
Transcriptional Regulation of ROS Homeostasis by the ERR Subfamily of Nuclear Receptors



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The ERRs
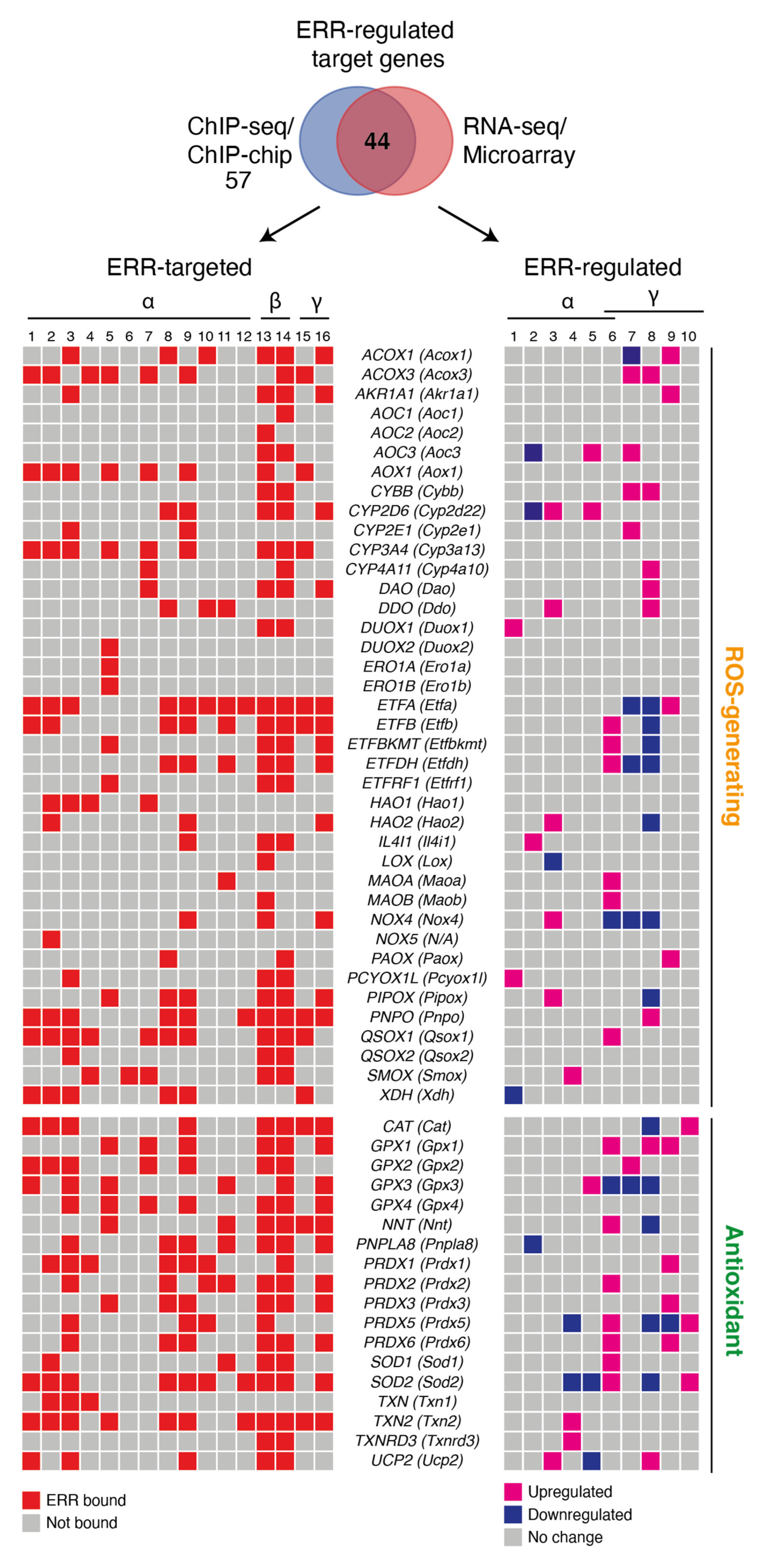
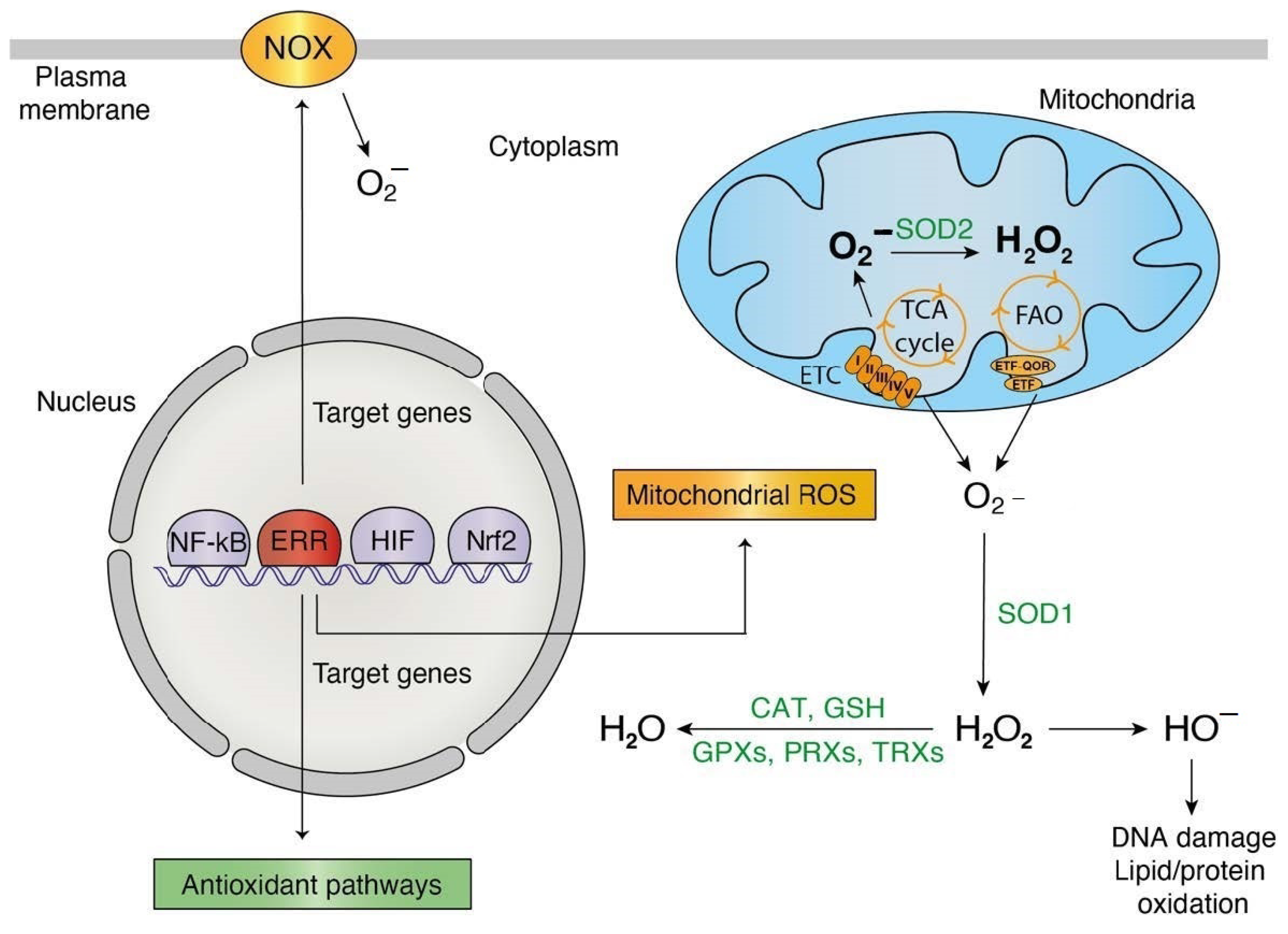
3. Transcriptional Control of ROS Metabolism by the ERRs
ROS-Generating ERR Targets


4. Influence of ROS in ERR Activity
5. ERR Activities and ROS during Oxidative Eustress and Distress
5.1. ERRs and Oxidative Eustress
5.2. ERRs in Oxidative Distress
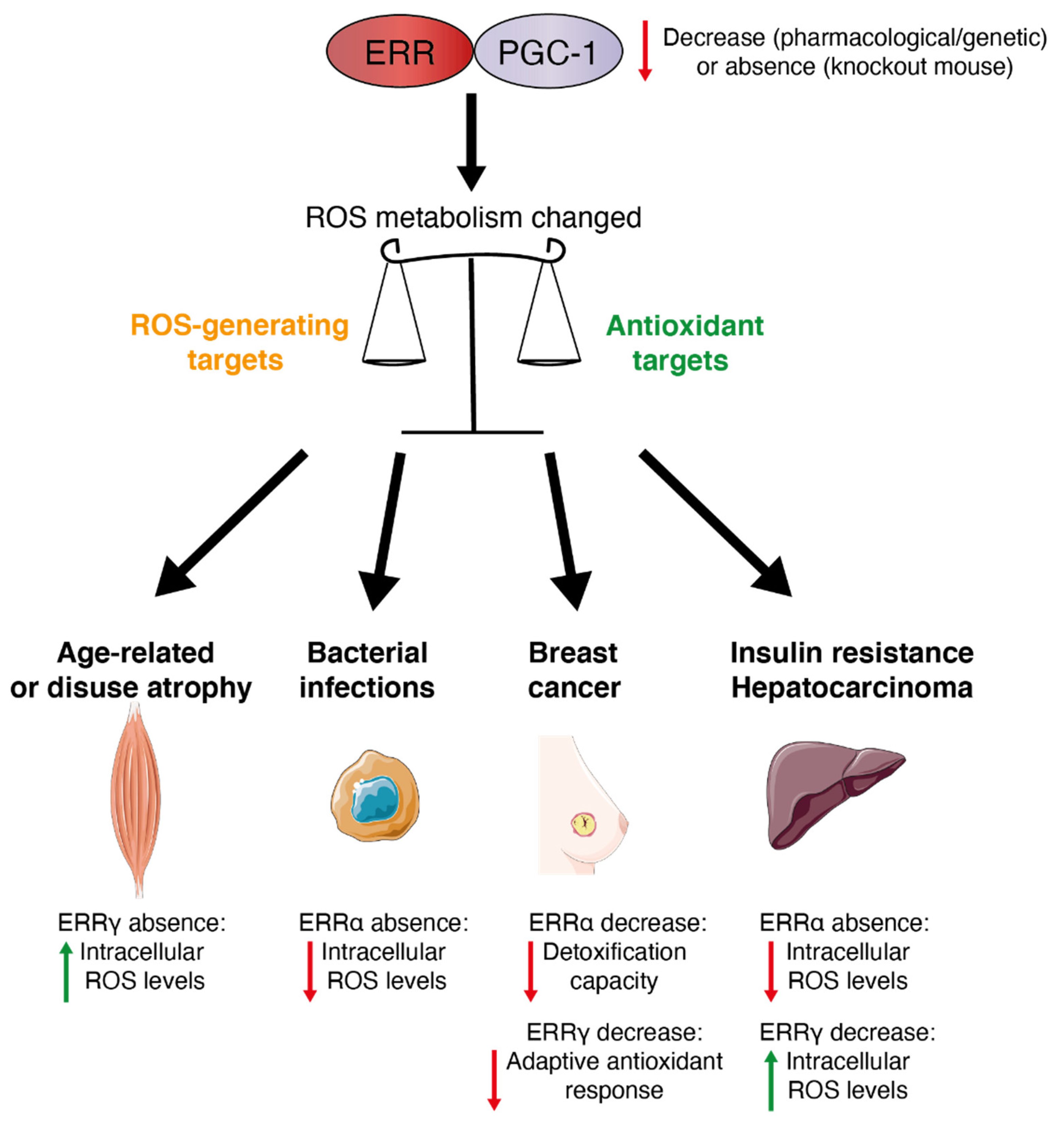
6. ERRs in Oxidative Stress-Related Physiopathology
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the Truth About Antioxidants: Mitohormesis Explains ROS-induced Health Benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Turpaev, K.T. Reactive Oxygen Species and Regulation of Gene Expression. Biochemistry 2002, 67, 281–292. [Google Scholar] [CrossRef]
- Giguère, V. Transcriptional Control of Energy Homeostasis by the Estrogen-related Receptors. Endocr. Rev. 2008, 29, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Giguère, V. Estrogen Related Receptors (ERRs): A New Dawn in Transcriptional Control of Mitochondrial Gene Networks. Mitochondrion 2011, 11, 544–552. [Google Scholar] [CrossRef]
- Deblois, G.; Smith, H.W.; Tam, I.S.; Gravel, S.P.; Caron, M.; Savage, P.; Labbé, D.P.; Bégin, L.R.; Tremblay, M.L.; Park, M.; et al. ERRα Mediates Metabolic Adaptations Driving Lapatinib Resistance in Breast Cancer. Nat. Commun. 2016, 7, 12156. [Google Scholar] [CrossRef] [PubMed]
- Vernier, M.; Dufour, C.R.; McGuirk, S.; Scholtes, C.; Li, X.; Bourmeau, G.; Kuasne, H.; Park, M.; St-Pierre, J.; Audet-Walsh, E.; et al. Estrogen-related Receptors are Targetable ROS sensors. Genes Dev. 2020, 34, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Giguère, V.; Yang, N.; Segui, P.; Evans, R.M. Identification of a New Class of Steroid Hormone Receptors. Nature 1988, 331, 91–94. [Google Scholar] [CrossRef]
- Eudy, J.D.; Yao, S.F.; Weston, M.D.; Ma-Edmonds, M.; Talmadge, C.B.; Cheng, J.J.; Kimberling, W.J.; Sumegi, J. Isolation of a Gene Encoding a Novel Member of the Nuclear Receptor Superfamily from the Critical Region of Usher Syndrome Type IIa at 1q41. Genomics 1998, 50, 382–384. [Google Scholar] [CrossRef]
- Hong, H.; Yang, L.; Stallcup, M.R. Hormone-independent Transcriptional Activation and Coactivator Binding by Novel Orphan Nuclear Receptor ERR3. J. Biol. Chem. 1999, 274, 22618–22626. [Google Scholar] [CrossRef] [PubMed]
- Heard, D.J.; Norby, P.L.; Holloway, J.; Vissing, H. Human ERRgamma, a Third member of the Estrogen Receptor-related Receptor (ERR) Subfamily of Orphan Nuclear Receptors: Tissue-specific Isoforms are Expressed During Development and in the Adult. Mol. Endocrinol. 2000, 14, 382–392. [Google Scholar] [CrossRef][Green Version]
- Tremblay, A.M.; Giguère, V. The NR3B Subgroup: An ovERRview. Nucl. Recept. Signal 2007, 5, e009. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; Hall, J.A.; Perry, M.C.; Laganière, J.; Ghahremani, M.; Park, M.; Hallett, M.; Giguère, V. Genome-wide Identification of Direct Target Genes Implicates Estrogen-related Receptor α as a Determinant of Breast Cancer Heterogeneity. Cancer Res. 2009, 69, 6149–6157. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.; Evans, R.M.; Blanchette, M.; Giguère, V. Genome-wide Orchestration of Cardiac Functions by Orphan Nucler Receptors ERRα and γ. Cell Metab. 2007, 5, 345–356. [Google Scholar] [CrossRef]
- Sladek, R.; Bader, J.-A.; Giguère, V. The Orphan Nuclear Receptor Estrogen-related Receptor α is a Transcriptional Regulator of the Human Medium-chain Acyl Coenzyme A Dehydrogenase Gene. Mol. Cell. Biol. 1997, 17, 5400–5409. [Google Scholar] [CrossRef]
