
Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
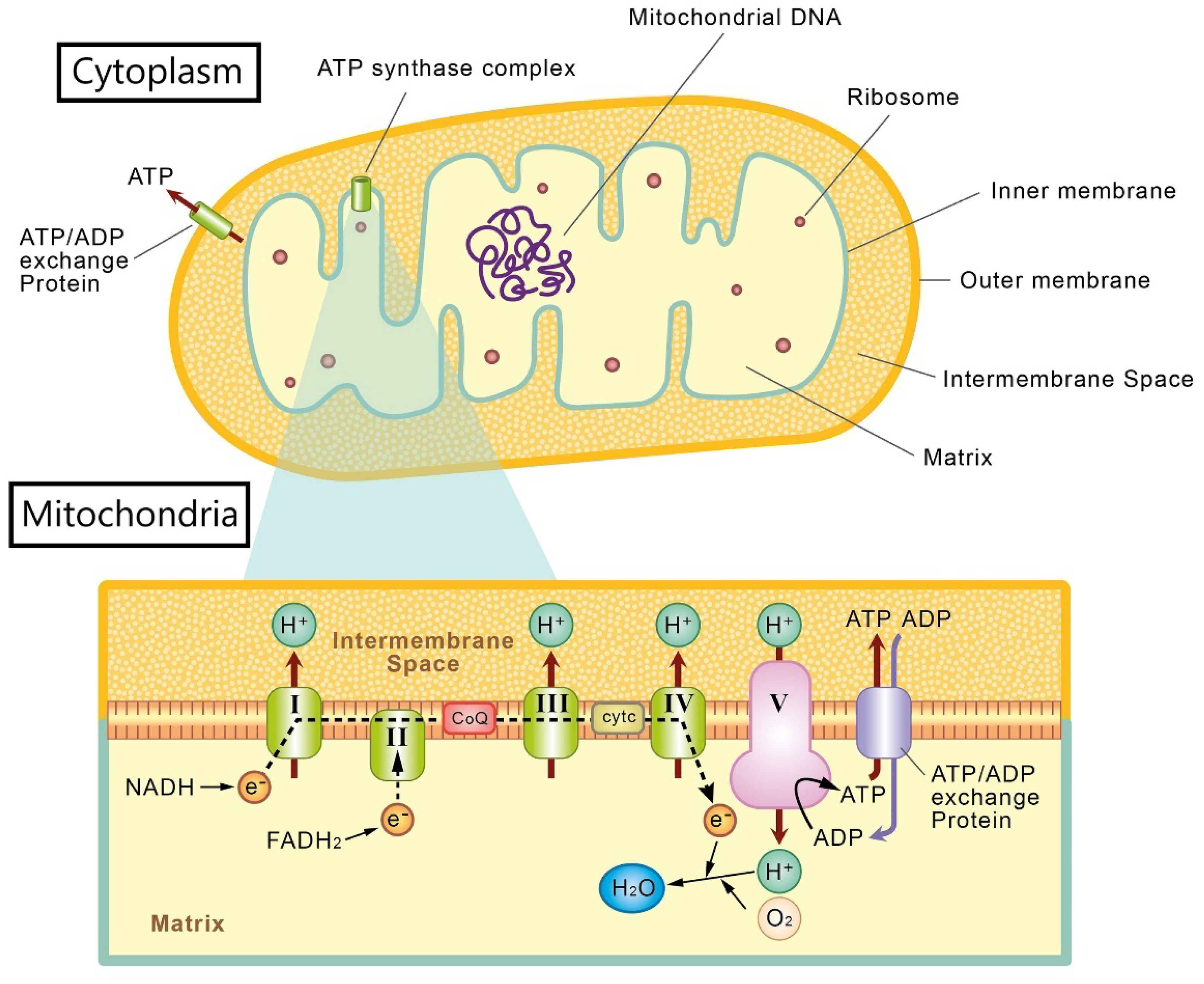
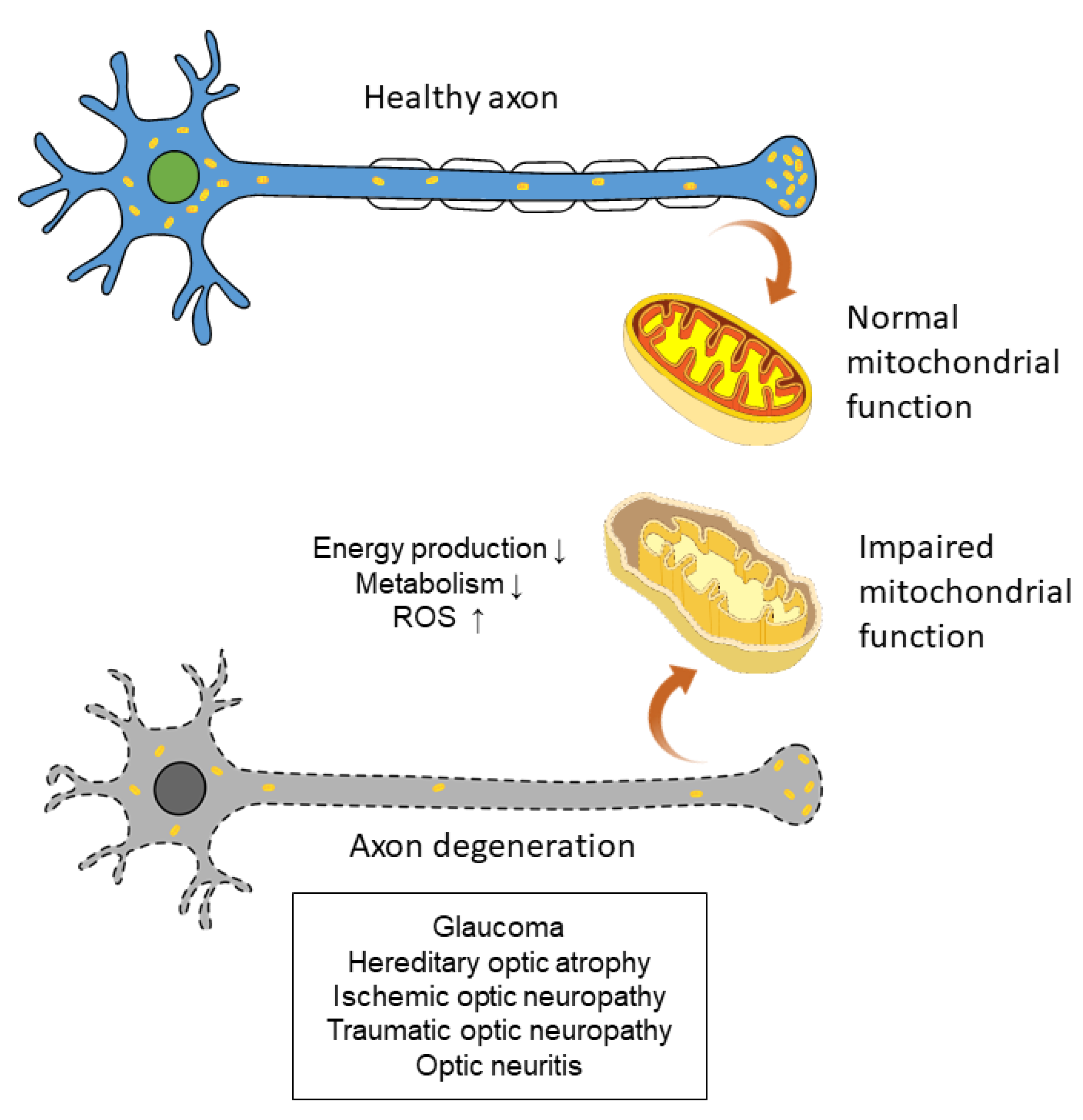
2. Mitochondria and Oxidative Stress in Retinal Ganglion Cells
3. Experimental Investigations of Oxidative Stress in Ocular Diseases with Retinal Ganglion Cell Degeneration
3.1. Glaucoma
3.2. Hereditary Optic Atrophy

{kind=link}
{kind=link}
| Publication | Study Model | Results |
|---|---|---|
| Wong et al., 2002 [100] | cybrid cells | Differentiation of LHON cells to neuronal forms resulted in significant increases in ROS production. |
| Beretta et al., 2004 [101] | cybrid cells | Impaired activity of the EAAT1 glutamate transporter and enhanced ROS production in LHON cells. |
| Danielson et al., 2005 [102] | cybrid cells | Increased levels of sorbitol, which has been linked to oxidative stress, were noted in LHON cells. |
| Floreani et al., 2005 [103] | cybrid cells | Decreased antioxidant defenses and increased oxidative stress in LHON cells. |
| Nguyen et al., 2011 [99] | mice | OPA1 gene mutations decreased antioxidant enzyme gene and protein expression. |
| Lin et al., 2012 [104] | mice | Increased ROS production in both mitochondrial and synaptosome analysis in the LHON mice model. |
3.3. Ischemic Optic Neuropathy
3.4. Traumatic Optic Neuropathy
3.5. Optic Neuritis
4. Experimental Models of Retinal Ganglion Cell Degeneration to Study Oxidative Stress
4.1. Glaucoma Model
4.2. Hereditary Optic Atrophy Model
4.3. Ischemic Optic Neuropathy Model
4.4. Traumatic Optic Neuropathy Model
4.5. Optic Neuritis Model
4.6. Induced Pluripotent Stem Cell-Derived Retinal Ganglion Cells
5. Potential Antioxidant Therapy for Retinal Ganglion Cell Degenerations
5.1. Antioxidants in Glaucoma
5.2. Antioxidants in Hereditary Optic Atrophy
5.3. Antioxidants in Ischemic Optic Neuropathy
5.4. Antioxidants in Traumatic Optic Neuropathy
5.5. Antioxidants in Optic Neuritis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Masland, R.H. The fundamental plan of the retina. Nat. Neurosci. 2001, 4, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Famiglietti, E.V., Jr.; Kolb, H. Structural basis for ON-and OFF-center responses in retinal ganglion cells. Science 1976, 194, 193–195. [Google Scholar] [CrossRef]
- Milosavljevic, N.; Storchi, R.; Eleftheriou, C.G.; Colins, A.; Petersen, R.S.; Lucas, R.J. Photoreceptive retinal ganglion cells control the information rate of the optic nerve. Proc. Natl. Acad. Sci. USA 2018, 115, E11817–E11826. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Lang, R.A. Retinal ganglion cell interactions shape the developing mammalian visual system. Development 2020, 147, dev196535. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef]
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Paniker, N.V.; Srivastava, S.K.; Beutler, E. Glutathione metabolism of the red cells. Effect of glutathione reductase deficiency on the stimulation of hexose monophosphate shunt under oxidative stress. Biochim. Biophys. Acta 1970, 215, 456–460. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET Imaging for Oxidative Stress in Neurodegenerative Disorders Associated with Mitochondrial Dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxidative Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Khatib, T.Z.; Martin, K.R. Protecting retinal ganglion cells. Eye 2017, 31, 218–224. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e415. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003, 10, 870–880. [Google Scholar] [CrossRef]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Barchiesi, A.; Vascotto, C. Transcription, Processing, and Decay of Mitochondrial RNA in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2221. [Google Scholar] [CrossRef]
- Chial, H.; Craig, J. mtDNA and mitochondrial diseases. Nat. Educ. 2008, 1, 217. [Google Scholar]
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916. [Google Scholar] [CrossRef] [PubMed]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. TIG 2018, 34, 682–692. [Google Scholar] [CrossRef]
- Sênos Demarco, R.; Uyemura, B.S.; D’Alterio, C.; Jones, D.L. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol. 2019, 21, 710–720. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Devasagayam, T.P.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804. [Google Scholar]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: Biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal 2010, 12, 1431–1470. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Li, Y.; Andereggen, L.; Yuki, K.; Omura, K.; Yin, Y.; Gilbert, H.Y.; Erdogan, B.; Asdourian, M.S.; Shrock, C.; de Lima, S.; et al. Mobile zinc increases rapidly in the retina after optic nerve injury and regulates ganglion cell survival and optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E209–E218. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Li, G.Y.; Ji, D.; Mortiboys, H.J.; Jackson, S. Light affects mitochondria to cause apoptosis to cultured cells: Possible relevance to ganglion cell death in certain optic neuropathies. J. Neurochem. 2008, 105, 2013–2028. [Google Scholar] [CrossRef]
- Sergeeva, E.G.; Rosenberg, P.A.; Benowitz, L.I. Non-Cell-Autonomous Regulation of Optic Nerve Regeneration by Amacrine Cells. Front Cell NeuroSci. 2021, 15, 666798. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.M.; Crawley, L.; Pahlitzsch, M.; Javaid, F.; Cordeiro, M.F. Glaucoma: The retina and beyond. Acta Neuropathol. 2016, 132, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Levkovitch-Verbin, H. Retinal ganglion cell apoptotic pathway in glaucoma: Initiating and downstream mechanisms. Prog. Brain Res. 2015, 220, 37–57. [Google Scholar] [CrossRef]
- Daniel, S.; Clark, A.F.; McDowell, C.M. Subtype-specific response of retinal ganglion cells to optic nerve crush. Cell Death Discov. 2018, 4, 7. [Google Scholar] [CrossRef]
- You, Y.; Gupta, V.K.; Li, J.C.; Klistorner, A.; Graham, S.L. Optic neuropathies: Characteristic features and mechanisms of retinal ganglion cell loss. Rev. Neurosci. 2013, 24, 301–321. [Google Scholar] [CrossRef]
- Agarwal, R.; Gupta, S.K.; Agarwal, P.; Saxena, R.; Agrawal, S.S. Current concepts in the pathophysiology of glaucoma. Indian J. Ophthalmol. 2009, 57, 257–266. [Google Scholar] [CrossRef]
- Alvarado, J.; Murphy, C.; Polansky, J.; Juster, R. Age-related changes in trabecular meshwork cellularity. Investig. Ophthalmol. Vis. Sci. 1981, 21, 714–727. [Google Scholar]
- Harada, C.; Noro, T.; Kimura, A.; Guo, X.; Namekata, K.; Nakano, T.; Harada, T. Suppression of Oxidative Stress as Potential Therapeutic Approach for Normal Tension Glaucoma. Antioxidants 2020, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Saccà, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef]
