
Endothelium as a Source and Target of H2S to Improve Its Trophism and Function



Abstract
1. Vascular Endothelium
2. Role of Endothelium in Physiology
3. Endothelial Dysfunction
4. Biochemistry of H2S Production
5. Molecular Signaling Activated by H2S into ECs
6. Cardiovascular Diseases Associated with Altered Levels of H2S
6.1. Hypertension
6.2. Diabetes
7. Molecular Mechanisms Regulated by H2S in Support of EC Function and Trophism
7.1. Antioxidant and Anti-Inflammatory Properties
7.2. Proangiogenic Effect
7.3. Wound Healing Promotion
8. Therapeutic Strategies to Improve H2S Concentration at Endothelial Level
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M. Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e26–e31. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial Cell Functions. J. Cell Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef]
- Boyce, S.; Lwaleed, B.; Kazmi, R. Homeostasis of hemostasis: The role of endothelium. Semin. Thromb. Hemost. 2015, 41, 549–555. [Google Scholar] [CrossRef]
- Good, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Ziche, M. Therapeutic implications of nitric oxide pathway in angiogenesis of tumors and inflammatory related disorders. In Therapeutic Application of Nitric Oxide in Cancer and Inflammatory Disorders; Morbidelli, L., Bonavida, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 65–91. [Google Scholar]
- Bełtowski, J. Synthesis, Metabolism, and Signaling Mechanisms of Hydrogen Sulfide: An Overview. Methods Mol. Biol. 2019, 2007, 1–8. [Google Scholar] [CrossRef]
- Kangussu, L.M.; Marzano, L.A.S.; Souza, C.F.; Dantas, C.C.; Miranda, A.S.; Simões E Silva, A.C. The Renin-Angiotensin System and the Cerebrovascular Diseases: Experimental and Clinical Evidence. Protein Pept. Lett. 2020, 27, 463–475. [Google Scholar] [CrossRef]
- Adams, L.; Franco, M.C.; Estevez, A.G. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. 2015, 240, 711–717. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044. [Google Scholar] [CrossRef]
- Parenti, A.; Morbidelli, L.; Ledda, F.; Granger, H.J.; Ziche, M. The bradykinin/B1 receptor promotes angiogenesis by upregulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. FASEB J. 2001, 15, 1487–1489. [Google Scholar] [CrossRef]
- Finetti, F.; Solito, R.; Morbidelli, L.; Giachetti, A.; Ziche, M.; Donnini, S. Prostaglandin E2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J. Biol. Chem. 2008, 283, 2139–2146. [Google Scholar] [CrossRef]
- Ziche, M.; Morbidelli, L.; Choudhuri, R.; Zhang, H.-T.; Donnini, S.; Granger, H.J.; Bicknell, R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Investig. 1997, 99, 2625–2664. [Google Scholar] [CrossRef]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef]
- Yan, M.S.; Marsden, P.A. Epigenetics in the Vascular Endothelium: Looking From a Different Perspective in the Epigenomics Era. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2297–2306. [Google Scholar] [CrossRef]
- Recchioni, R.; Marcheselli, F.; Antonicelli, R.; Mensà, E.; Lazzarini, R.; Procopio, A.D.; Olivieri, F. Epigenetic effects of physical activity in elderly patients with cardiovascular disease. Exp. Gerontol. 2017, 100, 17–27. [Google Scholar] [CrossRef]
- Turunen, M.P.; Ylä-Herttuala, S. Epigenetic regulation of key vascular genes and growth factors. Cardiovasc. Res. 2011, 90, 441–446. [Google Scholar] [CrossRef]
- Russell-Hallinan, A.; Watson, C.J.; O’Dwyer, D.; Grieve, D.J.; O’Neill, K.M. Epigenetic Regulation of Endothelial Cell Function by Nucleic Acid Methylation in Cardiac Homeostasis and Disease. Cardiovasc. Drugs Ther. 2020. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Haybar, H.; Shahrabi, S.; Rezaeeyan, H.; Shirzad, R.; Saki, N. Endothelial Cells: From Dysfunction Mechanism to Pharmacological Effect in Cardiovascular Disease. Cardiovasc. Toxicol. 2019, 19, 13–22. [Google Scholar] [CrossRef]
- Loscalzo, J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol. Haemost. Thromb. 2002, 32, 359–360. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Wenzel, P.; Wickramanayake, J.M.; Schuhmacher, S.; Jansen, T.; Lackner, K.J.; Torzewski, M.; Münzel, T. Nitrate tolerance as a model of vascular dysfunction: Roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol. Rep. 2009, 61, 33–48. [Google Scholar] [CrossRef]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of dysfunction in the ageing vasculature and role in age-related disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Trott, D.W.; Fadel, P.J. Inflammation as a mediator of arterial ageing. Exp. Physiol. 2019, 104, 1455–1471. [Google Scholar] [CrossRef]