- Kallen, J.; Lattmann, R.; Beerli, R.; Blechschmidt, A.; Blommers, M.J.; Geiser, M.; Ottl, J.; Schlaeppi, J.M.; Strauss, A.; Fournier, B. Crystal Structure of Human Estrogen-related Receptor Alpha in Complex with a Synthetic Inverse Agonist Reveals Its Novel Molecular Mechanism. J. Biol. Chem. 2007, 282, 23231–23239. [Google Scholar] [CrossRef]
- Greschik, H.; Flaig, R.; Renaud, J.P.; Moras, D. Structural Basis for the Deactivation of the Estrogen-related Receptor Gamma by Diethylstilbestrol or 4-hydroxytamoxifen and Determinants of Selectivity. J. Biol. Chem. 2004, 279, 33639–33646. [Google Scholar] [CrossRef]
- Patch, R.J.; Searle, L.L.; Kim, A.J.; De, D.; Zhu, X.; Askari, H.B.; O’Neill, J.C.; Abad, M.C.; Rentzeperis, D.; Liu, J.; et al. Identification of Diaryl Ether-based Ligands for Estrogen-related Receptor α as Potential Antidiabetic Agents. J. Med. Chem. 2011, 54, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, G.B.; Bergeron, D.; Giguère, V. 4-hydroxytamoxifen is an Isoform-specific Inhibitor of Orphan Estrogen-receptor-related (ERR) Nuclear Receptors β and γ. Endocrinology 2001, 142, 4572–4575. [Google Scholar] [CrossRef]
- Kim, J.; Im, C.Y.; Yoo, E.K.; Ma, M.J.; Kim, S.B.; Hong, E.; Chin, J.; Hwang, H.; Lee, S.; Kim, N.D.; et al. Identification of Selective ERRγ Inverse Agonists. Molecules 2016, 21, 80. [Google Scholar] [CrossRef]
- Crevet, L.; Vanacker, J.M. Regulation of the Expression of the Estrogen Related Receptors (ERRs). Cell. Mol. Life Sci. 2020, 77, 4573–4579. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Lou, G.; Zhao, L.; Xu, Z.; Zhang, Y.; He, F. miR-137 Targets Estrogen-related Receptor α and Impairs the Proliferative and Migratory Capacity of Breast Cancer Cells. PLoS ONE 2012, 7, e39102. [Google Scholar] [CrossRef]
- Lu, T.M.; Lu, W.; Zhao, L.J. MicroRNA-137 Affects Proliferation and Migration of Placenta Trophoblast Cells in Preeclampsia by Targeting ERRalpha. Reprod. Sci. 2017, 24, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.L.; Song, C.C.; Li, Y.F.; He, J.J.; Li, Y.L.; Zheng, X.L.; Yang, G.S. miR-125a Inhibits Porcine Preadipocytes Differentiation by Targeting ERRalpha. Mol. Cell. Biochem. 2014, 395, 155–165. [Google Scholar] [CrossRef]
- Lu, M.; Ding, K.; Zhang, G.; Yin, M.; Yao, G.; Tian, H.; Lian, J.; Liu, L.; Liang, M.; Zhu, T.; et al. MicroRNA-320a Sensitizes Tamoxifen-resistant Breast Cancer Cells to Tamoxifen by Targeting ARPP-19 and ERRgamma. Sci. Rep. 2015, 5, 8735. [Google Scholar] [CrossRef]
- Liu, R.H.; Meng, Q.; Shi, Y.P.; Xu, H.S. Regulatory Role of MicroRNA-320a in the Proliferation, Migration, Invasion, and Apoptosis of Trophoblasts and Endothelial Cells by Targeting Estrogen-related Receptor Gamma. J. Cell. Physiol. 2018, 234, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Du, J.; Shen, L.; Tan, Z.; Jiang, D.; Jiang, A.; Li, Q.; Tang, G.; Jiang, Y.; Wang, J.; et al. miR-204-5p Regulates C2C12 Myoblast Differentiation by Targeting MEF2C and ERR. Biomed. Pharmacother. 2018, 101, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Jiang, H.; Ma, D.; Nakaso, K.; Feng, J. Parkin Degrades Estrogen-related Receptors to Limit the Expression of Monoamine Oxidases. Hum. Mol. Genet. 2011, 20, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Chaveroux, C.; Eichner, L.J.; Dufour, C.R.; Shatnawi, A.; Khoutorsky, A.; Bourque, G.; Sonenberg, N.; Giguère, V. Molecular and Genetic Crosstalks Between mTOR and ERRα are Key Determinants of Rapamycin-induced Non-alcoholic Fatty Liver. Cell Metab. 2013, 17, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Li, B.; Li, Y.; Xia, K.; Aman, S.; Yang, Y.; Ahmad, B.; Zhao, B.; Wu, H. FBXL10 Promotes ERRalpha Protein Stability and Proliferation of Breast Cancer Cells by Enhancing the Mono-ubiquitylation of ERRalpha. Cancer Lett. 2021, 502, 108–119. [Google Scholar] [CrossRef]
