
Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus

 ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Design
2.3. Metabolic Parameters
2.4. Protein Extraction
2.5. Analysis of Tumor Necrosis Factor Alpha (TNF-Alpha)
2.6. Western Blotting
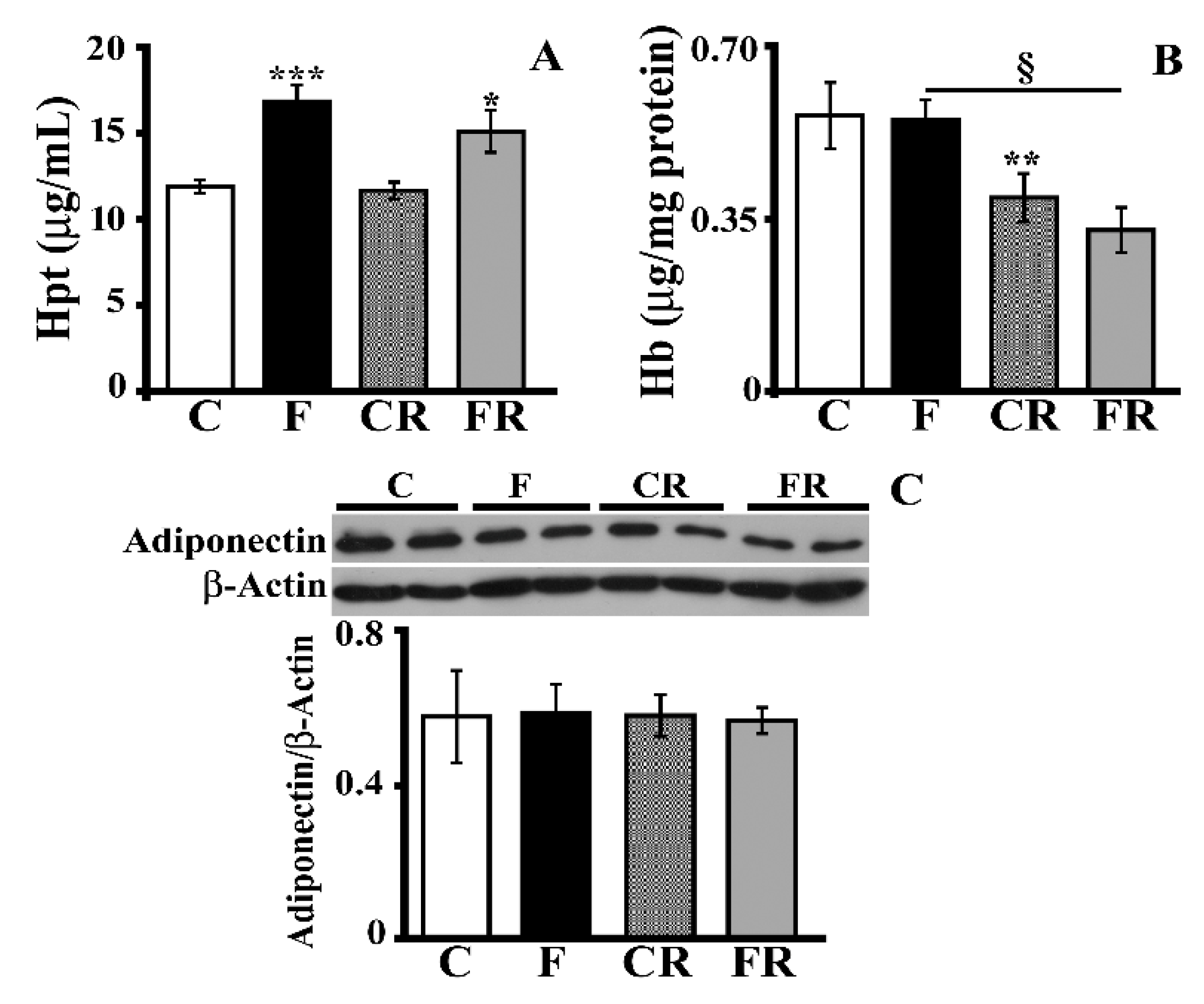
2.7. Haptoglobin (Hpt) and Hemoglobin (Hb) Evaluation
2.8. Mitochondrial Analyses in Rat Hippocampi
- FCRLI = CIL/CI&IIE;
- FCRLI&II = CI&IIL/CI&IIE;
- FCRPI = CIP/CI&IIE;
- FCRPI&II = CI&IIP/CI&IIE.
- In addition, flux control factors (FCFs) were calculated as follows:
- Coupling efficiency of oxidative phosphorylation = 1 − CI&IIL/CI&IIP;
- Excess capacity of the electron transport chain = 1 − CI&IIP/CI&IIE;
- FCF Complex I = 1 − (CIIE/CI &IIE);
- FCF Complex II = 1 − (CIP/CI + IIP).