- Khyzha, N.; Alizada, A.; Wilson, M.D.; Fish, J.E. Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol. Med. 2017, 23, 332–347. [Google Scholar] [CrossRef]
- Coco, C.; Sgarra, L.; Potenza, M.A.; Nacci, C.; Pasculli, B.; Barbano, R.; Parrella, P.; Montagnani, M. Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus? Int. J. Mol. Sci. 2019, 20, 2949. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Qiu, H.; Kim, S.; Jjingo, D.; Hoffman, R.; Kim, C.W.; Jang, I.; Son, D.J.; Kim, D.; Pan, C.; et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J. Clin. Investig. 2014, 124, 3187–3199. [Google Scholar] [CrossRef]
- Mongelli, A.; Atlante, S.; Bachetti, T.; Martelli, F.; Farsetti, A.; Gaetano, C. Epigenetic Signaling and RNA Regulation in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 509. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.M.; Wang, C.; Tigdi, J.; Si, X.; Dumpit, C.; Charles, S.; Gamage, A.; Moraes, T.J.; Lee, W.L. Influenza infects lung microvascular endothelium leading to microvascular leak: Role of apoptosis and claudin-5. PLoS ONE 2012, 7, e47323. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Goldenberg, N.M.; Lee, W.L. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antivir. Res. 2012, 93, 2–15. [Google Scholar] [CrossRef]
- Cross, D.; Drury, R.; Hill, J.; Pollard, A.J. Epigenetics in Sepsis: Understanding Its Role in Endothelial Dysfunction, Immunosuppression, and Potential Therapeutics. Front. Immunol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef]
- Tombor, L.S.; John, D.; Glaser, S.F.; Luxán, G.; Forte, E.; Furtado, M.; Rosenthal, N.; Baumgarten, N.; Schulz, M.H.; Wittig, J.; et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 2021, 12, 681. [Google Scholar] [CrossRef]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces ageing, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Nannelli, G.; Morbidelli, L.; Ziche, M.; Donnini, S. Endothelial aldehyde dehydrogenase 2 as a target to maintain vascular wellness and function in ageing. Biomedicines 2020, 8, 4. [Google Scholar] [CrossRef]
- Stromsnes, K.; Mas-Bargues, C.; Gambini, J.; Gimeno-Mallench, L. Protective Effects of Polyphenols Present in Mediterranean Diet on Endothelial Dysfunction. Oxid. Med. Cell. Longev. 2020, 2020, 2097096. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef]
- Yang, G.; Wang, R. H2S and Blood Vessels: An Overview. Handb. Exp. Pharmacol. 2015, 230, 85–110. [Google Scholar]
- Kanagy, N.L.; Szabo, C.; Papapetropoulos, A. Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 2017, 312, C537–C549. [Google Scholar] [CrossRef]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal. 2011, 15, 373–378. [Google Scholar] [CrossRef]
- Yu, X.H.; Cui, L.B.; Wu, K.; Zheng, X.L.; Cayabyab, F.S.; Chen, Z.W.; Tang, C.K. Hydrogen sulfide as a potent cardiovascular protective agent. Clin. Chim. Acta 2014, 437, 78–87. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Mistry, R.K.; Murray, T.V.A.; Prysyazhna, O.; Martin, D.; Burgoyne, J.R.; Santos, C.; Eaton, P.; Shah, A.M.; Brewer, A.C. Transcriptional regulation of cystathionine-γ-lyase in endothelial cells by NADPH oxidase 4-dependent signaling. J. Biol. Chem. 2016, 291, 1774–1788. [Google Scholar] [CrossRef]
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L.; et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 2015, 46, 192–203. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Leisegang, M.S.; Wittig, J.; Zukunft, S.; Kapasakalidi, A.; Fisslthaler, B.; Tsilimigras, D.; Zografos, G.; Filis, K.; et al. Shear stress regulates cystathionine γ lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. 2020, 28, 101379. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Sigala, F.; Wittig, I.; Heidler, J.; Zukunft, S.; Tsilimigras, D.I.; Randriamboavonjy, V.; Wittig, J.; Kojonazarov, B.; et al. Cystathionine gamma lyase sulfhydrates the RNA binding protein human antigen r to preserve endothelial cell function and delay atherogenesis. Circulation 2019, 139, 101–114. [Google Scholar] [CrossRef]
- Yuan, S.; Yurdagul, A., Jr.; Peretik, J.M.; Alfaidi, M.; Al Yafeai, Z.; Pardue, S.; Kevil, C.G.; Orr, A.W. Cystathionine γ-Lyase Modulates Flow-Dependent Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2126–2136. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Shi, S.; Gao, F.; Wu, J.; Dong, S.; Zhang, W.; Liu, Y.; Zhong, X. Calcium sensing receptor initiating cystathionine-gamma-lyase/hydrogen sulfide pathway to inhibit platelet activation in hyperhomocysteinemia rat. Exp. Cell Res. 2017, 358, 171–181. [Google Scholar] [CrossRef]