- Feilchenfeld, Z.; Yücel, Y.H.; Gupta, N. Oxidative injury to blood vessels and glia of the pre-laminar optic nerve head in human glaucoma. Exp. Eye Res. 2008, 87, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T. Ocular Blood Flow and Influencing Factors for Glaucoma. Asia-Pac. J. Ophthalmol. 2016, 5, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Gherghel, D.; Griffiths, H.R.; Hilton, E.J.; Cunliffe, I.A.; Hosking, S.L. Systemic reduction in glutathione levels occurs in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 877–883. [Google Scholar] [CrossRef]
- Gherghel, D.; Mroczkowska, S.; Qin, L. Reduction in blood glutathione levels occurs similarly in patients with primary-open angle or normal tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3333–3339. [Google Scholar] [CrossRef]
- Yuki, K.; Murat, D.; Kimura, I.; Tsubota, K. Increased serum total antioxidant status and decreased urinary 8-hydroxy-2′-deoxyguanosine levels in patients with normal-tension glaucoma. Acta OphthalMol. 2010, 88, e259–e264. [Google Scholar] [CrossRef]
- Himori, N.; Kunikata, H.; Shiga, Y.; Omodaka, K.; Maruyama, K.; Takahashi, H.; Nakazawa, T. The association between systemic oxidative stress and ocular blood flow in patients with normal-tension glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2016, 254, 333–341. [Google Scholar] [CrossRef]
- Kerrigan, L.A.; Zack, D.J.; Quigley, H.A.; Smith, S.D.; Pease, M.E. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch. Ophthalmol. 1997, 115, 1031–1035. [Google Scholar] [CrossRef]
- Quigley, H.A.; Nickells, R.W.; Kerrigan, L.A.; Pease, M.E.; Thibault, D.J.; Zack, D.J. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Investig. Ophthalmol. Vis. Sci. 1995, 36, 774–786. [Google Scholar]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Osborne, N.N. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp. Eye Res. 2010, 90, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Osborne, N.N. Oxidative-induced apoptosis to an immortalized ganglion cell line is caspase independent but involves the activation of poly(ADP-ribose)polymerase and apoptosis-inducing factor. Brain Res. 2008, 1188, 35–43. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sánez, D.A.; Keller Sarmiento, M.I.; Rosenstein, R.E. Retinal oxidative stress induced by high intraocular pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef]
- Ko, M.L.; Peng, P.H.; Ma, M.C.; Ritch, R.; Chen, C.F. Dynamic changes in reactive oxygen species and antioxidant levels in retinas in experimental glaucoma. Free Radic. Biol. Med. 2005, 39, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Cai, J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Harada, C.; Nakamura, K.; Quah, H.M.; Okumura, A.; Namekata, K.; Saeki, T.; Aihara, M.; Yoshida, H.; Mitani, A.; et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J. Clin. Investig. 2007, 117, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Reides, C.G.; Evelson, P.A.; Llesuy, S.F. Time course changes of oxidative stress markers in a rat experimental glaucoma model. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4635–4640. [Google Scholar] [CrossRef]
- Harada, C.; Namekata, K.; Guo, X.; Yoshida, H.; Mitamura, Y.; Matsumoto, Y.; Tanaka, K.; Ichijo, H.; Harada, T. ASK1 deficiency attenuates neural cell death in GLAST-deficient mice, a model of normal tension glaucoma. Cell Death Differ. 2010, 17, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Kimura, A.; Azuchi, Y.; Hashimoto, N.; Moriya-Ito, K.; Komaki, Y.; Lee, C.Y.; Okahara, N.; Guo, X.; et al. Normal tension glaucoma-like degeneration of the visual system in aged marmosets. Sci. Rep. 2019, 9, 14852. [Google Scholar] [CrossRef]
- Naguib, S.; Backstrom, J.R.; Gil, M.; Calkins, D.J.; Rex, T.S. Retinal oxidative stress activates the NRF2/ARE pathway: An early endogenous protective response to ocular hypertension. Redox Biol. 2021, 42, 101883. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639. [Google Scholar] [CrossRef] [PubMed]
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur. J. Ophthalmol. 2012, 22, 461–465. [Google Scholar] [CrossRef]
- Bi, R.; Logan, I.; Yao, Y.G. Leber Hereditary Optic Neuropathy: A Mitochondrial Disease Unique in Many Ways. Handb. Exp. Pharmacol. 2017, 240, 309–336. [Google Scholar] [CrossRef]
- Huoponen, K.; Vilkki, J.; Aula, P.; Nikoskelainen, E.K.; Savontaus, M.L. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am. J. Hum. Genet. 1991, 48, 1147–1153. [Google Scholar]
- Johns, D.R.; Neufeld, M.J.; Park, R.D. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 1551–1557. [Google Scholar] [CrossRef]
- Wallace, D.C.; Lott, M.T. Leber Hereditary Optic Neuropathy: Exemplar of an mtDNA Disease. Handb. Exp. Pharmacol. 2017, 240, 339–376. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, S.; Robinson, B.H. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H. Human complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim. Biophys. Acta 1998, 1364, 271–286. [Google Scholar] [CrossRef]
- Carelli, V.; Rugolo, M.; Sgarbi, G.; Ghelli, A.; Zanna, C.; Baracca, A.; Lenaz, G.; Napoli, E.; Martinuzzi, A.; Solaini, G. Bioenergetics shapes cellular death pathways in Leber’s hereditary optic neuropathy: A model of mitochondrial neurodegeneration. Biochim. Biophys. Acta 2004, 1658, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Carelli, V.; Martinuzzi, A.; Rugolo, M. Apoptotic cell death of cybrid cells bearing Leber’s hereditary optic neuropathy mutations is caspase independent. Ann. N. Y. Acad. Sci. 2003, 1010, 213–217. [Google Scholar] [CrossRef]
- Yen, M.Y.; Wang, A.G.; Wei, Y.H. Leber’s hereditary optic neuropathy: A multifactorial disease. Prog. Retin. Eye Res. 2006, 25, 381–396. [Google Scholar] [CrossRef]
- Kirches, E. LHON: Mitochondrial Mutations and More. Curr. Genom. 2011, 12, 44–54. [Google Scholar] [CrossRef]
- Rovcanin, B.; Jancic, J.; Pajic, J.; Rovcanin, M.; Samardzic, J.; Djuric, V.; Nikolic, B.; Ivancevic, N.; Novakovic, I.; Kostic, V. Oxidative Stress Profile in Genetically Confirmed Cases of Leber’s Hereditary Optic Neuropathy. J. Mol. Neurosci. MN 2021, 71, 1070–1081. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Van Bergen, N.J.; Chakrabarti, R.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial disorders and the eye. Eye Brain 2011, 3, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1712–1712.e1. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Fuhrmann, N. Dominant optic atrophy, OPA1, and mitochondrial quality control: Understanding mitochondrial network dynamics. Mol. Neurodegener. 2013, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Sun, S.; Erchova, I.; Sengpiel, F.; Votruba, M. Opa1 Deficiency Leads to Diminished Mitochondrial Bioenergetics With Compensatory Increased Mitochondrial Motility. Investig. Ophthalmol. Vis. Sci. 2020, 61, 42. [Google Scholar] [CrossRef]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Wong, A.; Cavelier, L.; Collins-Schramm, H.E.; Seldin, M.F.; McGrogan, M.; Savontaus, M.L.; Cortopassi, G.A. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 2002, 11, 431–438. [Google Scholar] [CrossRef]