2.9. Oxidative Stress Markers and Antioxidant Enzymes
2.10. Immunofluorescence Analysis
2.11. Statistical Analysis
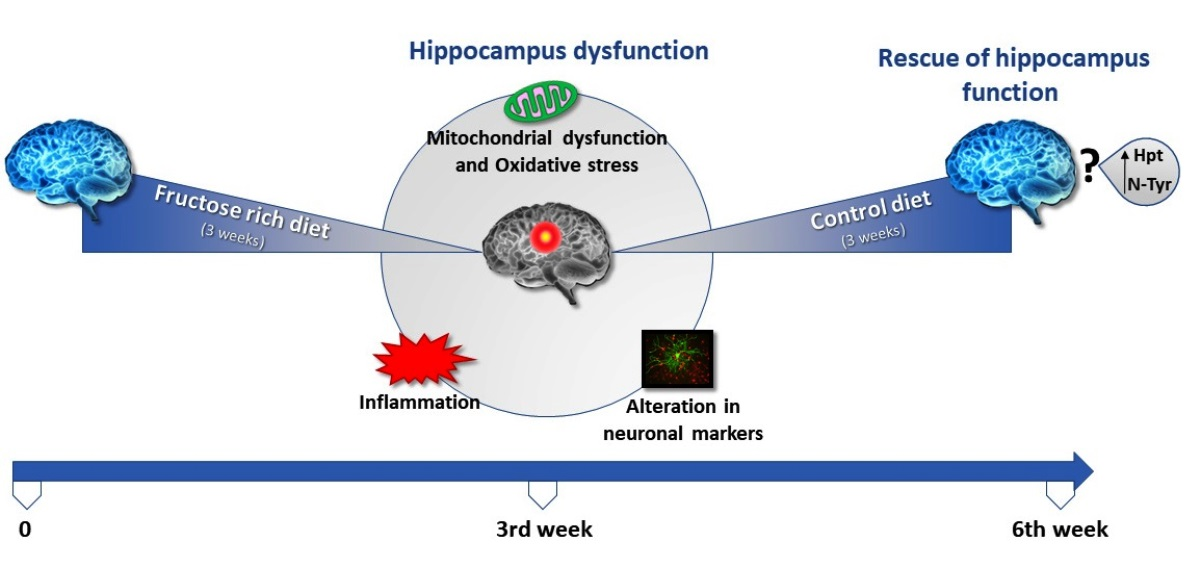
3. Results
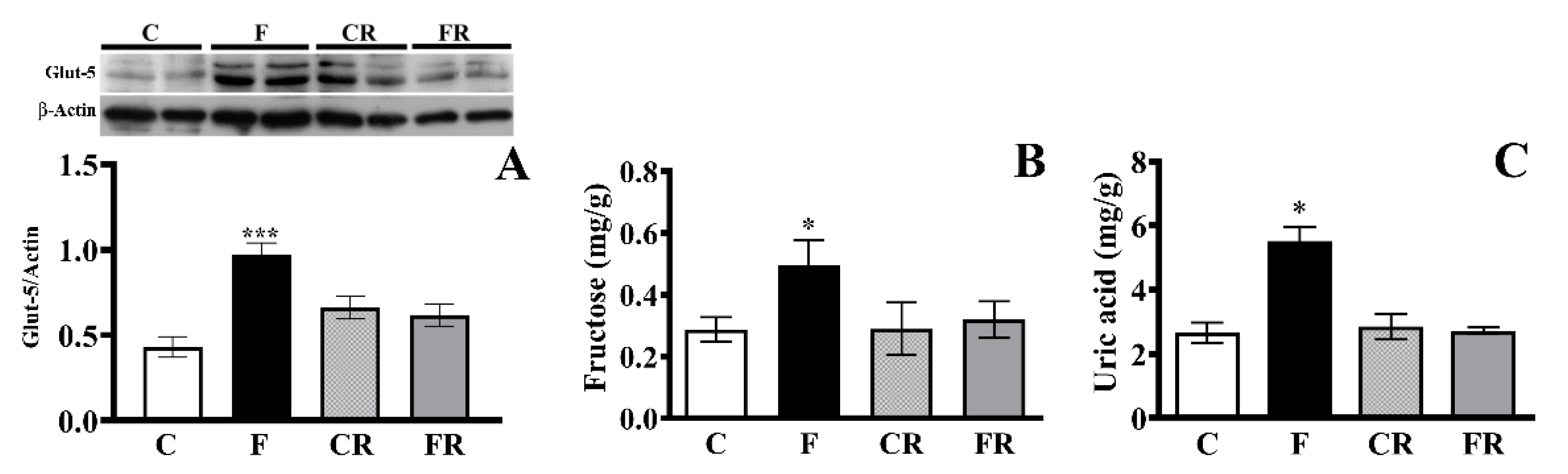
3.1. Glut-5 Expression, Fructose and Uric Acid Level in Hippocampus
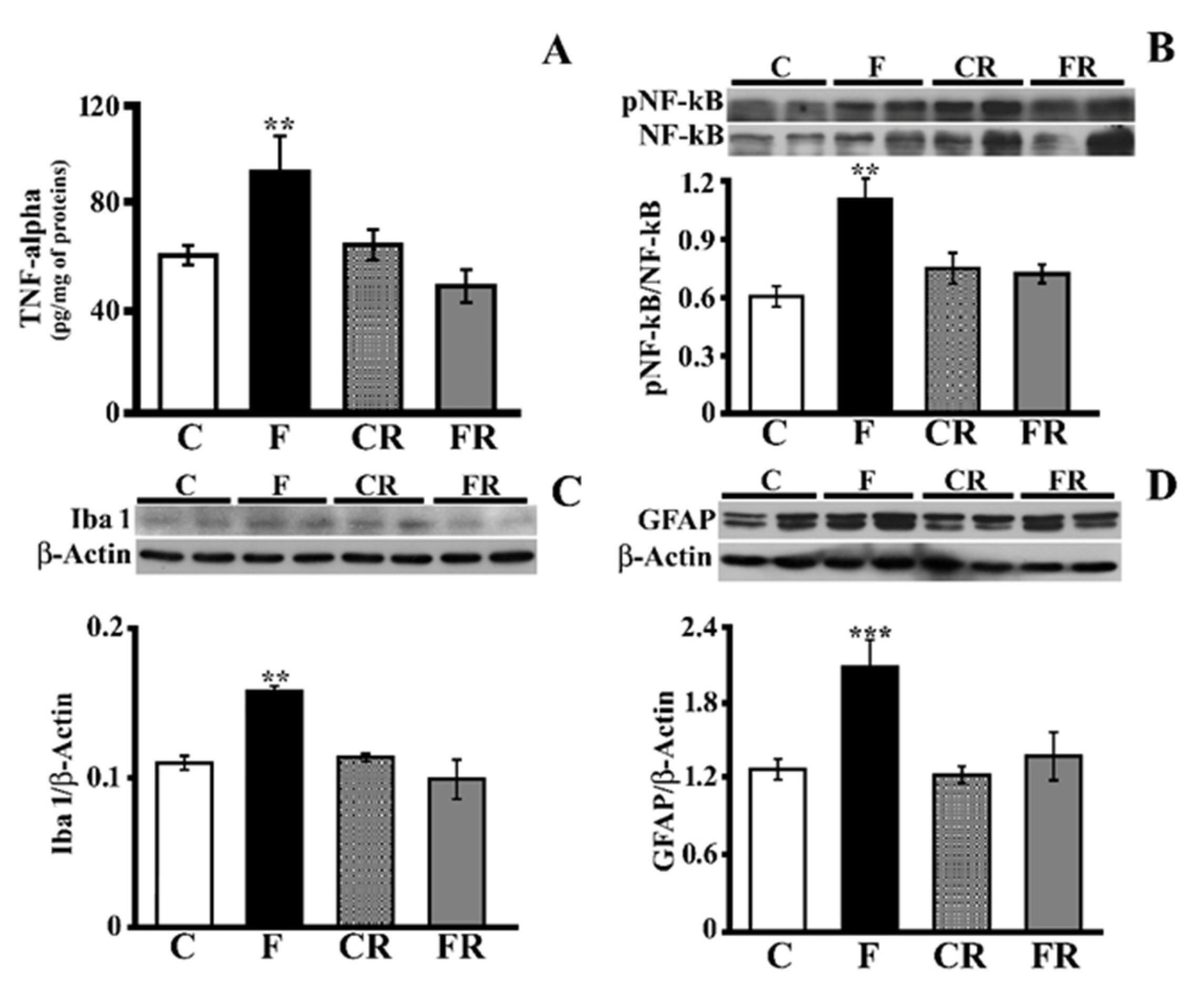
3.2. Markers of Hippocampal Inflammation
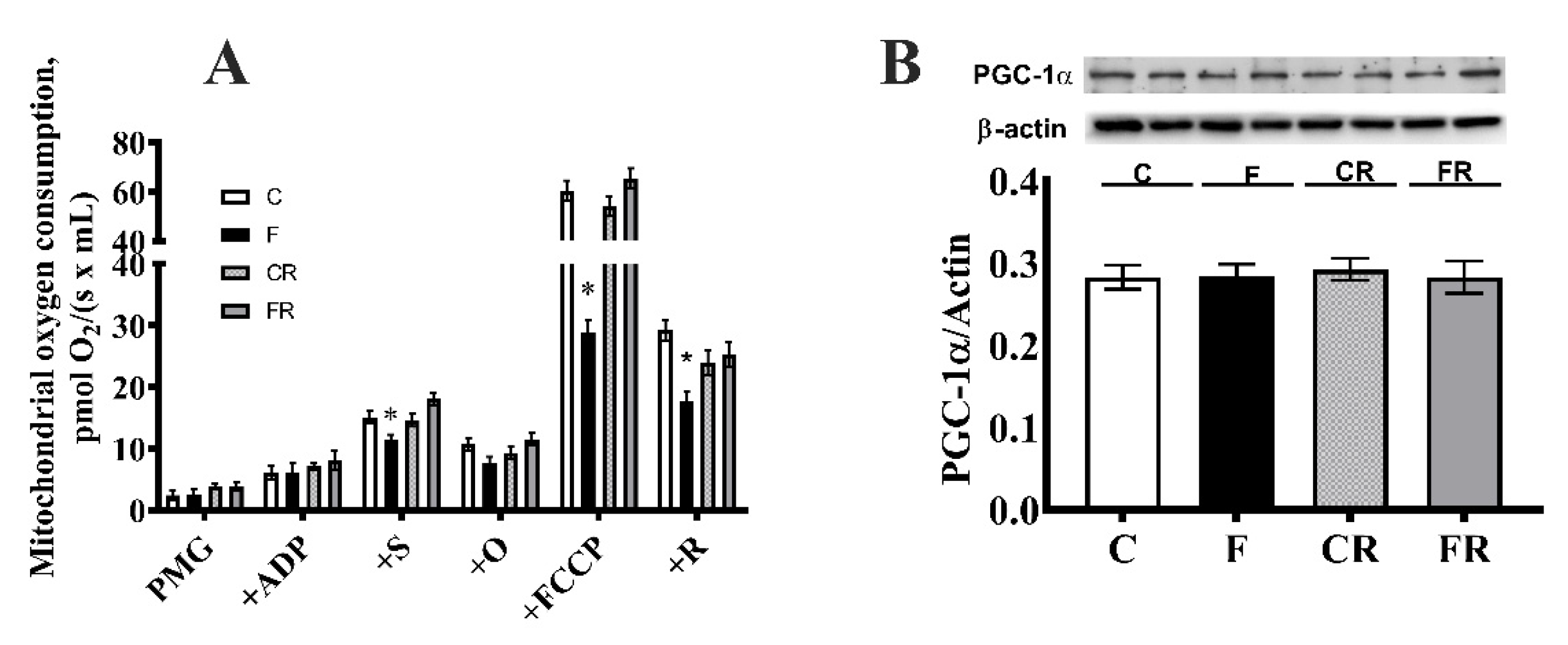
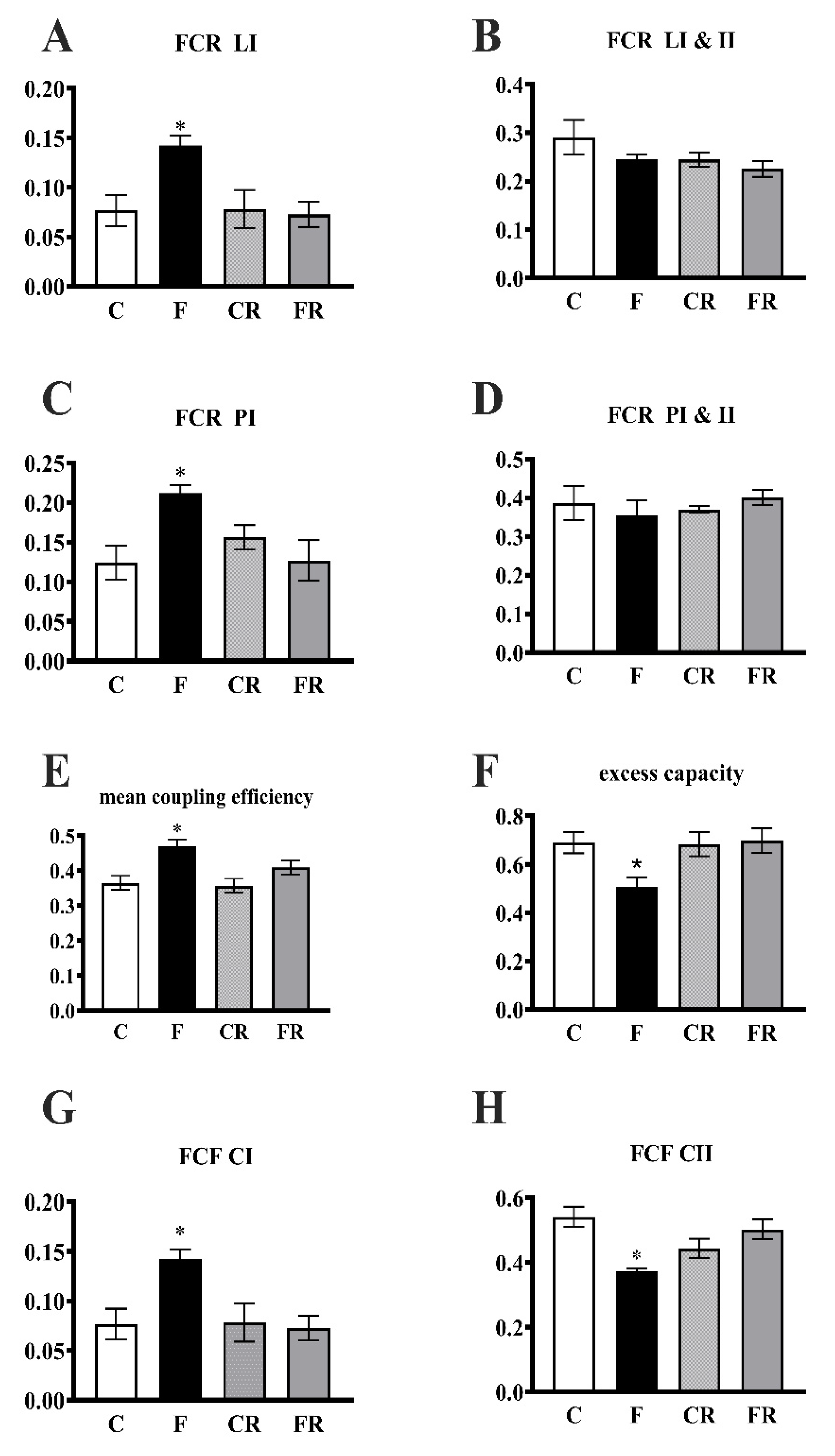
3.3. Electron Transport Chain Pathway
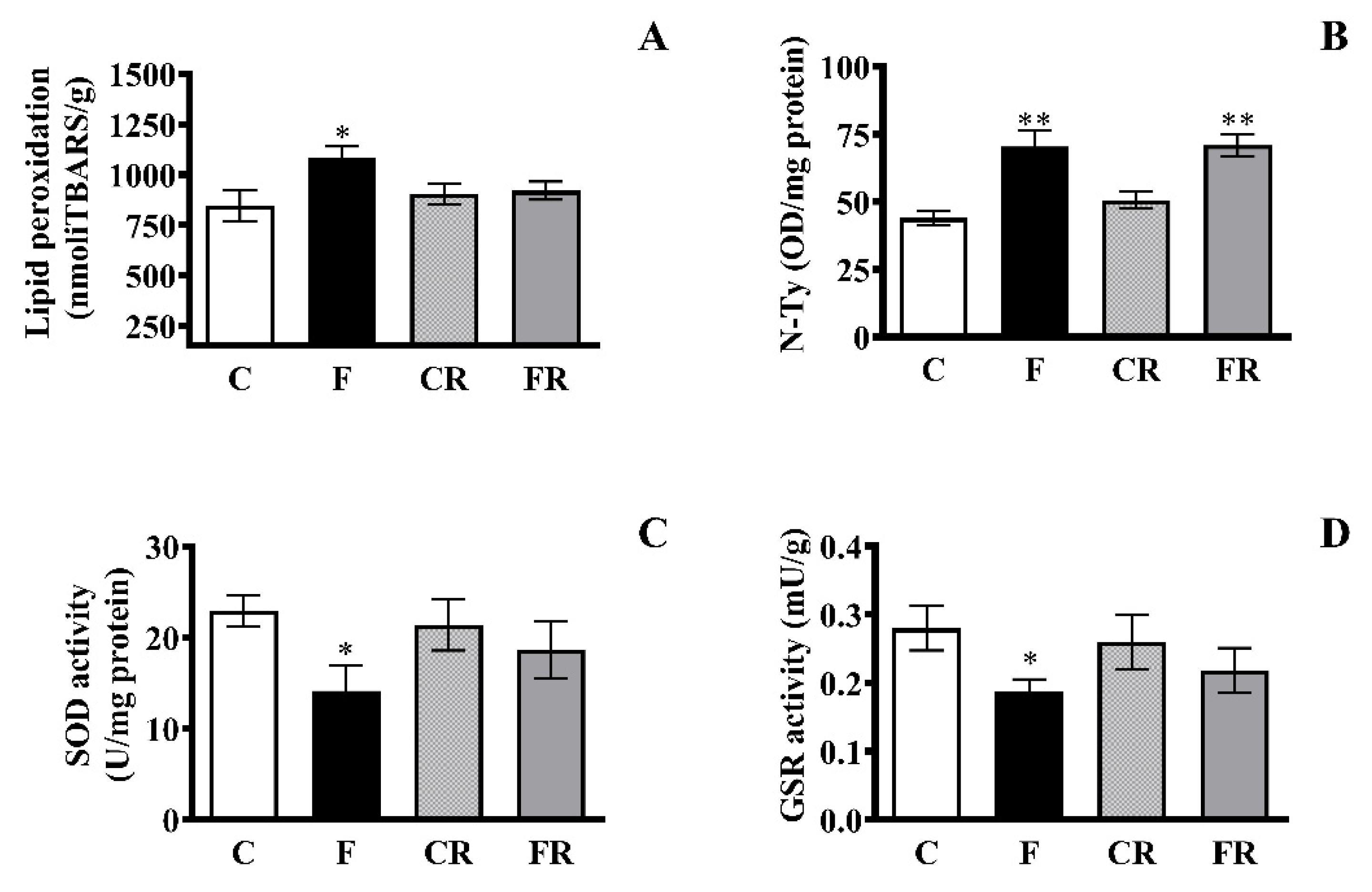
3.4. Markers of Oxidative Status
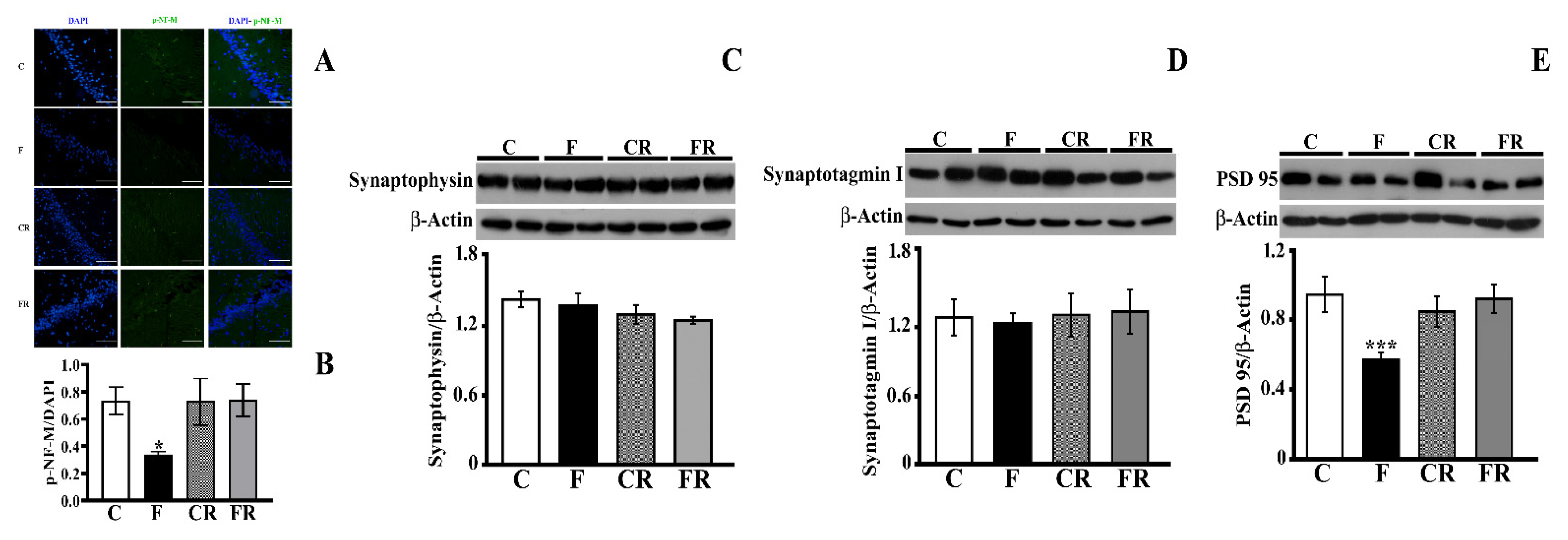
3.5. Analysis of Neurofilament M and Synaptic Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Soft drink consumption and obesity: It is all about fructose. Curr. Opin. Lipidol. 2010, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004, 292, 927–934. [Google Scholar] [CrossRef]
- Livesey, G.; Taylor, R. Fructose consumption and consequences for glycation, plasma triacylglycerol, and body weight: Metaanalyses and meta-regression models of intervention studies. Am. J. Clin. Nutr. 2008, 88, 1419–1437. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collino, M.; Rogazzo, M.; Medana, C.; Nigro, D.; Boccuzzi, G.; Aragno, M. Advanced glycation end products promote hepatosteatosis by interfering with SCAP-SREBP pathway in fructose-drinking mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G398–G407. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, M.S.; Iossa, S.; Cigliano, L. Sweet but Bitter: Focus on Fructose Impact on Brain Function in Rodent Models. Nutrients 2020, 13, 1. [Google Scholar] [CrossRef]
- Johnson, R.J.; Gomez-Pinilla, F.; Nagel, M.; Nakagawa, T.; Rodriguez-Iturbe, B.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Cerebral Fructose metabolism as a Potential Mechanism Driving Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 560865. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Ma, Y.; Hong, Y.; Hou, X.; Chen, J.; Shen, C.; Sun, M.; Shang, Y.; Dong, S.; Zeng, Z.; et al. Lycopene attenuates insulin signaling deficits, oxidative stress, neuroinflammation, and cognitive impairment in fructose-drinking insulin resistant rats. Neuropharmacology 2014, 86, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Van der Borght, K.; Köhnke, R.; Göransson, N.; Deierborg, T.; Brundin, P.; Erlanson-Albertsson, C.; Lindqvist, A. Reduced Neurogenesis in the Rat Hippocampus Following High Fructose Consumption. Regul. Pept. 2011, 167, 26–30. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as risk factor for neurodegeneration: Key role for carboxy methyllysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef]