- Badiei, A.; Gieseg, S.; Davies, S.; Izani Othman, M.; Bhatia, M. LPS up-regulates cystathionine g-lyase gene expression in primary human macrophages via NF-κB/ERK pathway. Inflamm. Allergy Drug Targets 2015, 14, 99–104. [Google Scholar] [CrossRef]
- Yang, G.; Pei, Y.; Teng, H.; Cao, Q.; Wang, R. Specificity protein-1 as a critical regulator of human cystathionine g-lyase in smooth muscle cells. J. Biol. Chem. 2011, 286, 26450–26460. [Google Scholar] [CrossRef]
- Taniguchi, S.; Kimura, T.; Umeki, T.; Kimura, Y.; Kimura, H.; Ishii, I.; Itoh, N.; Naito, Y.; Yamamoto, H.; Niki, I. Protein phosphorylation involved in the gene expression of the hydrogen sulphide producing enzyme cystathionine γ-lyase in the pancreatic β-cell. Mol. Cell Endocrinol. 2012, 350, 31–38. [Google Scholar] [CrossRef]
- Gonzalez Bosc, L.V.; Osmond, J.M.; Giermakowska, W.K.; Pace, C.E.; Riggs, J.L.; Jackson-Weaver, O.; Kanagy, N.L. NFAT regulation of cystathionine γ-lyase expression in endothelial cells is impaired in rats exposed to intermittent hypoxia. Am. J. Physiol. Heart Circ Physiol. 2017, 312, H791–H799. [Google Scholar] [CrossRef]
- Leucker, T.M.; Nomura, Y.; Kim, J.H.; Bhatta, A.; Wang, V.; Wecker, A.; Jandu, S.; Santhanam, L.; Berkowitz, D.; Romer, L.; et al. Cystathionine gamma-lyase protects vascular endothelium: A role for inhibition of histone deacetylase 6. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H711–H720. [Google Scholar] [CrossRef]
- Chi, Z.; Byeon, H.E.; Seo, E.; Nguyen, Q.T.; Lee, W.; Jeong, Y.; Choi, J.; Pandey, D.; Berkowitz, D.E.; Kim, J.H.; et al. Histone deacetylase 6 inhibitor tubastatin A attenuates angiotensin II-induced hypertension by preventing cystathionine γ-lyase protein degradation. Pharmacol. Res. 2019, 146, 104281. [Google Scholar] [CrossRef]
- Wang, G.G.; Chen, Q.Y.; Li, W.; Lu, X.H.; Zhao, X. Ginkgolide B increases hydrogen sulfide and protects against endothelial dysfunction in diabetic rats. Croat. Med. J. 2015, 56, 4–13. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef]
- Wang, R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009, 76, 700–704. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Ohno, K.; Okuda, K.; Uehara, T. Endogenous S-sulfhydration of PTEN helps protect against modification by nitric oxide. Biochem. Biophys. Res. Commun. 2015, 456, 245–249. [Google Scholar] [CrossRef]
- Zhao, K.; Ju, Y.; Li, S.; Altaany, Z.; Wang, R.; Yang, G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014, 15, 792–800. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2021, 143, 935–948. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef]
- Altaany, Z.; Ju, Y.; Yang, G.; Wang, R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014, 7, ra87. [Google Scholar] [CrossRef]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef]
- Stubbert, D.; Prysyazhna, O.; Rudyk, O.; Scotcher, J.; Burgoyne, J.R.; Eaton, P. Protein kinase G Iα oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 2014, 64, 1344–1351. [Google Scholar] [CrossRef]
- Liu, X.; Pan, L.; Zhuo, Y.; Gong, Q.; Rose, P.; Zhu, Y. Hypoxia-inducible factor-1alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol. Pharm. Bull. 2010, 33, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef]
- Kan, J.; Guo, W.; Huang, C.; Bao, G.; Zhu, Y.; Zhu, Y.Z. S-propargylcysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid. Redox Signal. 2014, 20, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, X.H.; Zhang, C.C.; Wang, M.J.; Xue, W.L.; Wu, D.D.; Ma, F.F.; Li, W.W.; Tao, B.B.; Zhu, Y.C. Hydrogen sulfide promotes angiogenesis by downregulating miR-640 via the VEGFR2/mTOR pathway. Am. J. Physiol. Cell Physiol. 2016, 31, C305–C317. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chakraborty, P.K.; Xiong, X.; Dwivedi, S.K.D.; Mustafi, S.B.; Leigh, N.R.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathionine g_-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 2016, 30, 441–456. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2020, 27, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef]
- Kutz, J.L.; Greaney, J.L.; Santhanam, L.; Alexander, L.M. Evidence for a functional vasodilatatory role for hydrogen sulphide in the human cutaneous microvasculature. J. Physiol. 2015, 593, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Bucci, M.; Vellecco, V.; Cantalupo, A.; Brancaleone, V.; Zhou, Z.; Evangelista, S.; Calderone, V.; Papapetropoulos, A.; Cirino, G. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc. Res. 2014, 102, 138–147. [Google Scholar] [CrossRef] [PubMed]
- D’Emmanuele di Villa Bianca, R.; Mitidieri, E.; Donnarumma, E.; Tramontano, T.; Brancaleone, V.; Cirino, G.; Bucci, M.; Sorrentino, R. Hydrogen sulfide is involved in dexamethasone-induced hypertension in rat. Nitric Oxide 2015, 46, 80–86. [Google Scholar] [CrossRef]