- Beretta, S.; Mattavelli, L.; Sala, G.; Tremolizzo, L.; Schapira, A.H.; Martinuzzi, A.; Carelli, V.; Ferrarese, C. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain A J. Neurol. 2004, 127, 2183–2192. [Google Scholar] [CrossRef]
- Danielson, S.R.; Carelli, V.; Tan, G.; Martinuzzi, A.; Schapira, A.H.; Savontaus, M.L.; Cortopassi, G.A. Isolation of transcriptomal changes attributable to LHON mutations and the cybridization process. Brain A J. Neurol. 2005, 128, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Floreani, M.; Napoli, E.; Martinuzzi, A.; Pantano, G.; De Riva, V.; Trevisan, R.; Bisetto, E.; Valente, L.; Carelli, V.; Dabbeni-Sala, F. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber’s hereditary optic neuropathy. FEBS J. 2005, 272, 1124–1135. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef]
- Biousse, V.; Newman, N.J. Ischemic Optic Neuropathies. N. Engl. J. Med. 2015, 372, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.R.; Arnold, A.C. Current concepts in the diagnosis, pathogenesis and management of nonarteritic anterior ischaemic optic neuropathy. Eye 2015, 29, 65–79. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, Y.J.; Kim, D.M. Increased expression of oxyproteins in the optic nerve head of an in vivo model of optic nerve ischemia. BMC Ophthalmol. 2012, 12, 63. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Li, W.; Yang, S. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro, E.S.O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Armstead, W.M.; Mirro, R.; Busija, D.W.; Leffler, C.W. Postischemic generation of superoxide anion by newborn pig brain. Am. J. Physiol. 1988, 255, H401–H403. [Google Scholar] [CrossRef] [PubMed]
- Oka, H.; Kanemitsu, H.; Nihei, H.; Nakayama, H.; Tamura, A.; Sano, K. Change of xanthine dehydrogenase and xanthine oxidase activities in rat brain following complete ischaemia. Neurol. Res. 1992, 14, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Kinuta, Y.; Kimura, M.; Itokawa, Y.; Ishikawa, M.; Kikuchi, H. Changes in xanthine oxidase in ischemic rat brain. J. Neurosurg. 1989, 71, 417–420. [Google Scholar] [CrossRef]
- Ono, T.; Tsuruta, R.; Fujita, M.; Aki, H.S.; Kutsuna, S.; Kawamura, Y.; Wakatsuki, J.; Aoki, T.; Kobayashi, C.; Kasaoka, S.; et al. Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res. 2009, 1305, 158–167. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Davies, K.J.; Delsignore, M.E.; Lin, S.W. Protein damage and degradation by oxygen radicals. II. Modification of amino acids. J. Biol. Chem. 1987, 262, 9902–9907. [Google Scholar] [CrossRef]
- Flammer, J.; Mozaffarieh, M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv. Ophthalmol. 2007, 52 (Suppl. S2), S162–S173. [Google Scholar] [CrossRef]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef]
- Errea, O.; Moreno, B.; Gonzalez-Franquesa, A.; Garcia-Roves, P.M.; Villoslada, P. The disruption of mitochondrial axonal transport is an early event in neuroinflammation. J. Neuroinflamm. 2015, 12, 152. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, X.; Wu, X.; Jiang, L.; Ahsan, A.; Ma, S.; Xiao, Z.; Han, F.; Qin, Z.H.; Hu, W.; et al. Somatic autophagy of axonal mitochondria in ischemic neurons. J. Cell Biol. 2019, 218, 1891–1907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Guo, Y.; Miller, N.R.; Bernstein, S.L. Optic nerve infarction and post-ischemic inflammation in the rodent model of anterior ischemic optic neuropathy (rAION). Brain Res. 2009, 1264, 67–75. [Google Scholar] [CrossRef]
- Bernardo-Colón, A.; Vest, V.; Clark, A.; Cooper, M.L.; Calkins, D.J.; Harrison, F.E.; Rex, T.S. Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Steinsapir, K.D.; Goldberg, R.A. Traumatic optic neuropathy. Surv. Ophthalmol. 1994, 38, 487–518. [Google Scholar] [CrossRef]
- Steinsapir, K.D. Traumatic optic neuropathy. Curr. Opin. Ophthalmol. 1999, 10, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Ruan, Y.W.; Ren, C.R.; Cui, Q.; So, K.F. Mechanisms of secondary degeneration after partial optic nerve transection. Neural Regen. Res. 2014, 9, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Farkas, O.; Povlishock, J.T. Cellular and subcellular change evoked by diffuse traumatic brain injury: A complex web of change extending far beyond focal damage. Prog. Brain Res. 2007, 161, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Bastakis, G.G.; Ktena, N.; Karagogeos, D.; Savvaki, M. Models and treatments for traumatic optic neuropathy and demyelinating optic neuritis. Dev. Neurobiol. 2019, 79, 819–836. [Google Scholar] [CrossRef]
- Cansler, S.M.; Evanson, N.K. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. J. Neurosci. Res. 2020, 98, 571–574. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A. Early events of secondary degeneration after partial optic nerve transection: An immunohistochemical study. J. Neurotrauma 2010, 27, 439–452. [Google Scholar] [CrossRef]
- Wells, J.; Kilburn, M.R.; Shaw, J.A.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Early in vivo changes in calcium ions, oxidative stress markers, and ion channel immunoreactivity following partial injury to the optic nerve. J. Neurosci. Res. 2012, 90, 606–618. [Google Scholar] [CrossRef]
- Ahmad, S.; Fatteh, N.; El-Sherbiny, N.M.; Naime, M.; Ibrahim, A.S.; El-Sherbini, A.M.; El-Shafey, S.A.; Khan, S.; Fulzele, S.; Gonzales, J.; et al. Potential role of A2A adenosine receptor in traumatic optic neuropathy. J. Neuroimmunol 2013, 264, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Payne, S.C.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Myelin sheath decompaction, axon swelling, and functional loss during chronic secondary degeneration in rat optic nerve. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6093–6101. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Velumian, A.A.; Fehlings, M.G. The role of excitotoxicity in secondary mechanisms of spinal cord injury: A review with an emphasis on the implications for white matter degeneration. J. Neurotrauma 2004, 21, 754–774. [Google Scholar] [CrossRef]
- Szymanski, C.R.; Chiha, W.; Morellini, N.; Cummins, N.; Bartlett, C.A.; O’Hare Doig, R.L.; Savigni, D.L.; Payne, S.C.; Harvey, A.R.; Dunlop, S.A.; et al. Paranode Abnormalities and Oxidative Stress in Optic Nerve Vulnerable to Secondary Degeneration: Modulation by 670 nm Light Treatment. PLoS ONE 2013, 8, e66448. [Google Scholar] [CrossRef]
- O’Hare Doig, R.L.; Bartlett, C.A.; Maghzal, G.J.; Lam, M.; Archer, M.; Stocker, R.; Fitzgerald, M. Reactive species and oxidative stress in optic nerve vulnerable to secondary degeneration. Exp. Neurol. 2014, 261, 136–146. [Google Scholar] [CrossRef]