- Tremblay, A.M.; Wilson, B.J.; Yang, X.J.; Giguère, V. Phosphorylation-dependent Sumoylation Regulates ERRα and γ Transcriptional Activity Through a Synergy Control Motif. Mol. Endocrinol. 2008, 22, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Vu, E.H.; Kraus, R.J.; Mertz, J.E. Phosphorylation-dependent Sumoylation of Estrogen-related Receptor α1. Biochemistry 2007, 46, 9795–9804. [Google Scholar] [CrossRef]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguère, V. An Acetylation Switch Modulates the Transcriptional Activity of Estrogen-related Recetpor α. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef]
- Misra, J.; Kim, D.K.; Jung, Y.S.; Kim, H.B.; Kim, Y.H.; Yoo, E.K.; Kim, B.G.; Kim, S.; Lee, I.K.; Harris, R.A.; et al. O-GlcNAcylation of Orphan Nuclear Receptor Estrogen-Related Receptor gamma Promotes Hepatic Gluconeogenesis. Diabetes 2016, 65, 2835–2848. [Google Scholar] [CrossRef]
- Tribollet, V.; Barenton, B.; Kroiss, A.; Vincent, S.; Zhang, L.; Forcet, C.; Cerutti, C.; Perian, S.; Allioli, N.; Samarut, J.; et al. miR-135a Inhibits the Invasion of Cancer Cells via Suppression of ERRalpha. PLoS ONE 2016, 11, e0156445. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, B.; Jiang, L.; Liu, J.; Han, S. MicroRNA-497 Downregulation Contributes to Cell Proliferation, Migration, and Invasion of Estrogen Receptor Alpha Negative Breast Cancer by Targeting Estrogen-related Receptor Alpha. Tumour Biol. 2016, 37, 13205–13214. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Perry, M.C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378* Mediates Metabolic Shift in Breast Cancer Cells via the PGC-1β/ERRα Transcriptional Pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Kopp, R.P.; Kelly, D.P. Peroxisome Proliferator-activated Receptor Coactivator-1α (PGC-1α) Coactivates the Cardiac-enriched Nuclear Receptors Estrogen-related Receptor-α and -γ. Identification of Novel Leucine-rich Interaction Motif within PGC-1α. J. Biol. Chem. 2002, 277, 40265–40274. [Google Scholar] [CrossRef]
- Kamei, Y.; Ohizumi, H.; Fujitani, Y.; Nemoto, T.; Tanaka, T.; Takahashi, N.; Kawada, T.; Miyoshi, M.; Ezaki, O.; Kakizuka, A. PPARgamma Coactivator 1beta/ERR Ligand 1 is an ERR Protein Ligand, Whose Expression Induces a High-energy Expenditure and Antagonizes Obesity. Proc. Natl. Acad. Sci. USA 2003, 100, 12378–12383. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Knutti, D.; Brogli, K.; Uhlmann, T.; Kralli, A. The Transcriptional Coactivator PGC-1 Regulates the Expression and Activity of the Orphan Nuclear Receptor Estrogen-related Receptor Alpha (ERRalpha). J. Biol. Chem. 2003, 278, 9013–9018. [Google Scholar] [CrossRef] [PubMed]
- Laganiere, J.; Tremblay, G.B.; Dufour, C.R.; Giroux, S.; Rousseau, F.; Giguere, V. A Polymorphic Autoregulatory Hormone Response Element in the Human Estrogen-related Receptor α (ERRα) Promoter Dictates Peroxisome Proliferator-activated Receptor α Coactivator-1α Control of ERRα Expressio. J. Biol. Chem. 2004, 279, 18504–18510. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Laganière, J.; Mehl, I.R.; Barish, G.D.; Chong, L.W.; Li, X.; Scheffler, I.E.; Mock, D.C.; Bataille, A.R.; Robert, F.; et al. Nuclear Receptor ERRα and Coactivator PGC-1β are Effectors of IFN-γ Induced Host Defense. Genes Dev. 2007, 21, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, E.; Papadopoli, D.J.; Gravel, S.P.; Yee, T.; Bridon, G.; Caron, M.; Bourque, G.; Giguère, V.; St-Pierre, J. The PGC-1α/ERRα Axis Represses One-carbon Metabolism and Promotes Sensitivity to Anti-folate Therapy in Breast Cancer. Cell Rep. 2016, 14, 920–931. [Google Scholar] [CrossRef]
- De Vitto, H.; Bode, A.M.; Dong, Z. The PGC-1/ERR Network and Its Role in Precision Oncology. NPJ Precis. Oncol. 2019, 3, 9. [Google Scholar] [CrossRef]