- Huang, B.; Chen, C.-T.; Chen, C.-S.; Wang, Y.-M.; Hsieh, H.-J.; Wang, D.L. Laminar shear flow increases hydrogen sulfide and activates a nitric oxide producing signaling cascade in endothelial cells. Biochem. Biophys. Res. Commun. 2015, 464, 1254–1259. [Google Scholar] [CrossRef]
- Hu, H.J.; Jiang, Z.S.; Qiu, J.; Zhou, S.H.; Liu, Q.M. Protective effects of hydrogen sulfide against angiotensin II-induced endoplasmic reticulum stress in HUVECs. Mol. Med. Rep. 2017, 15, 2213–2222. [Google Scholar] [CrossRef][Green Version]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Hart, J.L. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens. Res. 2015, 38, 13–20. [Google Scholar] [CrossRef]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, watersoluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Meng, G.; Ma, Y.; Xie, L.; Ferro, A.; Ji, Y. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br. J. Pharmacol. 2015, 172, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Tomasova, L.; Pavlovicova, M.; Malekova, L.; Misak, A.; Kristek, F.; Grman, M.; Cacanyiova, S.; Tomasek, M.; Tomaskova, Z.; Perry, A.; et al. Effects of AP39, a novel triphenylphosphonium derivatized anethole dithiolethione hydrogen sulfide donor, on rat haemodynamic parameters and chloride and calcium Cav3 and RyR2 channels. Nitric Oxide 2015, 46, 131–144. [Google Scholar] [CrossRef]
- Van Goor, H.; van den Born, J.C.; Hillebrands, J.-L.; Joles, J.A. Hydrogen sulfide in hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Liu, S.; Dai, J.; Yang, T.; Jiang, X.; Duan, X.; Wu, Y. Hydrogen sulfide defends against the cardiovascular risk of Nw-nitro-L-argininemethyl ester induced hypertension in rats via the nitric oxide/endothelial nitric oxide synthase pathway. Chin. Med. J. 2014, 127, 3751–3757. [Google Scholar] [CrossRef]
- Xue, H.; Zhou, S.; Xiao, L.; Guo, Q.; Liu, S.; Wu, Y. Hydrogen sulfide improves the endothelial dysfunction in renovascular hypertensive rats. Physiol. Res. 2015, 64, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Dong, J.H. Hydrogen sulfide improves endothelial dysfunction via downregulating BMP4/COX-2 pathway in rats with hypertension. Oxid. Med. Cell. Longev. 2016, 8128957. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Dong, J.H.; Teng, X.; Jin, S.; Xue, H.M.; Liu, S.Y.; Guo, Q.; Shen, W.; Ni, X.C.; Wu, Y.M. Hydrogen sulfide improves endothelial dysfunction in hypertension by activating peroxisome proliferator-activated receptor delta/endothelial nitric oxide synthase signaling. J. Hypertens. 2018, 36, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Teng, X.; Jin, S.; Dong, J.; Guo, Q.; Tian, D.; Wu, Y. Hydrogen sulfide improves endothelial dysfunction by inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2019, 37, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Possomato-Vieira, J.S.; Goncalves-Rizzi, V.H. Clinical and experimental evidences of hydrogen sulfide involvement in lead-induced hypertension. BioMed Res. Int. 2018, 2018, 4627391. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.J.; Pushpakumar, S.; Tyagi, S.C.; Sen, U. Homocysteine and hydrogen sulfide in epigenetic, metabolic and microbiota related renovascular hypertension. Pharmacol. Res. 2016, 113, 300–312. [Google Scholar] [CrossRef]
- Cheng, Z.; Kishore, R. Potential role of hydrogen sulfide in diabetes-impaired angiogenesis and ischemic tissue repair. Redox Biol. 2020, 101704. [Google Scholar] [CrossRef]
- Brancaleone, V.; Roviezzo, F.; Vellecco, V.; De Gruttola, L.; Bucci, M.; Cirino, G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br. J. Pharmacol. 2008, 155, 673–680. [Google Scholar] [CrossRef]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A., Jr. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef]
- Jin, S.; Pu, S.X.; Hou, C.L.; Ma, F.F.; Li, N.; Li, X.H.; Tan, B.; Tao, B.B.; Wang, M.J.; Zhu, Y.C. Cardiac H2S generation is reduced in ageing diabetic mice. Oxid. Med. Cell. Longev. 2015, 758358, 2015. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2012, 17, 68–80. [Google Scholar] [CrossRef]
- Suzuki, K.; Sagara, M.; Aoki, C.; Tanaka, S.; Aso, Y. Clinical implication of plasma hydrogen sulfide levels in Japanese patients with type 2 diabetes. Intern. Med. 2017, 56, 17–21. [Google Scholar] [CrossRef]