- Cummins, N.; Bartlett, C.A.; Archer, M.; Bartlett, E.; Hemmi, J.M.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Changes to mitochondrial ultrastructure in optic nerve vulnerable to secondary degeneration in vivo are limited by irradiation at 670 nm. BMC NeuroSci. 2013, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Levkovitch-Verbin, H.; Harris-Cerruti, C.; Groner, Y.; Wheeler, L.A.; Schwartz, M.; Yoles, E. RGC death in mice after optic nerve crush injury: Oxidative stress and neuroprotection. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4169–4174. [Google Scholar]
- Lieven, C.J.; Hoegger, M.J.; Schlieve, C.R.; Levin, L.A. Retinal ganglion cell axotomy induces an increase in intracellular superoxide anion. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Hoorbakht, H.; Bagherkashi, F. Optic neuritis, its differential diagnosis and management. Open OphthalMol. J. 2012, 6, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol. 2014, 13, 83–99. [Google Scholar] [CrossRef]
- Youl, B.D.; Turano, G.; Miller, D.H.; Towell, A.D.; MacManus, D.G.; Moore, S.G.; Jones, S.J.; Barrett, G.; Kendall, B.E.; Moseley, I.F.; et al. The pathophysiology of acute optic neuritis. An association of gadolinium leakage with clinical and electrophysiological deficits. Brain A J. Neurol. 1991, 114 Pt 6, 2437–2450. [Google Scholar] [CrossRef]
- Pau, D.; Al Zubidi, N.; Yalamanchili, S.; Plant, G.T.; Lee, A.G. Optic neuritis. Eye 2011, 25, 833–842. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Li, R.; Qiu, W.; Chang, Y.; Sun, X.; Fang, L.; Chen, C.; Yang, Y.; Lu, Z.; Hu, X.; et al. Association of serum gamma-glutamyltransferase and C-reactive proteins with neuromyelitis optica and multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 65–70. [Google Scholar] [CrossRef]
- Falardeau, J.; Fryman, A.; Wanchu, R.; Marracci, G.H.; Mass, M.; Wooliscroft, L.; Bourdette, D.N.; Murchison, C.F.; Hills, W.L.; Yadav, V. Oral lipoic acid as a treatment for acute optic neuritis: A blinded, placebo controlled randomized trial. Mult. Scler. J. Exp. Transl. Clin. 2019, 5, 2055217319850193. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A.; Hope, G.M.; Rao, N.A. Influence of antioxidant enzymes in reduction of optic disc edema in experimental optic neuritis. J. Free Radic. Biol. Med. 1986, 2, 349–357. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxidative Med. Cell. Longev. 2017, 2017, 2817252. [Google Scholar] [CrossRef]
- Khan, R.S.; Fonseca-Kelly, Z.; Callinan, C.; Zuo, L.; Sachdeva, M.M.; Shindler, K.S. SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front. Cell NeuroSci. 2012, 6, 63. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A.; Hope, G.M.; Rao, N.A. Antioxidant enzyme suppression of demyelination in experimental optic neuritis. Curr. Eye Res. 1989, 8, 467–477. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A.; Rao, N.A. Hydrogen peroxide localization in experimental optic neuritis. Arch. Ophthalmol. 1990, 108, 1614–1621. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Suppression of mitochondrial oxidative stress provides long-term neuroprotection in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 681–691. [Google Scholar] [CrossRef]
- Qi, X.; Sun, L.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. Long-term suppression of neurodegeneration in chronic experimental optic neuritis: Antioxidant gene therapy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513. [Google Scholar]
- Zhang, J.; Fang, F.; Li, L.; Huang, H.; Webber, H.C.; Sun, Y.; Mahajan, V.B.; Hu, Y. A Reversible Silicon Oil-Induced Ocular Hypertension Model in Mice. J. Vis. Exp. 2019, 153. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Romano, G.L.; Eandi, C.M.; Toro, M.D.; Rejdak, R.; Di Benedetto, G.; Lazzara, F.; Bernardini, R.; Drago, F.; Cantarella, G.; et al. Brimonidine is Neuroprotective in Animal Paradigm of Retinal Ganglion Cell Damage. Front. Pharmacol. 2021, 12, 705405. [Google Scholar] [CrossRef]
- Benozzi, J.; Nahum, L.P.; Campanelli, J.L.; Rosenstein, R.E. Effect of hyaluronic acid on intraocular pressure in rats. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2196–2200. [Google Scholar]
- Ueda, J.; Sawaguchi, S.; Hanyu, T.; Yaoeda, K.; Fukuchi, T.; Abe, H.; Ozawa, H. Experimental glaucoma model in the rat induced by laser trabecular photocoagulation after an intracameral injection of India ink. Jpn. J. Ophthalmol. 1998, 42, 337–344. [Google Scholar] [CrossRef]
- Grozdanic, S.D.; Betts, D.M.; Sakaguchi, D.S.; Allbaugh, R.A.; Kwon, Y.H.; Kardon, R.H. Laser-induced mouse model of chronic ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Chang, P.; Pennesi, M.E.; Yang, Z.; Zhang, J.; Li, D.; Wu, S.M.; Gross, R.L. Effects of elevated intraocular pressure on mouse retinal ganglion cells. Vis. Res. 2005, 45, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ederra, J.; Verkman, A.S. Mouse model of sustained elevation in intraocular pressure produced by episcleral vein occlusion. Exp. Eye Res. 2006, 82, 879–884. [Google Scholar] [CrossRef]
- Sheldon, W.G.; Warbritton, A.R.; Bucci, T.J.; Turturro, A. Glaucoma in food-restricted and ad libitum-fed DBA/2NNia mice. Lab. Anim. Sci. 1995, 45, 508–518. [Google Scholar]
- Libby, R.T.; Anderson, M.G.; Pang, I.H.; Robinson, Z.H.; Savinova, O.V.; Cosma, I.M.; Snow, A.; Wilson, L.A.; Smith, R.S.; Clark, A.F.; et al. Inherited glaucoma in DBA/2J mice: Pertinent disease features for studying the neurodegeneration. Vis. NeuroSci. 2005, 22, 637–648. [Google Scholar] [CrossRef]
- Chang, B.; Smith, R.S.; Hawes, N.L.; Anderson, M.G.; Zabaleta, A.; Savinova, O.; Roderick, T.H.; Heckenlively, J.R.; Davisson, M.T.; John, S.W. Interacting loci cause severe iris atrophy and glaucoma in DBA/2J mice. Nat. Genet. 1999, 21, 405–409. [Google Scholar] [CrossRef]
- John, S.W. Mechanistic insights into glaucoma provided by experimental genetics the cogan lecture. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Jun, A.S.; Brown, M.D.; Wallace, D.C. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc. Natl. Acad. Sci. USA 1994, 91, 6206–6210. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, E.; Bugiani, M.; Invernizzi, F.; de Souza, C.F.; Farina, L.; Carrara, F.; Lamantea, E.; Antozzi, C.; Confalonieri, P.; Sanseverino, M.T.; et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain A J. Neurol. 2007, 130, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain A J. Neurol. 2007, 130, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Chang, C.H.; Huang, T.L.; Huang, S.P.; Tsai, R.K. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in a rat model of anterior ischemic optic neuropathy (rAION). Exp. Eye Res. 2014, 118, 109–116. [Google Scholar] [CrossRef]