- Deblois, G.; Giguère, V. Oestrogen-related Receptors in Breast Cancer: Control of Cellular Metabolism and Beyond. Nat. Rev. Cancer 2013, 13, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; He, N.; Lin, C.S.; Wei, Z.; Hah, N.; Waizenegger, W.; He, M.X.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. ERRgamma Promotes Angiogenesis, Mitochondrial Biogenesis, and Oxidative Remodeling in PGC1alpha/beta-Deficient Muscle. Cell Rep. 2018, 22, 2521–2529. [Google Scholar] [CrossRef]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Chen, Q.; Gupta, N.; et al. PGC-1β-regulated Mitochondrial Biogenesis and Function in Myotubes is Mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Mehl, I.R.; Chong, L.W.; Nofsinger, R.R.; Evans, R.M. PGC-1β Controls Mitochondrial Metabolism to Modulate Circadian Activity, Adaptive Thermogenesis, and Hepatic Steatosis. Proc. Natl. Acad. Sci. USA 2007, 104, 5223–5228. [Google Scholar] [CrossRef] [PubMed]
- Vernier, M.; Giguère, V. Aging, Senescence and Mitochondria: The PGC-1/ERR Axis. J. Mol. Endocrinol. 2021, 66, R1–R14. [Google Scholar] [CrossRef]
- Deblois, G.; St-Pierre, J.; Giguère, V. The PGC-1/ERR Signaling Axis in Cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical Profiling of Nuclear Receptor Expression Reveals a Hierarchical Transcriptional Network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Huppunen, J.; Aarnisalo, P. Dimerization Modulates the Activity of the Orphan Nuclear Receptor ERRgamma. Biochem. Biophys. Res. Commun. 2004, 314, 964–970. [Google Scholar] [CrossRef]
- Audet-Walsh, E.; Giguère, V. The Multiple Universes of Estrogen-related Receptor α and γ in Metabolic Control and Related Diseases. Acta Pharmacol. Sin. 2015, 36, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Gantner, M.L.; Hazen, B.C.; Eury, E.; Brown, E.L.; Kralli, A. Complementary Roles of Estrogen-Related Receptors in Brown Adipocyte Thermogenic Function. Endocrinology 2016, 157, 4770–4781. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguère, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-Related Receptor α (ERRα) and ERRγ Are Essential Coordinators of Cardiac Metabolism and Function. Mol. Cell. Biol. 2015, 35, 1281–1298. [Google Scholar] [CrossRef]
- Brown, E.L.; Hazen, B.C.; Eury, E.; Wattez, J.S.; Gantner, M.L.; Albert, V.; Chau, S.; Sanchez-Alavez, M.; Conti, B.; Kralli, A. Estrogen-Related Receptors Mediate the Adaptive Response of Brown Adipose Tissue to Adrenergic Stimulation. iScience 2018, 2, 221–237. [Google Scholar] [CrossRef]
- Luo, J.; Sladek, R.; Carrier, J.; Bader, J.-A.; Richard, D.; Giguère, V. Reduced Fat Mass in Mice Lacking Orphan Nuclear Receptor Estrogen-related Receptor α. Mol. Cell. Biol. 2003, 23, 7947–7956. [Google Scholar] [CrossRef]
- Luo, J.; Sladek, R.; Bader, J.-A.; Rossant, J.; Giguère, V. Placental Abnormalities in Mouse Embryos Lacking Orphan Nuclear Receptor ERRβ. Nature 1997, 388, 778–782. [Google Scholar] [CrossRef]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguère, V.; et al. ERRγ Directs and Maintains the Transition to Oxidative Metabolism in the Post-natal Heart. Cell Metab 2007, 6, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Misra, J.; Kim, D.K.; Choi, H.S. ERRγ: A Junior Orphan with a Senior Role in Metabolism. Trends Endocrinol. Metab. 2017, 28, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Alaynick, W.A.; Way, J.M.; Wilson, S.A.; Benson, W.G.; Pei, L.; Downes, M.; Yu, R.; Jonker, J.W.; Holt, J.A.; Rajpal, D.K.; et al. ERRγ Regulates Cardiac, Gastric, and Renal Potassium Homeostasis. Mol. Endocrinol. 