- Geng, B.; Cai, B.; Liao, F.; Zheng, Y.; Zeng, Q.; Fan, X.; Gong, Y.; Yang, J.; Cui, Q.H.; Tang, C.; et al. Increase or decrease hydrogen sulfide exert opposite lipolysis, but reduce global insulin resistance in high fatty diet induced obese mice. PLoS ONE 2013, 8, e73892. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Alendy, M.J.; Ahmed, K.I.; Leeuwenburgh, C.; Julian, D. The hydrogen sulfide signaling system: Changes during aging and the benefits of caloric restriction. Age 2010, 32, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Módis, K.; Szczesny, B.; Brunyánszki, A.; Oláh, G.; Rios, E.C.S.; Yanagi, K.; Ahmad, A.; Papapetropoulos, A.; Szabo, C. Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/H2S pathway: Functional impairment by hyperglycemia and restoration by DL-lipoic acid. Mol. Med. 2015, 21, 1–14. [Google Scholar] [CrossRef]
- Suzuki, K.; Olah, G.; Modis, K.; Coletta, C.; Kulp, G.; Gerö, D.; Szoleczky, P.; Chang, T.; Zhou, Z.; Wu, L.; et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 13829–13834. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Wang, H.; Kho, S.H.; Rinkiko, S.; Sheng, X.; Shen, H.M.; Zhu, Y.Z. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE. 2013, 8, e53147. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Zhang, Y.; Yu, C.; Liu, Y.; Gao, L.; Zhao, J. Hydrogen sulfide protects against high-glucose-induced apoptosis in endothelial cells. J. Cardiovasc. Pharmacol. 2012, 59, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X.; et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef]
- Liu, N.; Wu, J.; Zhang, L.; Gao, Z.; Sun, Y.; Yu, M.; Zhao, Y.; Dong, S.; Lu, F.; Zhang, W. Hydrogen Sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J. Cell Mol. Med. 2017, 21, 3190–3203. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, S.; Chai, J.; Zhang, Y.; Ma, X.; Chen, J.; Guan, Q.; Wan, M.; Liu, Y. Hydrogen sulfide improves diabetic wound healing in ob/ob mice via attenuating inflammation. J. Diabetes Complicat. 2017, 31, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef]
- Liu, F.; Chen, D.D.; Sun, X.; Xie, H.H.; Yuan, H.; Jia, W.; Chen, A.F. Hydrogen sulfide improves wound healing via restoration of endothelial progenitor cell functions and activation of angiopoietin-1 in type 2 diabetes. Diabetes 2014, 63, 1763–1778. [Google Scholar] [CrossRef]
- Yang, C.T.; Chen, L.; Chen, W.L.; Li, N.; Chen, M.J.; Li, X.; Zheng, X.; Zhao, Y.Z.; Wu, Y.X.; Xian, M.; et al. Hydrogen sulfide primes diabetic wound to close through inhibition of NETosis. Mol. Cell. Endocrinol. 2019, 480, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Van den Born, J.C.; Mencke, R.; Conroy, S.; Zeebregts, C.J.; van Goor, H.; Hillebrands, J.L. Cystathionine γ-lyase is expressed in human atherosclerotic plaque microvessels and is involved in micro-angiogenesis. Sci. Rep. 2016, 6, 34608. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Li, E.; Mu, Q.; Zhang, Y.; Wei, X.; Li, H.; Cheng, L.; Zhang, B. Hydrogen sulfide inhalation decreases early blood-brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J. Cereb. Blood Flow Metab. 2015, 35, 494–500. [Google Scholar] [CrossRef]
- Wang, T.; Wang, L.; Zaidi, S.R.; Sammani, S.; Siegler, J.; Moreno-Vinasco, L.; Mathew, B.; Natarajan, V.; Garcia, J.G.N. Hydrogen sulfide attenuates particulate matter-induced human lung endothelial barrier disruption via combined reactive oxygen species scavenging and Akt activation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 491–496. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen sulfide and cellular redox homeostasis. Oxid. Med. Cell Longev. 2016, 6043038. [Google Scholar] [CrossRef]
- Gerö, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016, 113, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Wang, H.; Zhu, Y.Z. The drug developments of hydrogen sulfide on cardiovascular disease. Oxid. Med. Cell Longev. 2018, 2018, 4010395. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Wang, X.; Gao, L.; Chen, J.; Liu, Y.; Yu, C.; Zhang, N.; Zhang, X.; Zhao, J. Hydrogen sulfide suppresses high glucose-induced expression of intercellular adhesion molecule-1 in endothelial cells. J. Cardiovasc. Pharmacol. 2013, 62, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Liu, W.; Liu, Y.; Fan, Y.; Wang, X.; Yu, C.; Zhang, Y.; Wang, S.; Liu, J.; Zhao, J.; et al. High glucose induces the release of endothelin-1 through the inhibition of hydrogen sulfide production in HUVECs. Int. J. Mol. Med. 2015, 35, 810–814. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, J.; Chen, M.; Liu, D.; Guo, R.; Lin, K.; Deng, H.; Zhi, X.; Zhang, W.; Feng, J.; Wu, W. Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucose induced injury by inhibiting the necroptosis pathway. Int. J. Mol. Med. 2018, 41, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Liu, X.H.; Gong, Q.H.; Wu, D.; Zhu, Y.Z. Hydrogen sulfide attenuated tumor necrosis factor-α-induced inflammatory signaling and dysfunction in vascular endothelial cells. PLoS ONE 2011, 6, e19766. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Perna, A.F.; Sepe, I.; Lanza, D.; Capasso, R.; Zappavigna, S.; Capasso, G.; Caraglia, M.; Ingrosso, D. Hydrogen sulfide reduces cell adhesion and relevant inflammatory triggering by preventing ADAM17-dependent TNF-alpha activation. J. Cell Biochem. 2013, 114, 1536–1548. [Google Scholar] [CrossRef]