- Li, J.M.; Ge, C.X.; Xu, M.X.; Wang, W.; Yu, R.; Fan, C.Y.; Kong, L.D. Betaine recovers hypothalamic neural injury by inhibiting astrogliosis and inflammation in fructose-fed rats. Mol. Nutr. Food Res. 2015, 59, 189–202. [Google Scholar] [CrossRef]
- Wu, H.W.; Ren, L.F.; Zhou, X.; Han, D.W. A high-fructose diet induces hippocampal insulin resistance and exacerbates memory deficits in male Sprague-Dawley rats. Nutr. Neurosci. 2015, 18, 323–328. [Google Scholar] [CrossRef]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.P.; Bartness, T.J.; Mielke, J.G.; Parent, M.B. A high fructose diet impairs spatial memory in male rats. Neurobiol. Learn. Mem. 2009, 92, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Cigliano, L.; Spagnuolo, M.S.; Crescenzo, R.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Liverini, G.; Iossa, S. Short-Term Fructose Feeding Induces Inflammation and Oxidative Stress in the Hippocampus of Young and Adult Rats. Mol. Neurobiol. 2018, 55, 2869–2883. [Google Scholar] [CrossRef]
- Jiménez-Maldonado, A.; Ying, Z.; Byun, H.R.; Gomez-Pinilla, F. Short-term fructose ingestion affects the brain independently from establishment of metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Fierros-Campuzano, J.; Ballesteros-Zebadúa, P.; Manjarrez-Marmolejo, J.; Aguilera, P.; Méndez-Diaz, M.; Prospero-García, O.; Franco-Pérez, J. Irreversible hippocampal changes induced by high fructose diet in rats. Nutr. Neurosci. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.N.; Slining, M.M.; Popkin, B.M. Trends in dietary intake among US 2- to 6-year-old children, 1989–2008. J. Acad. Nutr. Diet. 2013, 113, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.M.; Kanoski, S.E. Blood-brain barrier disruption: Mechanistic links between Western diet consumption and dementia. Front. Aging Neurosci. 2014, 6, 88. [Google Scholar] [CrossRef]
- Clark, K.A.; Alves, J.M.; Jones, S.; Yunker, A.G.; Luo, S.; Cabeen, R.P.; Angelo, B.; Xiang, A.H.; Page, K.A. Dietary Fructose Intake and Hippocampal Structure and Connectivity during Childhood. Nutrients 2020, 12, 909. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.L.; Chan, K.; Jarrard, L.E.; Kanoski, S.E.; Clegg, D.J.; Benoit, S.C. Contributions of the Hippocampus and Medial Prefrontal Cortex to Energy and BodyWeight Regulation. Hippocampus 2009, 19, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Hannapel, R.C.; Henderson, Y.H.; Nalloor, R.; Vazdarjanova, A.; Parent, M.B. Ventral hippocampal neurons inhibit postprandial energy intake. Hippocampus 2017, 27, 274–284. [Google Scholar] [CrossRef]