- Wen, Y.T.; Huang, T.L.; Huang, S.P.; Chang, C.H.; Tsai, R.K. Early applications of granulocyte colony-stimulating factor (G-CSF) can stabilize the blood-optic-nerve barrier and ameliorate inflammation in a rat model of anterior ischemic optic neuropathy (rAION). Dis. Model. Mech. 2016, 9, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.N.; Kapupara, K.; Wen, Y.T.; Chen, Y.H.; Pan, I.H.; Tsai, R.K. Haematococcus pluvialis-Derived Astaxanthin Is a Potential Neuroprotective Agent against Optic Nerve Ischemia. Mar. Drugs 2020, 18, 85. [Google Scholar] [CrossRef]
- Liu, P.K.; Wen, Y.T.; Lin, W.; Kapupara, K.; Tai, M.; Tsai, R.K. Neuroprotective effects of low-dose G-CSF plus meloxicam in a rat model of anterior ischemic optic neuropathy. Sci. Rep. 2020, 10, 10351. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Guo, Y.; Kelman, S.E.; Flower, R.W.; Johnson, M.A. Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4153–4162. [Google Scholar] [CrossRef]
- Slater, B.J.; Mehrabian, Z.; Guo, Y.; Hunter, A.; Bernstein, S.L. Rodent anterior ischemic optic neuropathy (rAION) induces regional retinal ganglion cell apoptosis with a unique temporal pattern. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3671–3676. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Colón, A.; Vest, V.; Cooper, M.L.; Naguib, S.A.; Calkins, D.J.; Rex, T.S. Progression and Pathology of Traumatic Optic Neuropathy From Repeated Primary Blast Exposure. Front. NeuroSci. 2019, 13, 719. [Google Scholar] [CrossRef]
- DeJulius, C.R.; Bernardo-Colón, A.; Naguib, S.; Backstrom, J.R.; Kavanaugh, T.; Gupta, M.K.; Duvall, C.L.; Rex, T.S. Microsphere antioxidant and sustained erythropoietin-R76E release functions cooperate to reduce traumatic optic neuropathy. J. Control Release 2021, 329, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Hines-Beard, J.; Marchetta, J.; Gordon, S.; Chaum, E.; Geisert, E.E.; Rex, T.S. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp. Eye Res. 2012, 99, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, S.; Lee, C.; Kumar, A.; Arjunan, P.; Li, Y.; Zhang, F.; Li, X. An optic nerve crush injury murine model to study retinal ganglion cell survival. J. Vis. Exp. 2011, 50, 2685. [Google Scholar] [CrossRef]
- Huang, S.P.; Fang, K.T.; Chang, C.H.; Huang, T.L.; Wen, Y.T.; Tsai, R.K. Autocrine protective mechanisms of human granulocyte colony-stimulating factor (G-CSF) on retinal ganglion cells after optic nerve crush. Exp. Eye Res. 2016, 143, 132–140. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, Y.; Cheng, Q.; Yu, S.; Gao, Y.; Shu, Q.; Yang, C.; Sun, Y.; Wang, J.; Xu, F.; et al. The expression changes of myelin and lymphocyte protein (MAL) following optic nerve crush in adult rats retinal ganglion cells. J. Mol. Neurosci. MN 2014, 54, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ye, D.; Huang, R.; Xu, Y.; Lu, P.; Chen, H.; Huang, J. Down Syndrome Critical Region 1 Reduces Oxidative Stress-Induced Retinal Ganglion Cells Apoptosis via CREB-Bcl-2 Pathway. Investig. Ophthalmol. Vis. Sci. 2020, 61, 23. [Google Scholar] [CrossRef]
- Sarikcioglu, L.; Demir, N.; Demirtop, A. A standardized method to create optic nerve crush: Yasargil aneurysm clip. Exp. Eye Res. 2007, 84, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Chang, C.H.; Wang, H.Z. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp. Eye Res. 2008, 87, 242–250. [Google Scholar] [CrossRef]
- Ye, D.; Shi, Y.; Xu, Y.; Huang, J. PACAP Attenuates Optic Nerve Crush-Induced Retinal Ganglion Cell Apoptosis Via Activation of the CREB-Bcl-2 Pathway. J. Mol. Neurosci. MN 2019, 68, 475–484. [Google Scholar] [CrossRef]
- Vest, V.; Bernardo-Colón, A.; Watkins, D.; Kim, B.; Rex, T.S. Rapid Repeat Exposure to Subthreshold Trauma Causes Synergistic Axonal Damage and Functional Deficits in the Visual Pathway in a Mouse Model. J. Neurotrauma 2019, 36, 1646–1654. [Google Scholar] [CrossRef]
- Bricker-Anthony, C.; Hines-Beard, J.; Rex, T.S. Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4853–4862. [Google Scholar] [CrossRef]
- Cockerham, G.C.; Rice, T.A.; Hewes, E.H.; Cockerham, K.P.; Lemke, S.; Wang, G.; Lin, R.C.; Glynn-Milley, C.; Zumhagen, L. Closed-eye ocular injuries in the Iraq and Afghanistan wars. N. Engl. J. Med. 2011, 364, 2172–2173. [Google Scholar] [CrossRef]
- Chaudhary, P.; Marracci, G.; Yu, X.; Galipeau, D.; Morris, B.; Bourdette, D. Lipoic acid decreases inflammation and confers neuroprotection in experimental autoimmune optic neuritis. J. Neuroimmunol 2011, 233, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.; Helling, N.; Hilla, A.; Heskamp, A.; Issberner, A.; Hildebrandt, T.; Kohne, Z.; Küry, P.; Berndt, C.; Aktas, O.; et al. Early alpha-lipoic acid therapy protects from degeneration of the inner retinal layers and vision loss in an experimental autoimmune encephalomyelitis-optic neuritis model. J. Neuroinflamm. 2018, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharm. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Madrid, R.E. Chronic progressive experimental allergic encephalomyelitis (EAE) in adult guinea pigs. J. Neuropathol Exp. Neurol. 1983, 42, 243–255. [Google Scholar] [CrossRef]
- Aranda, M.L.; Dorfman, D.; Sande, P.H.; Rosenstein, R.E. Experimental optic neuritis induced by the microinjection of lipopolysaccharide into the optic nerve. Exp. Neurol. 2015, 266, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.L.; González Fleitas, M.F.; De Laurentiis, A.; Keller Sarmiento, M.I.; Chianelli, M.; Sande, P.H.; Dorfman, D.; Rosenstein, R.E. Neuroprotective effect of melatonin in experimental optic neuritis in rats. J. Pineal Res. 2016, 60, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yokoi, T.; Tamalu, F.; Watanabe, S.; Nishina, S.; Azuma, N. Generation of retinal ganglion cells with functional axons from human induced pluripotent stem cells. Sci. Rep. 2015, 5, 8344. [Google Scholar] [CrossRef]
- Ohlemacher, S.K.; Sridhar, A.; Xiao, Y.; Hochstetler, A.E.; Sarfarazi, M.; Cummins, T.R.; Meyer, J.S. Stepwise Differentiation of Retinal Ganglion Cells from Human Pluripotent Stem Cells Enables Analysis of Glaucomatous Neurodegeneration. Stem Cells 2016, 34, 1553–1562. [Google Scholar] [CrossRef]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 14520. [Google Scholar] [CrossRef]
- Teotia, P.; Chopra, D.A.; Dravid, S.M.; Van Hook, M.J.; Qiu, F.; Morrison, J.; Rizzino, A.; Ahmad, I. Generation of Functional Human Retinal Ganglion Cells with Target Specificity from Pluripotent Stem Cells by Chemically Defined Recapitulation of Developmental Mechanism. Stem Cells 2017, 35, 572–585. [Google Scholar] [CrossRef]
- Chavali, V.R.M.; Haider, N.; Rathi, S.; Vrathasha, V.; Alapati, T.; He, J.; Gill, K.; Nikonov, R.; Duong, T.T.; McDougald, D.S.; et al. Dual SMAD inhibition and Wnt inhibition enable efficient and reproducible differentiations of induced pluripotent stem cells into retinal ganglion cells. Sci. Rep. 2020, 10, 11828. [Google Scholar] [CrossRef]