- Mani, S.; Untereiner, A.; Wu, L.; Wang, R. Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid. Redox Signal. 2014, 20, 805–817. [Google Scholar] [CrossRef]
- Li, X.H.; Xue, W.L.; Wang, M.J.; Zhou, Y.; Zhang, C.C.; Sun, C.; Zhu, L.; Liang, K.; Chen, Y.; Tao, B.B.; et al. H2S regulates endothelial nitric oxide synthase protein stability by promoting microRNA-455-3p expression. Sci. Rep. 2017, 7, 44807. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, A.; Wang, Y. SIRT1 in Endothelial Cells as a Novel Target for the Prevention of Early Vascular Aging. J. Cardiovasc. Pharmacol. 2016, 67, 465–473. [Google Scholar] [CrossRef]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid. Redox Signal. 2019, 30, 184–197. [Google Scholar] [CrossRef]
- Lin, Y.; Zeng, H.; Gao, L.; Gu, T.; Wang, C.; Zhang, H. Hydrogen sulfide attenuates atherosclerosis in a partially ligated carotid artery mouse model via regulating angiotensin converting enzyme 2 expression. Front. Physiol. 2017, 8, 782. [Google Scholar] [CrossRef]
- Zhang, H.; Bai, Z.; Zhu, L.; Liang, Y.; Fan, X.; Li, J.; Wen, H.; Shi, T.; Zhao, Q.; Wang, Z. Hydrogen sulfide donors: Therapeutic potential in anti-atherosclerosis. Eur. J. Med. Chem. 2020, 205, 112665. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA. 2009, 106, 21972–21977. [Google Scholar] [CrossRef]
- Abdollahi Govar, A.; Törő, G.; Szaniszlo, P.; Pavlidou, A.; Bibli, S.I.; Thanki, K.; Resto, V.A.; Chao, C.; Hellmich, M.R.; Szabo, C.; et al. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br. J. Pharmacol. 2020, 177, 866–883. [Google Scholar] [CrossRef]
- Predmore, B.L.; Julian, D.; Cardounel, A.J. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism. Front. Physiol. 2011, 2, 104. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef]
- Monti, M.; Hyseni, I.; Pacini, A.; Monzani, E.; Casella, L.; Morbidelli, L. Cross-talk between endogenous H2S and NO accounts for vascular protective activity of the metal-nonoate Zn(PipNONO)Cl. Biochem. Pharmacol. 2018, 152, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Tao, B.B.; Liu, S.Y.; Zhang, C.C.; Fu, W.; Cai, W.J.; Wang, Y.; Shen, Q.; Wang, M.J.; Chen, Y.; Zhang, L.J.; et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox Signal. 2013, 19, 448–464. [Google Scholar] [CrossRef]
- Tao, B.B.; Cai, W.J.; Zhu, Y.C. H2S Is a Promoter of Angiogenesis: Identification of H2S “Receptors” and Its Molecular Switches in Vascular Endothelial Cells. Handb. Exp. Pharmacol. 2015, 230, 137–152. [Google Scholar] [PubMed]
- Meng, G.; Zhao, S.; Xie, L.; Han, Y.; Ji, Y. Protein S-sulfhydration by hydrogen sulfide in cardiovascular system. Br. J. Pharmacol. 2018, 175, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Dicay, M.; McKnight, W.; Martin, G.R. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007, 21, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Li, W. Hydrogen sulfide improves vessel formation of the ischemic adductor muscle and wound healing in diabetic db/db mice. Iran. J. Basic Med. Sci. 2019, 22, 1192–1197. [Google Scholar] [PubMed]
- Xu, M.; Hua, Y.; Qi, Y.; Meng, G.; Yang, S. Exogenous hydrogen sulphide supplement accelerates skin wound healing via oxidative stress inhibition and vascular endothelial growth factor enhancement. Exp. Dermatol. 2019, 28, 776–785. [Google Scholar] [CrossRef]
- Xue, W.L.; Chen, R.Q.; Zhang, Q.Q.; Li, X.H.; Cao, L.; Li, M.Y.; Li, Y.; Lin, G.; Chen, Y.; Wang, M.J.; et al. Hydrogen sulfide rescues high glucose-induced migration dysfunction in HUVECs by upregulating miR-126-3p. Am. J. Physiol. Cell Physiol. 2020, 318, C857–C869. [Google Scholar] [CrossRef] [PubMed]
- Piragine, E.; Calderone, V. Pharmacological modulation of the hydrogen sulfide (H2S) system by dietary H2 S-donors: A novel promising strategy in the prevention and treatment of type 2 diabetes mellitus. Phytother. Res. 2020. [Google Scholar] [CrossRef]
- Calderone, V.; Martelli, A.; Testai, L.; Citi, V.; Breschi, M.C. Using hydrogen sulfide to design and develop drugs. Expert Opin. Drug Discov. 2016, 11, 163–175. [Google Scholar] [CrossRef]
