahara, N.; Papapetropoulos, A.; Bertrand, J.; Cirino, G.; So, A.; Busso, N. The protective role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway against experimental osteoarthritis. Arthritis Res. 2020, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Ying, J.; Xiang, L.; Zhang, C. The biologic effect of hydrogen sulfide and its function in various diseases. Medicine 2018, 97, e13065. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell. Mol. Life Sci. 2017, 74, 1391–1412. [Google Scholar] [CrossRef]
- Meijide-Failde, R.; Blanco, F.J. Hydrogen Sulfide and Inflammatory Joint Diseases. Curr. Drug Targets 2017, 18, 1641–1652. [Google Scholar] [CrossRef]
- Zavaczki, E.; Jeney, V.; Agarwal, A.; Zarjou, A.; Oros, M.; Katkó, M.; Varga, Z.; Balla, G.; Balla, J. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011, 80, 731–739. [Google Scholar] [CrossRef]
- Sikura, K.; Potor, L.; Szerafin, T.; Oros, M.; Nagy, P.; Méhes, G.; Hendrik, Z.; Zarjou, A.; Agarwal, A.; Posta, N.; et al. Hydrogen sulfide inhibits calcification of heart valves; implications for calcific aortic valve disease. Br. J. Pharmacol. 2019, 177, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liao, F.; Lin, X.; Zheng, F.; Fan, J.; Cui, Q.; Yang, J.; Geng, B.; Cai, J. Cystathionine gamma-Lyase-Hydrogen Sulfide Induces Runt-Related Transcription Factor 2 Sulfhydration, Thereby Increasing Osteoblast Activity to Promote Bone Fracture Healing. Antioxid. Redox Signal. 2017, 27, 742–753. [Google Scholar] [CrossRef]
- Gupta, S.; Kühnisch, J.; Mustafa, A.; Lhotak, S.; Schlachterman, A.; Slifker, M.J.; Klein-Szanto, A.; High, K.A.; Austin, R.C.; Kruger, W.D. Mouse models of cystathionine beta-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 2009, 23, 883–893. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef]
- Trocha, K.M.; Kip, P.; Tao, M.; MacArthur, M.R.; Trevino-Villarreal, J.H.; Longchamp, A.; Toussaint, W.; Lambrecht, B.N.; de Vries, M.R.; Quax, P.H.A.; et al. Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine gamma-lyase. Cardiovasc. Res. 2020, 116, 416–428. [Google Scholar] [CrossRef]
- Akahoshi, N.; Kobayashi, C.; Ishizaki, Y.; Izumi, T.; Himi, T.; Suematsu, M.; Ishii, I. Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum. Mol. Genet. 2008, 17, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef]
- Pritzker, K.; Gay, S.; Jimenez, S.; Ostergaard, K.; Pelletier, J.-P.; Revell, P.; Salter, D.; Berg, W.V.D. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef]
- Gregory, C.; Gunn, W.; Peister, A.; Prockop, D.J. An Alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal. Biochem. 2004, 329, 77–84. [Google Scholar] [CrossRef]
- Prudhommeaux, F.; Schiltz, C.; Lioté, F.; Hina, A.; Champy, R.; Bucki, B.; Ortiz-Bravo, E.; Meunier, A.; Rey, C.; Bardin, T. Variation in the inflammatory properties of basic calcium phosphate crystals according to crystal type. Arthritis Rheum. 1996, 39, 1319–1326. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ea, H.K.; Chobaz, V.; van Lent, P.; Lioté, F.; So, A.; Busso, N. Dispensable role of myeloid differentiation primary response gene 88 (MyD88) and MyD88-dependent toll-like receptors (TLRs) in a murine model of osteoarthritis. Jt. Bone Spine 2014, 81, 320–324. [Google Scholar] [CrossRef]