- Langer, K.B.; Ohlemacher, S.K.; Phillips, M.J.; Fligor, C.M.; Jiang, P.; Gamm, D.M.; Meyer, J.S. Retinal Ganglion Cell Diversity and Subtype Specification from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1282–1293. [Google Scholar] [CrossRef]
- VanderWall, K.B.; Vij, R.; Ohlemacher, S.K.; Sridhar, A.; Fligor, C.M.; Feder, E.M.; Edler, M.C.; Baucum, A.J., 2nd; Cummins, T.R.; Meyer, J.S. Astrocytes Regulate the Development and Maturation of Retinal Ganglion Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 12, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Manafi, N.; Shokri, F.; Achberger, K.; Hirayama, M.; Mohammadi, M.H.; Noorizadeh, F.; Hong, J.; Liebau, S.; Tsuji, T.; Quinn, P.M.J.; et al. Organoids and organ chips in ophthalmology. Ocul. Surf. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Fligor, C.M.; Lavekar, S.S.; Harkin, J.; Shields, P.K.; VanderWall, K.B.; Huang, K.C.; Gomes, C.; Meyer, J.S. Extension of retinofugal projections in an assembled model of human pluripotent stem cell-derived organoids. Stem Cell Rep. 2021, 16, 2228–2241. [Google Scholar] [CrossRef]
- Wagstaff, P.E.; Ten Asbroek, A.; Ten Brink, J.B.; Jansonius, N.M.; Bergen, A.A.B. An alternative approach to produce versatile retinal organoids with accelerated ganglion cell development. Sci. Rep. 2021, 11, 1101. [Google Scholar] [CrossRef]
- Tucker, B.A.; Solivan-Timpe, F.; Roos, B.R.; Anfinson, K.R.; Robin, A.L.; Wiley, L.A.; Mullins, R.F.; Fingert, J.H. Duplication of TBK1 Stimulates Autophagy in iPSC-derived Retinal Cells from a Patient with Normal Tension Glaucoma. J. Stem Cell Res. 2014, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Teotia, P.; Van Hook, M.J.; Wichman, C.S.; Allingham, R.R.; Hauser, M.A.; Ahmad, I. Modeling Glaucoma: Retinal Ganglion Cells Generated from Induced Pluripotent Stem Cells of Patients with SIX6 Risk Allele Show Developmental Abnormalities. Stem Cells 2017, 35, 2239–2252. [Google Scholar] [CrossRef]
- Wong, R.C.B.; Lim, S.Y.; Hung, S.S.C.; Jackson, S.; Khan, S.; Van Bergen, N.J.; De Smit, E.; Liang, H.H.; Kearns, L.S.; Clarke, L.; et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 2017, 9, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Wang, A.G.; Chen, Y.T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.C.; Hwang, D.K.; Yang, Y.P.; Shen, C.N.; Lee, H.C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Yarmishyn, A.A.; Yang, Y.P.; Lu, P.C.; Chou, S.J.; Wang, M.L.; Lin, T.C.; Hwang, D.K.; Chou, Y.B.; Chen, S.J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Nguyen, P.N.N.; Lin, T.C.; Yarmishyn, A.A.; Chen, W.S.; Hwang, D.K.; Chiou, G.Y.; Lin, T.W.; Chien, C.S.; Tsai, C.Y.; et al. Glutamate Stimulation Dysregulates AMPA Receptors-Induced Signal Transduction Pathway in Leber’s Inherited Optic Neuropathy Patient-Specific hiPSC-Derived Retinal Ganglion Cells. Cells 2019, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Oh-ishi, S.; Hayashi, I.; Hayashi, M.; Yamaki, K.; Utsunomiya, I. Pharmacological demonstration of inflammatory mediators using experimental inflammatory models: Rat pleurisy induced by carrageenin and phorbol myristate acetate. Dermatologica 1989, 179 (Suppl. S1), 68–71. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Diaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernandez, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with an optic atrophy ‘plus’ phenotype due to a mutation in the OPA1 gene. Stem Cell Res. 2016, 16, 673–676. [Google Scholar] [CrossRef][Green Version]
- Zurita-Diaz, F.; Galera-Monge, T.; Moreno-Izquierdo, A.; Corton, M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human DOA ‘plus’ iPSC line, IISHDOi003-A, with the mutation in the OPA1 gene: C.1635C>A.; p.Ser545Arg. Stem Cell Res. 2017, 24, 81–84. [Google Scholar] [CrossRef]
- Sladen, P.E.; Perdigao, P.R.L.; Salsbury, G.; Novoselova, T.; van der Spuy, J.; Chapple, J.P.; Yu-Wai-Man, P.; Cheetham, M.E. CRISPR-Cas9 correction of OPA1 c.1334G>A: P.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Mol. Nucleic Acids 2021, 26, 432–443. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrosio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Kawase, K.; Funato, M.; Seki, J.; Kawase, C.; Ohuchi, K.; Kameyama, T.; Ando, S.; Sato, A.; Morozumi, W.; et al. Effect of Timolol on Optineurin Aggregation in Transformed Induced Pluripotent Stem Cells Derived From Patient with Familial Glaucoma. Investig. OphthalMol. Vis. Sci. 2018, 59, 2293–2304. [Google Scholar] [CrossRef]
- VanderWall, K.B.; Huang, K.C.; Pan, Y.; Lavekar, S.S.; Fligor, C.M.; Allsop, A.R.; Lentsch, K.A.; Dang, P.; Zhang, C.; Tseng, H.C.; et al. Retinal Ganglion Cells With a Glaucoma OPTN(E50K) Mutation Exhibit Neurodegenerative Phenotypes when Derived from Three-Dimensional Retinal Organoids. Stem Cell Rep. 2020, 15, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spanò, A.; Cavaliere, F.; Rombolà, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal damage caused by high intraocular pressure-induced transient ischemia is prevented by coenzyme Q10 in rat. Int. Rev. Neurobiol. 2007, 82, 397–406. [Google Scholar] [CrossRef]
- Ozdemir, G.; Tolun, F.I.; Gul, M.; Imrek, S. Retinal oxidative stress induced by intraocular hypertension in rats may be ameliorated by brimonidine treatment and N-acetyl cysteine supplementation. J. Glaucoma 2009, 18, 662–665. [Google Scholar] [CrossRef]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef]
- Chou, T.H.; Romano, G.L.; Amato, R.; Porciatti, V. Nicotinamide-Rich Diet in DBA/2J Mice Preserves Retinal Ganglion Cell Metabolic Function as Assessed by PERG Adaptation to Flicker. Nutrients 2020, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Maruyama, K.; Himori, N.; Omodaka, K.; Yokoyama, Y.; Shiga, Y.; Morin, R.; Nakazawa, T. The novel Rho kinase (ROCK) inhibitor K-115: A new candidate drug for neuroprotective treatment in glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7126–7136. [Google Scholar] [CrossRef]
- Akaiwa, K.; Namekata, K.; Azuchi, Y.; Guo, X.; Kimura, A.; Harada, C.; Mitamura, Y.; Harada, T. Edaravone suppresses retinal ganglion cell death in a mouse model of normal tension glaucoma. Cell Death Dis. 2017, 8, e2934. [Google Scholar] [CrossRef]
- Haroon, M.F.; Fatima, A.; Schöler, S.; Gieseler, A.; Horn, T.F.; Kirches, E.; Wolf, G.; Kreutzmann, P. Minocycline, a possible neuroprotective agent in Leber’s hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11,778 mutation. Neurobiol. Dis. 2007, 28, 237–250. [Google Scholar] [CrossRef]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Martinuzzi, A.; Carelli, V.; Rugolo, M. Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids. Investig. Ophthalmol. Vis. Sci. 2008, 49, 671–676. [Google Scholar] [CrossRef]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012, 69, 331–338. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain A J. Neurol. 2011, 134, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Rizzo, G.; Carbonelli, M.; De Negri, A.M.; Sadun, F.; Carta, A.; Guerriero, S.; Simonelli, F.; et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain A J. Neurol. 2011, 134, e188. [Google Scholar] [CrossRef]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain A J. Neurol. 2013, 136, e230. [Google Scholar] [CrossRef] [PubMed]