- Crescenzo, R.; Spagnuolo, M.S.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Gatto, C.; Iossa, S.; Cigliano, L. Effect of Initial Aging and High-Fat/High-Fructose Diet on Mitochondrial Bioenergetics and Oxidative Status in Rat Brain. Mol. Neurobiol. 2019, 56, 7651–7663. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, M.S.; Bergamo, P.; Crescenzo, R.; Iannotta, L.; Treppiccione, L.; Iossa, S.; Cigliano, L. Brain Nrf2 pathway, autophagy, and synaptic function proteins are modulated by a short-term fructose feeding in young and adult rats. Nutr. Neurosci. 2020, 23, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, M.S.; Pallottini, V.; Mazzoli, A.; Iannotta, L.; Tonini, C.; Morone, B.; Ståhlman, M.; Crescenzo, R.; Strazzullo, M.; Iossa, S.; et al. A Short-Term Western Diet Impairs Cholesterol Homeostasis and Key Players of Beta Amyloid Metabolism in Brain of Middle-Aged Rats. Mol. Nutr. Food Res. 2020, 64, e2000541. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Maresca, B.; Mollica, M.P.; Cavaliere, G.; Cefaliello, C.; Trinchese, G.; Esposito, M.G.; Scudiero, R.; Crispino, M.; Abrescia, P.; et al. Haptoglobin increases with age in rat hippocampus and modulates Apolipoprotein E mediated cholesterol trafficking in neuroblastoma cell lines. Front. Cell Neurosci. 2014, 8, 212. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Donizetti, A.; Iannotta, L.; Aliperti, V.; Cupidi, C.; Bruni, A.C.; Cigliano, L. Brain-derived neurotrophic factor modulates cholesterol homeostasis and Apolipoprotein E synthesis in human cell models of astrocytes and neurons. J. Cell Physiol. 2018, 233, 6925–6943. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Maresca, B.; La Marca, V.; Carrizzo, A.; Veronesi, C.; Cupidi, C.; Piccoli, T.; Maletta, R.G.; Bruni, A.C.; Abrescia, P.; et al. Haptoglobin interacts with apolipoprotein E and beta-amyloid and influences their crosstalk. ACS Chem. Neurosci. 2014, 5, 837–847. [Google Scholar] [CrossRef]
- Mazzoli, A.; Spagnuolo, M.S.; Gatto, C.; Nazzaro, M.; Cancelliere, R.; Crescenzo, R.; Iossa, S.; Cigliano, L. Adipose Tissue and Brain Metabolic Responses to Western Diet-Is There a Similarity between the Two? Int. J. Mol. Sci. 2020, 21, 786. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Cigliano, L.; Abrescia, P. The binding of haptoglobin to apolipoprotein AI: Influence of hemoglobin and concanavalin A. Biol. Chem. 2003, 384, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Zangrandi, L.; Schwarzer, C.; Gnaiger, E. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high- resolution respirometry. Mitochondrion 2015, 25, 104–112. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis. IOROBOROS MiPNet Publications, 4th ed.; Steiger Druck GmbH: Axams, Austria, 2014; ISBN 978-973-9502399-9502398-9502390. [Google Scholar]