2010, 24, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial Complex II and Reactive Oxygen Species in Disease and Therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Charest-Marcotte, A.; Dufour, C.R.; Wilson, B.J.; Tremblay, A.M.; Eichner, L.J.; Arlow, D.H.; Mootha, V.K.; Giguère, V. The Homeobox Protein Prox1 is a Negative Modulator of ERRα/PGC-1α Bioenergetic Functions. Genes Dev. 2010, 24, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguère, V.; Kelly, D.P. Estrogen-related Receptor α Directs Peroxisome Proliferator-activated Receptor α Signaling in the Transcriptional Control of Energy Metabolism in Cardiac and Skeletal Muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; McDonnell, D.P. Molecular Pathways: The Metabolic Regulator Estrogen-related Receptor Alpha as a Therapeutic Target in Cancer. Clin. Cancer Res. 2012, 18, 6089–6095. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The Estrogen-related Receptor Alpha (ERRalpha) Functions in PPARgamma Coactivator 1alpha (PGC-1alpha)-induced Mitochondrial Biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Handschin, C.; Arlow, D.; Xie, X.; St Pierre, J.; Sihag, S.; Yang, W.; Altshuler, D.; Puigserver, P.; Patterson, N.; et al. Erralpha and Gabpa/b Specify PGC-1alpha-dependent Oxidative Phosphorylation Gene Expression That is Altered in Diabetic Muscle. Proc. Natl. Acad. Sci. USA 2004, 101, 6570–6575. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Li, X.; Lindsley, L.; Wang, X.; Shaughnessy, S.; Daniels, T.G.; Szustakowski, J.; Nirmala, N.R.; Wu, Z.; Stevenson, S.C. Estrogen-related Receptor Alpha is Essential for the Expression of Antioxidant Protection Genes and Mitochondrial Function. Biochem. Biophys. Res. Commun. 2007, 357, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Fan, W.; Downes, M.; Yu, R.T.; Jonker, J.W.; Alaynick, W.A.; Banayo, E.; Karunasiri, M.S.; Lorca, S.; Evans, R.M. Exercise and PGC-1α-independent Synchronization of Type I Muscle Metabolism and Vasculature by ERRγ. Cell Metab. 2011, 13, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.-J.; Levasseur, M.-P.; Dufour, C.R.; Perry, M.-C.; Giguère, V. Loss of Estrogen-related Receptor α Promotes Hepatocellular Carcinogenesis Development via Metabolic and Inflammatory Disturbances. Proc. Natl. Acad. Sci. USA 2013, 110, 17975–17980. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Nuclear Receptor Signaling and Cardiac Energetics. Circ. Res. 2004, 95, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid Hormone Receptor and ERRα Coordinately Regulate Mitochondrial Fission, Mitophagy, Biogenesis, and Function. Sci. Signal. 2018, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.M.; Baker, K.D.; Lam, G.; Evans, J.; Thummel, C.S. The Drosophila Estrogen-related Receptor Directs a Metabolic Switch That Supports Developmental Growth. Cell Metab. 2011, 13, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Beebe, K.; Robins, M.M.; Hernandez, E.J.; Lam, G.; Horner, M.A.; Thummel, C.S. Drosophila Estrogen-related Receptor Directs a Transcriptional Switch That Supports Adult Glycolysis and Lipogenesis. Genes Dev. 2020, 34, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH Oxidases: An Overview from Structure to Innate Immunity-associated Pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of Fatty Acids is the Source of Increased Mitochondrial Reactive Oxygen Species Production in Kidney Cortical Tubules in Early Diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Kelly, D.P. A Role for Estrogen-related Receptor Alpha in the Control of Mitochondrial Fatty Acid Beta-oxidation During Brown Adipocyte Differentiation. J. Biol. Chem. 1997, 272, 31693–31699. [Google Scholar] [CrossRef]