- Barboni, P.; Valentino, M.L.; La Morgia, C.; Carbonelli, M.; Savini, G.; De Negri, A.; Simonelli, F.; Sadun, F.; Caporali, L.; Maresca, A.; et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain A J. Neurol. 2013, 136, e231. [Google Scholar] [CrossRef]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Amore, G.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 2020, 7, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg-Cohen, N.; Dadon-Bar-El, S.; Hasanreisoglu, M.; Avraham-Lubin, B.C.; Dratviman-Storobinsky, O.; Cohen, Y.; Weinberger, D. Possible neuroprotective effect of brimonidine in a mouse model of ischaemic optic neuropathy. Clin. Exp. Ophthalmol. 2009, 37, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.T.; Huang, C.W.; Liu, C.P.; Chen, C.H.; Tu, C.M.; Hwang, C.S.; Chen, Y.H.; Chen, W.R.; Lin, K.L.; Ho, Y.C.; et al. Inhibition of Retinal Ganglion Cell Loss By a Novel ROCK Inhibitor (E212) in Ischemic Optic Nerve Injury Via Antioxidative and Anti-Inflammatory Actions. Investig. Ophthalmol. Vis. Sci. 2021, 62, 21. [Google Scholar] [CrossRef]
- Sanjari, N.; Pakravan, M.; Nourinia, R.; Esfandiari, H.; Hafezi-Moghadam, A.; Zandi, S.; Nakao, S.; Shah-Heidari, M.H.; Jamali, A.; Yaseri, M.; et al. Intravitreal Injection of a Rho-Kinase Inhibitor (Fasudil) for Recent-Onset Nonarteritic Anterior Ischemic Optic Neuropathy. J. Clin. Pharm. 2016, 56, 749–753. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Bartlett, C.A.; Evill, L.; Rodger, J.; Harvey, A.R.; Dunlop, S.A. Secondary degeneration of the optic nerve following partial transection: The benefits of lomerizine. Exp. Neurol. 2009, 216, 219–230. [Google Scholar] [CrossRef]
- Naguib, S.; Bernardo-Colón, A.; Cencer, C.; Gandra, N.; Rex, T.S. Galantamine protects against synaptic, axonal, and vision deficits in experimental neurotrauma. Neurobiol. Dis. 2020, 134, 104695. [Google Scholar] [CrossRef]
- Cho, H.K.; Kim, S.; Lee, E.J.; Kee, C. Neuroprotective Effect of Ginkgo Biloba Extract Against Hypoxic Retinal Ganglion Cell Degeneration In Vitro and In Vivo. J. Med. Food 2019, 22, 771–778. [Google Scholar] [CrossRef]
- Kang, T.K.; Le, T.T.; Kim, K.A.; Kim, Y.J.; Lee, W.B.; Jung, S.H. Roots of Lithospermum erythrorhizon promotes retinal cell survival in optic nerve crush-induced retinal degeneration. Exp. Eye Res. 2021, 203, 108419. [Google Scholar] [CrossRef]
- Gaydar, V.; Ezrachi, D.; Dratviman-Storobinsky, O.; Hofstetter, S.; Avraham-Lubin, B.C.; Goldenberg-Cohen, N. Reduction of apoptosis in ischemic retinas of two mouse models using hyperbaric oxygen treatment. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7514–7522. [Google Scholar] [CrossRef]
- Zhang, H.K.; Ye, Y.; Li, K.J.; Zhao, Z.N.; He, J.F. Gypenosides Prevent H(2)O(2)-Induced Retinal Ganglion Cell Apoptosis by Concurrently Suppressing the Neuronal Oxidative Stress and Inflammatory Response. J. Mol. Neurosci. MN 2020, 70, 618–630. [Google Scholar] [CrossRef]
- Guo, X.; Harada, C.; Namekata, K.; Kimura, A.; Mitamura, Y.; Yoshida, H.; Matsumoto, Y.; Harada, T. Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2696–2703. [Google Scholar] [CrossRef]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed]
- Sena, D.F.; Lindsley, K. Neuroprotection for treatment of glaucoma in adults. Cochrane Database Syst. Rev. 2017, 1, Cd006539. [Google Scholar] [CrossRef]
- Abbhi, V.; Saini, L.; Mishra, S.; Sethi, G.; Kumar, A.P.; Piplani, P. Design and synthesis of benzimidazole-based Rho kinase inhibitors for the treatment of glaucoma. Bioorg. Med. Chem. 2017, 25, 6071–6085. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Netarsudil Ophthalmic Solution 0.02%: First Global Approval. Drugs 2018, 78, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Amore, G.; Romagnoli, M.; Carbonelli, M.; Barboni, P.; Carelli, V.; La Morgia, C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 2021, 81, 57–86. [Google Scholar] [CrossRef] [PubMed]
- Theodorou-Kanakari, A.; Karampitianis, S.; Karageorgou, V.; Kampourelli, E.; Kapasakis, E.; Theodossiadis, P.; Chatziralli, I. Current and Emerging Treatment Modalities for Leber’s Hereditary Optic Neuropathy: A Review of the Literature. Adv. Ther. 2018, 35, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Lamorgese, A.; Piersantelli, M.; Pagliarani, S.; Benvenuti, F.; Canestrari, F. Oxidative stress and antioxidant status in patients undergoing prolonged exposure to hyperbaric oxygen. Clin. Biochem. 2004, 37, 312–317. [Google Scholar] [CrossRef] [PubMed]


| Publication | Study Model | Results |
|---|---|---|
| Moreno et al., 2004 [69] | Rat | Decreased total retinal superoxide dismutase and catalase activities in increased intraocular pressure induced by hyaluronic acid injection to the anterior chamber. |
| Ko et al., 2005 [70] | Rat | ROS level and activity of antioxidant enzymes increased in elevation of intraocular pressure by cauterization of 3 episcleral veins. |
| Tezel et al., 2005 [71] | Rat | Increased protein oxidation levels in eyes with elevated intraocular pressure by hypertonic saline injections into episcleral veins. |
| Harada et al., 2007 [72] | Mice | Glutamate/aspartate transporter knockout mice had decreased glutathione level and demonstrated pathological features of NTG. |
| Ferreira et al., 2010 [73] | Rat | ROS levels increased in elevation of intraocular pressure by cauterization of 2 episcleral veins. |
| Harada et al., 2010 [74] | Mice | Deficiency of apoptosis signal-regulating kinase 1, an enzyme, leads to stress-induced RGCs apoptosis, preventing optic nerve degeneration in the NTG model. |
| Noro et al., 2019 [75] | Common marmoset | In the aged marmosets presented with glaucoma-like characteristics, increased expression of 4-hydroxy-2-nonenal in the inner retina and blood, and decreased glutathione in blood were found. |
| Naguib et al., 2021 [76] | Mice | ROS levels increased up to 5 weeks following IOP elevation and inhibition of nuclear factor E2-related factor 2 (Nrf2) gene, which participated in antioxidation pathway, leading to earlier axon degeneration. |
| Publication | Study Model | Results |
|---|---|---|
| Levkovitch-Verbin et al., 2000 [140] | Mice | Overexpressing superoxide dismutase, which metabolized ROS, increased RGCs survival in eyes with crush injury of the optic nerve. |