- Spagnuolo, M.S.; Mollica, M.P.; Maresca, B.; Cavaliere, G.; Cefaliello, C.; Trinchese, G.; Scudiero, R.; Crispino, M.; Cigliano, L. High fat diet and inflammation—Modulation of haptoglobin level in rat brain. Front. Cell Neurosci. 2015, 9, 479. [Google Scholar] [CrossRef]
- Crescenzo, R.; Mazzoli, A.; Cancelliere, R.; Bianco, F.; Giacco, A.; Liverini, G.; Dulloo, A.G.; Iossa, S. Polyunsaturated Fatty Acids Stimulate De novo Lipogenesis and Improve Glucose Homeostasis during Refeeding with High Fat Diet. Front. Physiol. 2017, 8, 178. [Google Scholar] [CrossRef]
- Flohè, L.; Otting, F. Superoxide dismutase assay. Meth. Enzymol. 1974, 105, 93–104. [Google Scholar]
- Carlberg, I.; Mannervik, B. Glutathione Reductase. Meth. Enzymol. 1958, 113, 484–485. [Google Scholar]
- Cigliano, L.; Spagnuolo, M.S.; Abrescia, P. Quantitative variations of the isoforms in haptoglobin 1-2 and 2-2 individual phenotypes. Arch. Biochem. Biophys. 2003, 416, 227–237. [Google Scholar] [CrossRef]
- Bloemer, J.; Pinky, P.D.; Govindarajulu, M.; Hong, H.; Judd, R.; Amin, R.H.; Moore, T.; Dhanasekaran, M.; Reed, M.N.; Suppiramaniam, V. Role of Adiponectin in Central Nervous System Disorders. Neural Plast. 2018, 2018, 4593530. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals and Antioxidants in the Year 2000: A Historical Look to the Future. Ann. N. Y. Acad. Sci. 2000, 899, 136–147. [Google Scholar] [CrossRef]
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration. Exp. Neurol. 2020, 328, 113247. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.H.; Ahn, J.H.; Park, J.; Kim, I.H.; Cho, J.H.; Shin, B.N.; Lee, T.K.; Kim, H.; Song, M.; et al. Effects of chronic scopolamine treatment on cognitive impairment and neurofilament expression in the mouse hippocampus. Mol. Med. Rep. 2018, 17, 1625–1632. [Google Scholar] [CrossRef]
- Xie, S.; Girshick, R.; Dollar, P.; Tu, Z.; He, K. Aggregated Residual Transformations for Deep Neural Networks. arXiv 2017, arXiv:1611.05431v2, 1492–1500. [Google Scholar]
- Courtney, N.A.; Bao, H.; Briguglio, J.S.; Chapman, E.R. Synaptotagmin 1 clamps synaptic vescicle fusion in mammalian neurons independent of clompexin. Nat. Commun. 2019, 10, 4076. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef]
- Shu, H.J.; Isenberg, K.; Cormier, R.J.; Benz, A.; Zorumski, C.F. Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience 2006, 140, 889–895. [Google Scholar] [CrossRef]