- Villena, J.A.; Hock, M.B.; Chang, W.Y.; Barcas, J.E.; Giguère, V.; Kralli, A. Orphan Nuclear Receptor Estrogen-related Receptor Alpha is Essential for Adaptive Thermogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 1418–1423. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically Associating Domains are Stable Units of Replication-timing Regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef]
- Yan, J.; Enge, M.; Whitington, T.; Dave, K.; Liu, J.; Sur, I.; Schmierer, B.; Jolma, A.; Kivioja, T.; Taipale, M.; et al. Transcription Factor Binding in Human Cells Occurs in Dense Clusters Formed Around Cohesin Anchor Sites. Cell 2013, 154, 801–813. [Google Scholar] [CrossRef]
- Consortium, E.P. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Tremblay, A.M.; Dufour, C.R.; Ghahremani, M.; Reudelhuber, T.L.; Giguère, V. Physiological Genomics Identifies Estrogen-related Receptor α as a Regulator of Renal Sodium and Potassium Homeostasis and the Renin-angiotensin Pathway. Mol. Endocrinol. 2010, 24, 22–32. [Google Scholar] [CrossRef]
- Saul, M.C.; Seward, C.H.; Troy, J.M.; Zhang, H.; Sloofman, L.G.; Lu, X.; Weisner, P.A.; Caetano-Anolles, D.; Sun, H.; Zhao, S.D.; et al. Transcriptional Regulatory Dynamics Drive Coordinated Metabolic and Neural Response to Social Challenge in Mice. Genome Res. 2017, 27, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.e420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lupino, K.; Wilkins, B.J.; Qiu, C.; Liu, J.; Omura, Y.; Allred, A.L.; McDonald, C.; Susztak, K.; Barish, G.D.; et al. Genomic Integration of ERRgamma-HNF1beta Regulates Renal Bioenergetics and Prevents Chronic Kidney Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E4910–E4919. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.C.; Dufour, C.R.; Tam, I.S.; B’Chir, W.; Giguère, V. Estrogen-related Receptor-α Coordinates Transcriptional Programs Essential for Exercise Tolerance and Muscle Fitness. Mol. Endocrinol. 2014, 28, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Matsuura, T.R.; Wan, S.; Ryba, D.M.; Kim, J.U.; Won, K.J.; Lai, L.; Petucci, C.; Petrenko, N.; Musunuru, K.; et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res. 2020, 126, 1685–1702. [Google Scholar] [CrossRef]
- Audet-Walsh, E.; Yee, T.; McGuirk, S.; Vernier, M.; Ouellet, C.; St-Pierre, J.; Giguère, V. Androgen-dependent Repression of ERRγ Reprograms Metabolism in Prostate Cancer. Cancer Res. 2017, 77, 378–389. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New Roles in Redox Signaling for an Old Antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A. Thioredoxin and Redox Signaling: Roles of the Thioredoxin System in Control of Cell Fate. Arch. Biochem. Biophys. 2017, 617, 101–105. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant Responses and Cellular Adjustments to Oxidative Stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS Homeostasis and Metabolism: A Dangerous Liason in Cancer Cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Jezek, J.; Jaburek, M.; Zelenka, J.; Jezek, P. Mitochondrial Phospholipase A2 Activated by Reactive oxygen Species in Heart Mitochondria Induces Mild Uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [CrossRef]
- Jaburek, M.; Jezek, J.; Zelenka, J.; Jezek, P. Antioxidant Activity by a Synergy of Redox-sensitive Mitochondrial Phospholipase A2 and Uncoupling Protein-2 in Lung and Spleen. Int. J. Biochem. Cell Biol. 2013, 45, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Dlaskova, A.; Zelenka, J.; Jaburek, M.; Jezek, P. H(2)O(2)-Activated Mitochondrial Phospholipase iPLA(2)gamma Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein-Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreatic Beta-cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual Dependence of Foxo3a and PGC-1alpha in the Induction of Oxidative Stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha Regulates the Mitochondrial Antioxidant Defense System in Vascular Endothelial Cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α Buffers ROS-mediated Removal of Mitochondria During Myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Tedeschi, P.M.; Bertino, J.R. Overexpression of the Mitochondrial Folate and Glycine-serine Pathway: A New Determinant of Methotrexate Selectivity in Tumors. Cancer Res. 2013, 73, 478–482. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Hopkins, B.L.; Neumann, C.A. Redoxins as Gatekeepers of the Transcriptional Oxidative Stress Response. Redox Biol. 2019, 21, 101104. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Carter, E.L.; Ragsdale, S.W. Modulation of Nuclear Receptor Function by Cellular Redox Poise. J. Inorg. Biochem. 2014, 133, 92–103. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, W.; Hekimi, S. A Mitochondrial Superoxide Signal Triggers Increased Longevity in Caenorhabditis Elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786.e5. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Hales, K.G.; Fuller, M.T. Developmentally Regulated Mitochondrial Fusion Mediated by a Conserved, Novel, Predicted GTPase. Cell 1997, 90, 121–129. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-related Protein, is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha Expression are Increased in Human Skeletal Muscle After Physical Exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacin, M.; Zorzano, A. Evidence for a Mitochondrial Regulatory Pathway Defined by Peroxisome Proliferator-activated Receptor-gamma Coactivator-1 Alpha, Estrogen-related Receptor-alpha, and Mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Tsushida, K.; Tanabe, K.; Masuda, K.; Tanimura, S.; Miyake, H.; Arata, Y.; Sugiyama, H.; Wada, J. Estrogen-related Receptor α is Essential for Maintaining Mitochondrial Integrity in Cisplatin-induced Acute Kidney Injury. Biochem. Biophys. Res. Commun. 2018, 498, 918–924. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple Dynamin Family Members Collaborate to Drive Mitochondrial Division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related Protein Drp1 is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Song, J.; Chen, X.; Li, S.; Yu, J.; Xia, W.; Ding, G.; Zhang, Y.; Jia, Z.; Zhang, A.; et al. Estrogen-related Receptor-alpha Mediates Puromycin Aminonucleoside-induced Mesangial Cell Apoptosis and Inflammatory Injury. Am. J. Physiol. Renal. Physiol. 2019, 316, F906–F913. [Google Scholar] [CrossRef]
- Wang, M.; Yang, G.; Jiang, X.; Lu, D.; Mei, H.; Chen, B. Peroxisome Proliferator-Activated Receptor-gamma Coactivator-1alpha (PGC-1alpha) Regulates the Expression of B-Cell Lymphoma/Leukemia-2 (Bcl-2) and Promotes the Survival of Mesenchymal Stem Cells (MSCs) via PGC-1alpha/ERRalpha Interaction in the Absence of Serum, Hypoxia, and High Glucose Conditions. Med. Sci. Monit. 2017, 23, 3451–3460. [Google Scholar] [CrossRef]
- Huang, M.; Chen, L.; Mao, X.; Liu, G.; Gao, Y.; You, X.; Gao, M.; Sehouli, J.; Sun, P. ERRalpha Inhibitor Acts as a Potential Agonist of PPARgamma to Induce Cell Apoptosis and Inhibit Cell Proliferation in Endometrial Cancer. Aging 2020, 12, 23029–23046. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 Transcription Factors--similar but not Identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Ao, A.; Wang, H.; Kamarajugadda, S.; Lu, J. Involvement of Estrogen-related Receptors in Transcriptional Response to Hypoxia and Growth of Solid Tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 7821–7826. [Google Scholar] [CrossRef]