| Lieven et al., 2006 [141] | Rat RGCs culture | Increased intracellular superoxide levels in retinal cell culture from rat eyes underwent optic nerve crush. |
| Fitzgerald et al., 2010 [131] | Rat | Increased oxidative stress associated enzyme (manganese superoxide dismutase) in the TON model with optic nerve transection. |
| Wells et al., 2012 [132] | Rat | Increased calcium flux and oxidative stress markers, and decreased catalase activity were found in TON model with optic nerve transection. |
| Ahmed et al., 2013 [133] | Mice | Increased intraocular ROS levels were found in TON mice induced by giving pressure posterior to the globe. |
| Szymanski et al., 2013 [137] O’Hare Doig et al., 2014 [138] | Rat | Increased ROS in secondary degeneration of TON in the model with partial optic nerve transection. |
| Bernardo-Colón et al., 2018 [124] | Mice | Increased retinal superoxide and decreased superoxide dismutases-2 in eyes injured by over-pressure air waves. |
| Publication | Study Model | Results |
|---|---|---|
| Guy et al., 1989 [152] | Guinea pig | Less demyelination in optic nerve was found with antioxidant enzyme catalase intraperitoneal administration in experimental allergic encephalomyelitis (EAE) model. |
| Guy et al., 1990 [153] | Guinea pigs | Hydrogen peroxide reaction products were found in the retrobulbar optic nerve and optic nerve head of EAE model. |
| Qi et al., 2007 [154,155] | Mice | Elevated ROS was found after antigenic sensitization of the EAE model. Increased RGCs loss was noted in the group with superoxide dismutases-2 suppression. |
| Larabee et al., 2016 [156] | Mice | More severe optic nerve inflammation and visual deficit were found in EAE model with knockout antioxidant transcription factor (nuclear factor-E2-related factor). |
| Antioxidants | Disease | Study Subjects | Findings |
|---|---|---|---|
| Coenzyme Q10 [222,223] | Glaucoma | DBA/2J mice; rat with ocular hypertension by intracameral saline injection | Diet supplemented with coenzyme Q10 could reduce oxidative stress mediated RGCs apoptosis. |
| N-Acetyl cysteine [224] | Glaucoma | Rat with induced ocular hypertension by sodium hyaluronate intracameral injection | Reduced retinal oxidative stress marker, malondialdehyde, caused by high intraocular pressure in rats with topical brimonidine tartrate eye drop installation and intraperitoneal N-acetyl cysteine injection. |
| Lipoic acid [225] | Glaucoma | DBA/2J mice | Increased expression of anti-oxidative genes and proteins, and decreased RGCs loss. |
| Vitamin B3 (nicotinamide) [226] | Glaucoma | DBA/2J mice | Increased RGC density, RGC soma size, and intensity of mitochondrial staining. Better electrical activity in pattern electroretinogram than control mice. |
| Rho kinase inhibitor K-115 [227] | Glaucoma | C57BL/6 mice | Decreased production of ROS and oxidation of lipids. |
| Edaravone [228] | Glaucoma | Normal tension glaucoma EAAC1-deficient mice | Reduced retinal oxidative stress and RGCs death. |
| Minocycline [229] | LHON | Cybrid cell with mtDNA 11778 mutation in teratoma cells | Increased the survival and conserved mitochondrial membrane potential of cybrid cells with overloading oxidative stress induced by thapsigargin. |
| Glutathione [230] | LHON | Cybrid cell with mtDNA 11778, 3640, and 14484 mutations in osteosarcoma cells | Prolonged survival in cells with induced oxidative injury by tert-butyl hydroperoxide and rotenone. |
| EPI-743 [231] | LHON | Human | Stop progression and improve vision in patients with LHON. |
| Idebenone [232,233,234] | LHON | Human | Prevent visual impairment and promote visual recovery in patients with LHON. |
| Idebenone [235,236] | DOA | Human | Improving and stabilizing visual function in patients with DOA. |
| Brimonidine [237] | NAION | Mice with photosensitization above the optic nerve head | Intraperitoneal injection of brimonidine decreased RGCs cell loss and oxidative stress. |
| Rho kinase inhibitor E212 [238] | NAION | Rat with laser induced optic nerve ischemia | Increased retinal superoxide dismutase activity and decreased ROS production by intravitreal injection. |
| Fasudil [239] | NAION | Human | Improvement of visual acuity in NAION patients with intravitreal injection. |
| Lomerizine [240] | TON | Rat with partial optic nerve transection | The calcium channel blocker reduced manganese superoxide dismutase expression and prevented secondary RGCs death. |
| Galantamine [241] | TON | Mice with eye blast injury | Reduced oxidative stress markers and inflammatory response. |
| Vit E [124] | TON | Mice with eye blast injury | High vitamin E diet prevented RGCs loss and decreased level of oxidative stress. |
| Ginkgo biloba [242] | TON | Rat with optic nerve clamping and RGCs cells with exogenous oxidative stress (H2O2) | Prolonged RGCs survival and in both in vitro and in vivo studies. |
| Lithospermum erythrorhizon [243] | TON | Mice with optic nerve crush | Protected RGCs from oxidative stress-induced cell death and reduced ROS production. |
| Hyperbaric oxygen treatment [244] | TON | Mice with optic nerve crush | Reduction of RGCs loss after hyperbaric oxygen therapy. |
| ROS-degradable propylene sulfide [180] | TON | Mice with eye blast injury | Reduction of inflammation and oxidative stress when combining with erythropoietin-R76E. |
| Lipoic acid [192,193] | ON | Experimental autoimmune encephalomyelitis mice | Decreased inflammation and prevented RGCs against oxidative damages. |
| Melatonin [197] | ON | Rat with lipopolysaccharide injection to optic nerve | Decreased microglial reactivity, demyelination, RGCs loss, and oxidative damages. |
| Gypenosides [245] | ON | Rat RGCs with exogenous oxidative stress (H2O2) | Reduced ROS production and inflammatory response and prevented RGCs from oxidative induced apoptosis. |
| Spermidine [246] | ON | Experimental autoimmune encephalomyelitis mice | Reduced demyelination H2O2-induced RGC damage. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, E.Y.-C.; Liu, P.-K.; Wen, Y.-T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.-K.; Tsai, R.-K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948. https://doi.org/10.3390/antiox10121948
Kang EY-C, Liu P-K, Wen Y-T, Quinn PMJ, Levi SR, Wang N-K, Tsai R-K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants. 2021; 10(12):1948. https://doi.org/10.3390/antiox10121948
Chicago/Turabian StyleKang, Eugene Yu-Chuan, Pei-Kang Liu, Yao-Tseng Wen, Peter M. J. Quinn, Sarah R. Levi, Nan-Kai Wang, and Rong-Kung Tsai. 2021. "Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration" Antioxidants 10, no. 12: 1948. https://doi.org/10.3390/antiox10121948
APA StyleKang, E. Y.-C., Liu, P.-K., Wen, Y.-T., Quinn, P. M. J., Levi, S. R., Wang, N.-K., & Tsai, R.-K. (2021). Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants, 10(12), 1948. https://doi.org/10.3390/antiox10121948