- Oppelt, S.A.; Zhang, W.; Tolan, D.R. Specific regions of the brain are capable of fructose metabolism. Brain Res. 2017, 1657, 312–322. [Google Scholar] [CrossRef]
- Meakin, P.J.; Fowler, M.J.; Rathbone, A.J.; Allen, L.M.; Ransom, B.R.; Ray, D.E.; Brown, A.M. Fructose metabolism in the adult mouse optic nerve, a central white matter tract. J. Cereb. Blood Flow Metab. 2007, 27, 86–99. [Google Scholar] [CrossRef]
- Wu, K.L.; Hung, C.Y.; Chan, J.Y.; Wu, C.-W. An increase in adenosine-5’-triphosphate (ATP) content in rostral ventrolateral medulla is engaged in the high fructose diet-induced hypertension. J. Biomed. Sci. 2014, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andrés-Hernando, A.; Ishimoto, T.; Sanchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef]
- Cicerchi, C.; Li, N.; Kratzer, J.; Garcia, G.; Roncal-Jimenez, C.A.; Tanabe, K.; Hunter, B.; Rivard, C.J.; Sautin, Y.Y.; Gaucher, E.A.; et al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: Evolutionary implications of the uricase loss in hominids. FASEB J. 2014, 28, 3339–3350. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Lanaspa, M.A. Sugar, uric acid and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Shao, X.; Gu, H.; Wang, Z.; Chai, X.; Tian, Y.; Shi, G. Highly selective electrochemical strategy for monitoring of cerebral Cu2+ based on a carbon Dot-TPEA hybridized surface. Anal. Chem. 2013, 85, 418–425. [Google Scholar] [CrossRef]
- Shao, X.; Lu, W.; Gao, F.; Li, D.; Hu, J.; Li, Y.; Zuo, Z.; Jie, H.; Zhao, Y.; Cen, X. Uric Acid Induces Cognitive Dysfunction through Hippocampal Inflammation in Rodents and Humans. J. Neurosci. 2016, 36, 10990–11005. [Google Scholar] [CrossRef]
- Salvatore, A.; Cigliano, L.; Bucci, E.M.; Corpillo, D.; Velasco, S.; Carlucci, A.; Pedone, C.; Abrescia, P. Haptoglobin binding to apolipoprotein A-I prevents damage from hydroxyl radicals on its stimulatory activity of the enzyme lecithin-cholesterol acyl-transferase. Biochemistry 2007, 46, 11158–11168. [Google Scholar] [CrossRef]
- Maresca, B.; Spagnuolo, M.S.; Cigliano, L. Haptoglobin modulates beta-amyloid uptake by U-87 MG astrocyte cell line. J. Mol. Neurosci. 2015, 56, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, M.S.; Cigliano, L.; Maresca, B.; Pugliese, C.R.; Abrescia, P. Identification of plasma haptoglobin forms which loosely bind hemoglobin. Biol. Chem. 2011, 392, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Cigliano, L.; Pugliese, C.R.; Spagnuolo, M.S.; Palumbo, R.; Abrescia, P. Haptoglobin binds the antiatherogenic protein apolipoprotein E—Impairment of apolipoprotein E stimulation of both lecithin:cholesterol acyltransferase activity and cholesterol uptake by hepatocytes. FEBS J. 2009, 276, 6158–6171. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Cigliano, L.; Mazzoli, A.; Cancelliere, R.; Carotenuto, R.; Tussellino, M.; Liverini, G.; Iossa, S. Early Effects of a Low Fat, Fructose-Rich Diet on Liver Metabolism, Insulin Signaling, and Oxidative Stress in Young and Adult Rats. Front. Physiol. 2018, 9, 411. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Ruiz, C.R.; Lebson, L.; Selenica, M.L.; Rizer, J.; Hunt, J.B., Jr.; Rojiani, R.; Reid, P.; Kammath, S.; Nash, K.; et al. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 2013, 34, 1610–1620. [Google Scholar] [CrossRef]
- Roberts, R.A.; Smith, R.A.; Safe, S.; Szabo, C.; Tjalkens, R.B.; Robertson, F.M. Toxicological and pathophysiological roles of reactive oxygen and nitrogen species. Toxicology 2010, 276, 85–94. [Google Scholar] [CrossRef]
- Agrawal, R.; Noble, E.; Vergnes, L.; Ying, Z.; Reue, K.; Gomez-Pinilla, F. Dietary fructose aggravates the pathobiology of traumatic brain injury by influencing energy homeostasis and plasticity. J. Cereb. Blood Flow Metab. 2016, 36, 941–953. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef] [PubMed]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.F.; Hirsch, E.; Hantraye, P.; et al. Involvement of Mitochondrial Complex II Defects in Neuronal Death Produced by N-Terminus Fragment of Mutated Huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar] [CrossRef]
- Skulachev, V.P. Uncoupling: New approaches to an old problem of bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124. [Google Scholar] [CrossRef]
- Herce-Pagliai, C.; Kotecha, S.; Shuker, D.E.G. Analytical Methods for 3-Nitrotyrosine as a Marker of Exposure to Reactive Nitrogen Species: A Review. Nitric Oxide 1998, 2, 324–336. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Dörrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. Elife 2018, 7, e34202. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Myeounghoon, C.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh Moghaddam, A.; Jelodar, S.K.; Ranjbar, M. Hesperetin nanoparticles attenuate anxiogenic-like behavior and cerebral oxidative stress through the upregulation of antioxidant enzyme expression in experimental dementia of Alzheimer’s type. Neurol. Res. 2020, 42, 477–486. [Google Scholar] [CrossRef]