- Singha, S.; Kim, D.; Moon, H.; Wang, T.; Kim, K.H.; Shin, Y.H.; Jung, J.; Seo, E.; Lee, S.-J.; Ahn, K.H. Toward a Selective, Sensitive, Fast-Responsive, and Biocompatible Two-Photon Probe for Hydrogen Sulfide in Live Cells. Anal. Chem. 2014, 87, 1188–1195. [Google Scholar] [CrossRef]
- Ma, C.-H.; Chiua, Y.C.; Wu, C.-H.; Jou, I.-M.; Tu, Y.-K.; Hung, C.-H.; Hsieh, P.-L.; Tsai, K.-L. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: A possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018, 15, 504–512. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Bryan, S.; Khaper, N.; Mani, S.; Wang, R. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [PubMed]
- Karger, A.; Steffen, B.T.; Nomura, S.O.; Guan, W.; Garg, P.K.; Szklo, M.; Budoff, M.J.; Tsai, M.Y. Association between Homocysteine and Vascular Calcification Incidence, Prevalence, and Progression in the MESA Cohort. J. Am. Hear. Assoc. 2020, 9, e013934. [Google Scholar] [CrossRef] [PubMed]
- Van Campenhout, A.; Moran, C.S.; Parr, A.; Clancy, P.; Rush, C.; Jakubowski, H.; Golledge, J. Role of homocysteine in aortic calcification and osteogenic cell differentiation. Atherosclerosis 2009, 202, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Pan, C.-S.; Geng, B.; Zhao, J.; Yu, F.; Pang, Y.-Z.; Tang, C.-S.; Qi, Y.-F. Hydrogen sulfide ameliorates vascular calcification induced by vitamin D3 plus nicotine in rats1. Acta Pharmacol. Sin. 2006, 27, 299–306. [Google Scholar] [CrossRef]
- Pasch, A.; Schaffner, T.; Huynh-Do, U.; Frey, B.M.; Frey, F.J.; Farese, S. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008, 74, 1444–1453. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.B.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Su, Y.; Liu, D.; Liu, Y.; Zhang, C.; Wang, J.; Wang, S. Physiologic Levels of Endogenous Hydrogen Sulfide Maintain the Proliferation and Differentiation Capacity of Periodontal Ligament Stem Cells. J. Periodontol. 2015, 86, 1276–1286. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, R.; Liu, X.; Zhou, Y.; Qu, C.; Kikuiri, T.; Wang, S.; Zandi, E.; Du, J.; Ambudkar, I.S.; et al. Hydrogen Sulfide Maintains Mesenchymal Stem Cell Function and Bone Homeostasis via Regulation of Ca2+ Channel Sulfhydration. Cell Stem Cell 2014, 15, 66–78. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, Y.; Lu, K.; Zhang, H.; Fan, X. GYY4137 promotes bone formation in a rabbit distraction osteogenesis model: A preliminary report. J. Oral. Maxillofac. Surg. 2015, 73, 732.e1–732.e6. [Google Scholar] [CrossRef]
- Grassi, F.; Tyagi, A.M.; Calvert, J.W.; Gambari, L.; Walker, L.D.; Yu, M.; Robinson, J.; Malik, A.; Lisignoli, G.; Vaccaro, C.; et al. Hydrogen Sulfide Is a Novel Regulator of Bone Formation Implicated in the Bone Loss Induced by Estrogen Deficiency. J. Bone Miner. Res. 2015, 31, 949–963. [Google Scholar] [CrossRef]
- Robert, K.; Maurin, N.; Vayssettes, C.; Siauve, N.; Janel, N. Cystathionine beta synthase deficiency affects mouse endochondral ossification. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2005, 282, 1–7. [Google Scholar]
- Levasseur, R. Bone tissue and hyperhomocysteinemia. Jt. Bone Spine 2009, 76, 234–240. [Google Scholar] [CrossRef]