- Hartle, M.D.; Pluth, M.D. A practical guide to working with H2S at the interface of chemistry and biology. Chem. Soc. Rev. 2016, 45, 6108–6117. [Google Scholar] [CrossRef]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef]
- Grambow, E.; Mueller-Graf, F.; Delyagina, E.; Frank, M.; Kuhla, A.; Vollmar, B. Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 2014, 25, 166–174. [Google Scholar] [CrossRef]
- Lilyanna, S.; Peh, M.T.; Liew, O.W.; Wang, P.; Moore, P.K.; Richards, A.M.; Martinez, E.C. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. J. Mol. Cell. Cardiol. 2015, 87, 27–37. [Google Scholar] [CrossRef]
- Kuschman, H.P.; Palczewski, M.B.; Thomas, D.D. Nitric oxide and hydrogen sulfide: Sibling rivalry in the family of epigenetic regulators. Free Radic. Biol. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Chang, L.L.; Tran, B.; Dai, T.; Zhong, R.; Mao, Y.C.; Zhu, Y.Z. ZYZ-803, a novel hydrogen sulfide-nitric oxide conjugated donor, promotes angiogenesis via cross- talk between STAT3 and CaMKII. Acta Pharmacol. Sin 2020, 41, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G.; et al. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G., Sr.; Gojon, G., Jr.; Giordano, T.; Krum, H. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef]
- Rushing, A.M.; Donnarumma, E.; Polhemus, D.J.; Au, K.R.; Victoria, S.E.; Schumacher, J.D.; Li, Z.; Jenkins, J.S.; Lefer, D.J.; Goodchild, T.T. Effects of a novel hydrogen sulfide prodrug in a porcine model of acute limb ischemia. J. Vasc. Surg. 2019, 69, 1924–1935. [Google Scholar] [CrossRef]
- Monti, M.; Terzuoli, E.; Ziche, M.; Morbidelli, L. The sulphydryl containing ACE inhibitor Zofenoprilat protects coronary endothelium from Doxorubicin-induced apoptosis. Pharmacol. Res. 2013, 76, 171–181. [Google Scholar] [CrossRef]
- Terzuoli, E.; Monti, M.; Vellecco, V.; Bucci, M.; Cirino, G.; Ziche, M.; Morbidelli, L. Characterization of zofenoprilat as an inducer of functional angiogenesis through increased H2 S availability. Br. J. Pharmacol. 2015, 172, 2961–2973. [Google Scholar] [CrossRef]
- Monti, M.; Terzuoli, E.; Ziche, M.; Morbidelli, L. H2S dependent and independent anti-inflammatory activity of zofenoprilat in cells of the vascular wall. Pharmacol. Res. 2016, 113, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef]
- Zhao, A.S.; Zou, D.; Wang, H.H.; Han, X.; Yang, P.; Huang, N. Hydrogen sulphide-releasing aspirin enhances cell capabilities of anti-oxidative lesions and anti- inflammation. Med. Gas Res. 2019, 9, 145–152. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; OKeefe, J.H.; McCarty, M.F. Boosting endogenous production of vasoprotective hydrogen sulfide via supplementation with taurine and N-acetylcysteine: A novel way to promote cardiovascular health. Open Heart 2017, 4, e000600. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, B.; Li, Y.; Sun, F.; Li, P.; Xia, W.; Zhou, X.; Li, Q.; Wang, X.; Chen, J.; et al. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension 2016, 67, 541–549. [Google Scholar] [CrossRef]
- Gutman, J.B.L.; Kongshavn, P.A.L. Cysteine/cystine-rich undenatured whey protein supplement in patients’ pressure ulcers outcomes: An open label study. J. Wound Care. 2019, 28 (Suppl. S7), S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef]
- Sharma, D.K.; Manral, A.; Saini, V.; Singh, A.; Srinivasan, B.P.; Tiwari, M. Novel diallyldisulfide analogs ameliorate cardiovascular remodeling in rats with L-NAME-induced hypertension. Eur. J. Pharmacol. 2012, 691, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Garikipati, V.N.; Nickoloff, E.; Wang, C.; Polhemus, D.J.; Zhou, J.; Benedict, C.; Khan, M.; Verma, S.K.; Rabinowitz, J.E.; et al. Restoration of hydrogen sulfide production in diabetic mice improves reparative function of bone marrow cells. Circulation 2016, 134, 1467–1483. [Google Scholar] [CrossRef]
- Yang, H.B.; Liu, H.M.; Yan, J.C.; Lu, Z.Y. Effect of diallyl trisulfide on ischemic tissue injury and revascularization in a diabetic mouse model. J. Cardiovasc. Pharmacol. 2018, 71, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Citi, V.; Piragine, E.; Pagnotta, E.; Ugolini, L.; Di Cesare Mannelli, L.; Testai, L.; Ghelardini, C.; Lazzeri, L.; Calderone, V.; Martelli, A. Anticancer properties of erucin, an H2S-releasing isothiocyanate, on human pancreatic adenocarcinoma cells (AsPC-1). Phytother. Res. 2019, 33, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef]