- Lee, M.K.; Cleveland, D.W. Neuronal intermediate filaments. Annu. Rev. Neurosci. 1996, 19, 187–217. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.; Hassinger, L.; Sihag, R.K.; Cleveland, D.W.; Mohan, P.; Nixon, R.A. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J. Cell Biol. 2000, 151, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Yabe, J.T.; Chan, W.K.; Chylinski, T.M.; Lee, S.; Pimenta, A.F.; Shea, T.B. The predominant form in which neurofilament subunits undergo axonal transport varies during axonal initiation, elongation, and maturation. Cell Motil. Cytoskelet. 2001, 48, 61–83. [Google Scholar] [CrossRef]
- Julien, J.P. Neurofilament functions in health and disease. Curr. Opin. Neurobiol. 1999, 9, 554–560. [Google Scholar] [CrossRef]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef]
- Vickers, J.C.; Kirkcaldie, M.T.; Phipps, A.; King, A.E. Alterations in neurofilaments and the transformation of the cytoskeleton in axons may provide insight into the aberrant neuronal changes of Alzheimer’s disease. Brain Res. Bull. 2016, 126 Pt 3, 324–333. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients, g/100 g | Control Diet | Fructose Diet |
|---|---|---|
| Standard Chow a | 50.5 | 50.5 |
| Sunflower Oil | 1.5 | 1.5 |
| Casein | 9.2 | 9.2 |
| Alphacel | 9.8 | 9.8 |
| Cornstarch | 20.4 | - |
| Fructose | - | 20.4 |
| Water | 6.4 | 6.4 |
| AIN-76 mineral mix | 1.6 | 1.6 |
| AIN-76 vitamin mix | 0.4 | 0.4 |
| Choline | 0.1 | 0.1 |
| Methionine | 0.1 | 0.1 |
| Energy content and composition | ||
| Gross Energy Density (kJ/g) | 17.2 | 17.2 |
| ME content (kJ/g) b | 11.1 | 11.1 |
| Proteins (% ME) | 29.0 | 29.0 |
| Lipids (% ME) | 10.6 | 10.6 |
| Carbohydrates (% ME) | 60.4 | 60.4 |
| Of which | ||
| Fructose | - | 30.0 |
| Starch | 52.8 | 22.8 |
| Sugars | 7.6 | 7.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzoli, A.; Spagnuolo, M.S.; Nazzaro, M.; Gatto, C.; Iossa, S.; Cigliano, L. Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus. Antioxidants 2021, 10, 487. https://doi.org/10.3390/antiox10030487
Mazzoli A, Spagnuolo MS, Nazzaro M, Gatto C, Iossa S, Cigliano L. Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus. Antioxidants. 2021; 10(3):487. https://doi.org/10.3390/antiox10030487
Chicago/Turabian StyleMazzoli, Arianna, Maria Stefania Spagnuolo, Martina Nazzaro, Cristina Gatto, Susanna Iossa, and Luisa Cigliano. 2021. "Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus" Antioxidants 10, no. 3: 487. https://doi.org/10.3390/antiox10030487
APA StyleMazzoli, A., Spagnuolo, M. S., Nazzaro, M., Gatto, C., Iossa, S., & Cigliano, L. (2021). Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus. Antioxidants, 10(3), 487. https://doi.org/10.3390/antiox10030487