- Wang, J.; Hegele, R.A. Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH). Hum. Genet. 2003, 112, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Masood, W.S.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-H.; Tang, C.-H.; Hung, S.-Y.; Liu, S.-H.; Lin, Y.-M.; Fu, W.-M.; Yang, R.-S. Upregulation of heme oxygenase-1 inhibits the maturation and mineralization of osteoblasts. J. Cell. Physiol. 2009, 222, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Al-Shawafi, H.A.; Sultana, S.; Makita, S.; Sohda, M.; Oda, K. Disulfide bonds are critical for tissue-nonspecific alkaline phosphatase function revealed by analysis of mutant proteins bearing a C201-Y or C489-S substitution associated with severe hypophosphatasia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 581–588. [Google Scholar] [CrossRef]
- Burguera, E.F.; Vela-Anero, A.; Magalhaes, J.; Meijide-Failde, R.; Blanco, F.J. Effect of hydrogen sulfide sources on inflammation and catabolic markers on interleukin 1beta-stimulated human articular chondrocytes. Osteoarthr. Cartilage. 2014, 22, 1026–1035. [Google Scholar] [CrossRef]
- Kloesch, B.; Liszt, M.; Steiner, G.; Broll, J. Inhibitors of p38 and ERK1/2 MAPkinase and hydrogen sulphide block constitutive and IL-1beta-induced IL-6 and IL-8 expression in the human chondrocyte cell line C-28/I2. Rheumatol. Int. 2012, 32, 729–736. [Google Scholar] [CrossRef]
- Sung Hsieh, H.H.; Chung, M.T.; Allen, R.M.; Ranganathan, K.; Habbouche, J.; Cholok, D.; Butts, J.; Kaura, A.; Tiruvannamalai-Annamalai, R.; Breuler, C.; et al. Evaluation of Salivary Cytokines for Diagnosis of both Trauma-Induced and Genetic Heterotopic Ossification. Front. Endocrinol. 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| Cbs | AGC AAC CCT TTG GCA CAC TA | CTT ATC CAC CAC CGC CCT G |
| Cse | GCC AGT CCT CGG GTT TTG AA | TTG TGG TGT AAT CGC TGC CT |
| Coll2 | ACA CTT TCC AAC CGC AGT CA | GGG AGG ACG GTT GGG TAT CA |
| Coll10 | AAA CGC CCA CAG GCA TAA AG | CAA CCC TGG CTC TCC TTG G |
| Runx2 | GGG AAC CAA GAA GGC ACA GA | TGG AGT GGA TGG ATG GGG AT |
| Sox9 | AAG ACT CTG GGC AAG CTC TGG A | TTG TCC GTT CTT CAC CGA CTT CCT |
| Gapdh | CTC ATG ACC ACA GTC CAT GC | CAC ATT GGG GGT AGG AAC AC |
| Mmp-3 | ATA CGA GGG CAC GAG GAG | AGA AGT AGA GAA ACC CAA ATG CT |
| Mmp-13 | GCA GTT CCA AAG GCT ACA AC | GCT GGG TCA CAC TTC TCT G |
| 3-Mst | CTG GGA AAC GGG GAG CG | GCT CGG AAA AGT TGC GGG |
| Tbp | CTT GAA ATC ATC CCT GCG AG | CGC TTT CAT TAA ATT CTT GAT GGT C |
| Timp-1 | CCC ACA AGT CCC AGA ACC GCA G | GCA GGC AAG CAA AGT GAC GGC |
| Timp-3 | TCC TAG ACC CAG TTC CAT ATA CAC TTC | TTG GAC TTC TGC CAA CTT CCT T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasi, S.; Ehirchiou, D.; Bertrand, J.; Castelblanco, M.; Mitchell, J.; Ishii, I.; So, A.; Busso, N. The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage. Antioxidants 2021, 10, 1433. https://doi.org/10.3390/antiox10091433
Nasi S, Ehirchiou D, Bertrand J, Castelblanco M, Mitchell J, Ishii I, So A, Busso N. The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage. Antioxidants. 2021; 10(9):1433. https://doi.org/10.3390/antiox10091433
Chicago/Turabian StyleNasi, Sonia, Driss Ehirchiou, Jessica Bertrand, Mariela Castelblanco, James Mitchell, Isao Ishii, Alexander So, and Nathalie Busso. 2021. "The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage" Antioxidants 10, no. 9: 1433. https://doi.org/10.3390/antiox10091433
APA StyleNasi, S., Ehirchiou, D., Bertrand, J., Castelblanco, M., Mitchell, J., Ishii, I., So, A., & Busso, N. (2021). The Gasotransmitter Hydrogen Sulfide (H2S) Prevents Pathologic Calcification (PC) in Cartilage. Antioxidants, 10(9), 1433. https://doi.org/10.3390/antiox10091433