- Miao, X.; Bai, Y.; Sun, W.; Cui, W.; Xin, Y.; Wang, Y.; Tan, Y.; Miao, L.; Fu, Y.; Su, G.; et al. Sulforaphane prevention of diabetes-induced aortic damage was associated with the up-regulation of Nrf2 and its down-stream antioxidants. Nutr. Metab. 2012, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.E.; Terschluesen, A.M.; Rimbach, G. Health promoting effects of brassica-derived phytochemicals: From chemopreventive and anti-inflammatory activities to epigenetic regulation. Oxid. Med. Cell. Longev. 2013, 964539. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Lin, A.H.; Liu, C.T.; Tsai, C.W.; Chang, I.S.; Chen, H.W.; Lii, C.K. Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFκB activation. Mol. Nutr. Food Res. 2013, 57, 1918–1930. [Google Scholar] [CrossRef]
- Martelli, A.; Citi, V.; Testai, L.; Brogi, S.; Calderone, V. Organic Isothiocyanates as Hydrogen Sulfide Donors. Antioxid. Redox Signal. 2020, 32, 110–144. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Piragine, E.; Citi, V.; Testai, L.; Pagnotta, E.; Ugolini, L.; Lazzeri, L.; Di Cesare Mannelli, L.; Manzo, O.L.; Bucci, M.; et al. Erucin exhibits vasorelaxing effects and antihypertensive activity by H2S-releasing properties. Br. J. Pharmacol. 2020, 177, 824–835. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Testai, L.; Marino, A.; Breschi, M.C.; Calderone, V. Hydrogen sulfide releasing capacity of natural isothiocyanates: Is it a reliable explanation for the multiple biological effects of Brassicaceae? Planta Med. 2014, 80, 610–613. [Google Scholar] [CrossRef]
- Sestito, S.; Pruccoli, L.; Runfola, M.; Citi, V.; Martelli, A.; Saccomanni, G.; Calderone, V.; Tarozzi, A.; Rapposelli, S. Design and synthesis of H2S-donor hybrids: A new treatment for Alzheimer’s disease? Eur. J. Med. Chem. 2019, 184, 111745. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, L.; An, T.; Xian, M.; Luckanagul, J.A.; Su, Z.; Lin, Y.; Wang, Q. A hydrogen sulfide-releasing alginate dressing for effective wound healing. Acta Biomater. 2020, 104, 85–94. [Google Scholar] [CrossRef]
- Wu, J.; Chen, A.; Zhou, Y.; Zheng, S.; Yang, Y.; An, Y.; Xu, K.; He, H.; Kang, J.; Luckanagul, J.A.; et al. Novel H2S-Releasing hydrogel for wound repair via in situ polarization of M2 macrophages. Biomaterials 2019, 222, 119398. [Google Scholar] [CrossRef]
- Gambari, L.; Amore, E.; Raggio, R.; Bonani, W.; Barone, M.; Lisignoli, G.; Grigolo, B.; Motta, A.; Grassi, F. Hydrogen sulfide-releasing silk fibroin scaffold for bone tissue engineering. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 102, 471–482. [Google Scholar] [CrossRef]
- Amedei, A.; Morbidelli, L. Circulating Metabolites Originating from Gut Microbiota Control Endothelial Cell Function. Molecules 2019, 24, 3992. [Google Scholar] [CrossRef] [PubMed]
- Morbidelli, L.; Donnini, S.; Ziche, M. Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardio-Oncology 2016, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.P.; Henderson, K.; Spencer, A.; Sligar, A.D.; Baker, A.B. Therapeutic strategies for enhancing angiogenesis in wound healing. Adv. Drug Deliv. Rev. 2019, 146, 97–125. [Google Scholar] [CrossRef]
- Citi, V.; Martelli, A.; Brancaleone, V.; Brogi, S.; Gojon, G.; Montanaro, R.; Morales, G.; Testai, L.; Calderone, V. Anti-inflammatory and antiviral roles of hydrogen sulfide: Rationale for considering H2S donors in COVID-19 therapy. Br. J. Pharmacol. 2020, 177, 4931–4941. [Google Scholar] [CrossRef] [PubMed]



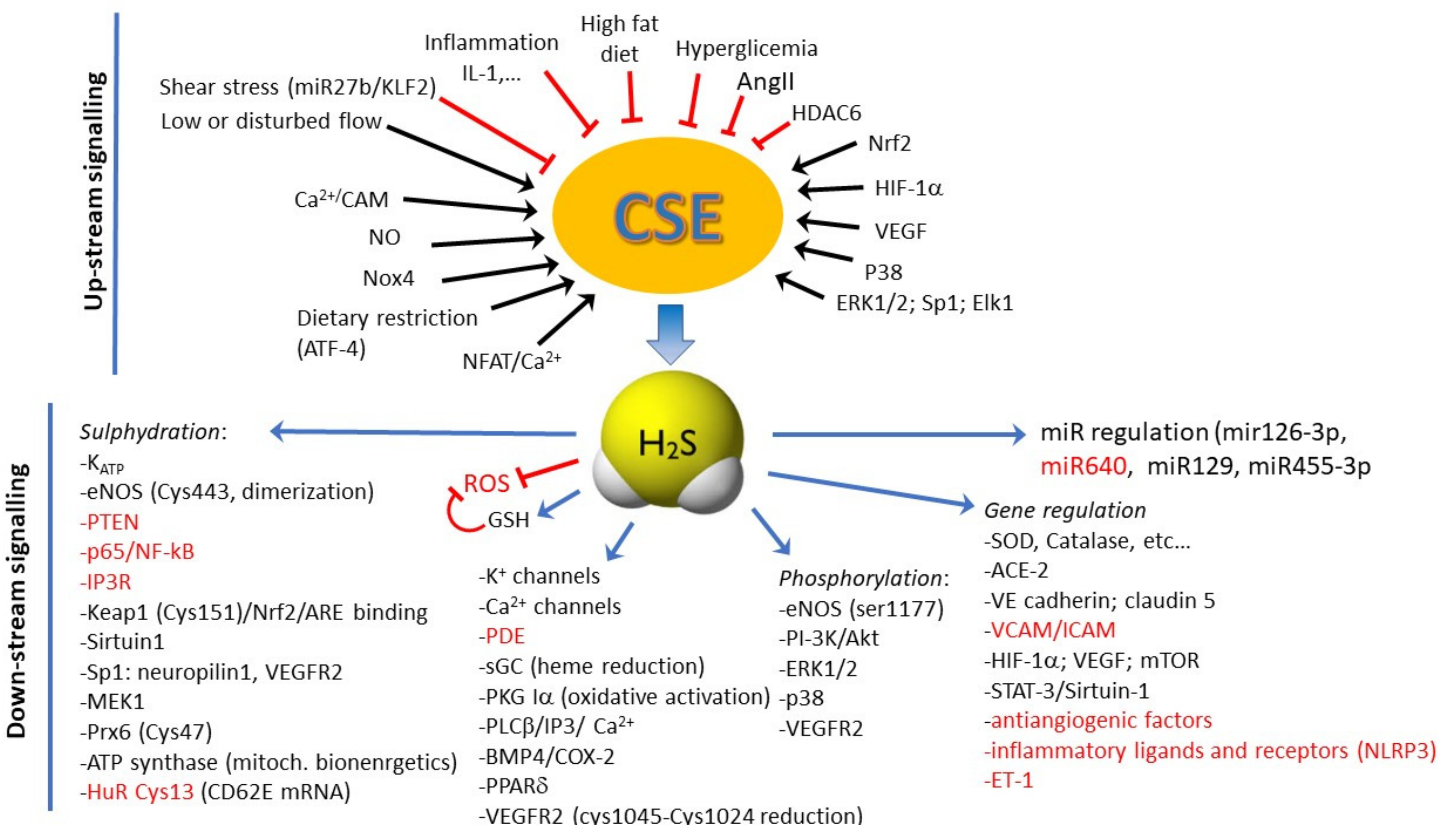{kind=link}
{kind=link}
| Identifier Year (Location) | Condition or Disease | Drug | Phase | Status Results |
|---|---|---|---|---|
| NCT01232257 2011 (The Netharlands) | HYPERTENSION (Chronic Kidney Disease, Chronic Kidney Failure, End Stage Kidney Disease, End Stage Renal Disease) | N-acetylcysteine (NAC) | Phase 3 | Completed No results posted |
| NCT03179163 2020 (USA) | HYPERTENSION | Captopril | Phase 1/2 | Recruiting |
| NCT03410537 2018 (China) | DIABETES TYPE 2 (Lower Extremity Artery Disease) | Taurine vs. Placebo | ND | Recruiting No results posted |
| None | ATHEROSCLEROSIS/ THROMBOSIS | - | - | - |
| None | ANGIOGENESIS/ WOUND HEALING | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccone, V.; Genah, S.; Morbidelli, L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants 2021, 10, 486. https://doi.org/10.3390/antiox10030486
Ciccone V, Genah S, Morbidelli L. Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants. 2021; 10(3):486. https://doi.org/10.3390/antiox10030486
Chicago/Turabian StyleCiccone, Valerio, Shirley Genah, and Lucia Morbidelli. 2021. "Endothelium as a Source and Target of H2S to Improve Its Trophism and Function" Antioxidants 10, no. 3: 486. https://doi.org/10.3390/antiox10030486
APA StyleCiccone, V., Genah, S., & Morbidelli, L. (2021). Endothelium as a Source and Target of H2S to Improve Its Trophism and Function. Antioxidants, 10(3), 486. https://doi.org/10.3390/antiox10030486