- Zou, C.; Yu, S.; Xu, Z.; Wu, D.; Ng, C.F.; Yao, X.; Yew, D.T.; Vanacker, J.M.; Chan, F.L. ERRalpha Augments HIF-1 Signalling by Directly Interacting with HIF-1alpha in Normoxic and Hypoxic Prostate Cancer Cells. J. Pathol. 2014, 233, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Padmanabha, D.; Gentile, L.B.; Dumur, C.I.; Beckstead, R.B.; Baker, K.D. HIF- and Non-HIF-regulated Hypoxic Responses Require the Estrogen-related Receptor in Drosophila Melanogaster. PLoS Genet. 2013, 9, e1003230. [Google Scholar] [CrossRef]
- Yuk, J.M.; Kim, T.S.; Kim, S.Y.; Lee, H.M.; Han, J.; Dufour, C.R.; Kim, J.K.; Jin, H.S.; Yang, C.S.; Park, K.S.; et al. Orphan Nuclear Receptor ERRα Controls Macrophage Metabolic Signaling and A20 Expression to Negatively Regulate TLR-Induced Inflammation. Immunity 2015, 43, 80–91. [Google Scholar] [CrossRef]
- Hamidian, A.; von Stedingk, K.; Munksgaard Thoren, M.; Mohlin, S.; Pahlman, S. Differential Regulation of HIF-1alpha and HIF-2alpha in Neuroblastoma: Estrogen-related Receptor Alpha (ERRalpha) Regulates HIF2A Transcription and Correlates to Poor Outcome. Biochem. Biophys. Res. Commun. 2015, 461, 560–567. [Google Scholar] [CrossRef]
- Rasbach, K.A.; Gupta, R.K.; Ruas, J.L.; Wu, J.; Naseri, E.; Estall, J.L.; Spiegelman, B.M. PGC-1alpha Regulates a HIF2alpha-dependent Switch in Skeletal Muscle Fiber Types. Proc. Natl. Acad. Sci. USA 2010, 107, 21866–21871. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, Y.K.; Byun, J.K.; Kim, M.K.; Kang, Y.N.; Kim, S.H.; Lee, S.; Jang, B.K.; Park, K.G. Estrogen-related Receptor Gamma is Upregulated in Liver Cancer and Its Inhibition Suppresses Liver Cancer Cell Proliferation via Induction of p21 and p27. Exp. Mol. Med. 2016, 48, e213. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yang, C.S.; Lee, H.M.; Kim, J.K.; Kim, Y.S.; Kim, Y.R.; Kim, J.S.; Kim, T.S.; Yuk, J.M.; Dufour, C.R.; et al. ESRRA (Estrogen-related Receptor α) is a Key Coordinator of Transcriptional and Post-translational Activation of Autophagy to Promote Innate Host Defense. Autophagy 2018, 14, 152–168. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Auwerx, J.; Huss, J.M. Impaired Myogenesis in Estrogen-related Receptor Gamma (ERRgamma)-deficient Skeletal Myocytes Due to Oxidative Stress. FASEB J. 2013, 27, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Marin-Royo, G.; Rodriguez, C.; Le Pape, A.; Jurado-Lopez, R.; Luaces, M.; Antequera, A.; Martinez-Gonzalez, J.; Souza-Neto, F.V.; Nieto, M.L.; Martinez-Martinez, E.; et al. The Role of Mitochondrial Oxidative Stress in the Metabolic Alterations in Diet-induced Obesity in Rats. FASEB J. 2019, 33, 12060–12072. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive Oxygen Species Have a Causal Role in Multiple Forms of Insulin Resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Dufour, C.R.; Levasseur, M.-P.; Pham, N.H.H.; Eichner, L.J.; Wilson, B.J.; Charest-Marcotte, A.; Duguay, D.; Poirier-Héon, J.-F.; Cermakian, N.; Giguère, V. Genomic Convergence Among ERRα, Prox1 and Bmal1 in the Control of Metabolic Clock Outputs. PLoS Genet. 2011, 7, e1002143. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholtes, C.; Giguère, V. Transcriptional Regulation of ROS Homeostasis by the ERR Subfamily of Nuclear Receptors. Antioxidants 2021, 10, 437. https://doi.org/10.3390/antiox10030437
Scholtes C, Giguère V. Transcriptional Regulation of ROS Homeostasis by the ERR Subfamily of Nuclear Receptors. Antioxidants. 2021; 10(3):437. https://doi.org/10.3390/antiox10030437
Chicago/Turabian StyleScholtes, Charlotte, and Vincent Giguère. 2021. "Transcriptional Regulation of ROS Homeostasis by the ERR Subfamily of Nuclear Receptors" Antioxidants 10, no. 3: 437. https://doi.org/10.3390/antiox10030437
APA StyleScholtes, C., & Giguère, V. (2021). Transcriptional Regulation of ROS Homeostasis by the ERR Subfamily of Nuclear Receptors. Antioxidants, 10(3), 437. https://doi.org/10.3390/antiox10030437