Abstract
Magnesium whitlockite (Mg-WH) has emerged as a promising biomaterial for bone regeneration due to its compositional similarity to natural bone minerals. This study aimed to systematically modify a dissolution–precipitation synthesis method to produce Mg-WH granules with tailored morphologies and controlled phase compositions for possible use in bone regeneration applications. Three distinct precursor granules were prepared by mixing varying amounts of ammonium dihydrogen phosphate and magnesium hydrogen phosphate with calcium sulfate. The precursors were then transformed into biphasic and single-phase Mg-WH granules by means of immersion in magnesium- and phosphate-containing solutions under controlled conditions. The X-ray diffraction results demonstrated that biphasic materials containing Mg-WH and either calcium-deficient hydroxyapatite (CDHA) or dicalcium phosphate anhydrous (DCPA) formed after 24 h of synthesis, depending on the synthesis conditions. Prolonging the reaction time to 48 h resulted in complete transformation into single-phase Mg-WH granules. Fourier-transform infrared spectroscopy confirmed the presence of functional groups characteristic of Mg-WH, CDHA, and DCPA in the intermediate products. The spectra also indicated the absence of precursor phases and the progressive elimination of secondary phases as the reaction time increased. Scanning electron microscopy analyses revealed notable morphological transformations from the raw granules to the product granules, with the latter exhibiting interlocked spherical and rod-like particles composed of fine Mg-WH rhombohedral crystals. N2 adsorption–desorption analyses exposed significant differences in the surface properties of the synthesized granules. By varying precursor, reaction solution compositions, and reaction times, the study elucidated the phase evolution mechanisms and demonstrated their impact on the structural, morphological, and surface properties of Mg-WH granules.
1. Introduction
Traumatic injuries affect patients’ quality of life and pose challenges for healthcare and biomedical research [1]. These conditions often lead to chronic pain, reduced mobility, and long-term physical limitations, while also placing a substantial burden on healthcare systems. The aging population in some regions and the growing demographics in others have contributed to an increased prevalence of bone-related disorders, highlighting the rising demand for bone implants [2].
Synthetic bioceramic calcium phosphate (CP) materials are sought for their biodegradability and their potential to promote bone tissue growth (osteogenesis) [3]. Traditionally, CP materials are synthesized at elevated temperatures, which often results in high crystallinity that can limit their biological performance [4]. Moreover, certain bioceramics, such as magnesium whitlockite (Mg-WH, Ca18Mg2(HPO4)2(PO4)12), degrade under such high-temperature conditions, limiting their applicability [5,6]. To address these challenges, dissolution–precipitation synthesis has gained prominence [7]. This method enables the fabrication of CP materials at lower temperatures, ensuring better control over crystallinity while preserving the desired shape of the precursor material [8]. Crucially, this approach enables the production of granules, which are directly suitable for bone grafting procedures. Although dissolution–precipitation synthesis holds promise for producing CP materials with favorable properties for biomedical use, further biological validation is required to confirm their clinical potential [9,10].
Recent studies have highlighted the potential advances of synthetic Mg-WH in bone regeneration, suggesting it may offer improved osteoinductive properties to widely used hydroxyapatite [11,12]. Mg-WH is reported to exhibit greater stability in acidic environments created by osteoclast activity, potentially supporting bone remodeling processes. In preclinical studies, Mg-WH granules have shown promising results in promoting bone formation without causing inflammatory responses [13,14]. Furthermore, there are indications that Mg-WH may have additional beneficial properties, such as anti-inflammatory activity and potential anticancer effects, as reported by Maximiano et al. [15]. Recent advancements in the processing of transparent materials, including ultrafast laser welding techniques, have further demonstrated the importance of controlled surface morphology and precise material engineering to achieve high-performance functionalities in complex environments [16].
It is well recognized that the morphology and structural characteristics of CP materials critically influence their in vivo performance, including osseointegration, resorption behavior, and bone tissue regeneration efficiency [17]. Surface area, porosity, crystallinity, and grain size are key parameters that affect these outcomes. For example, Ghanaati et al. demonstrated that variations in the physical properties of β-TCP-based bone substitutes can significantly influence both the inflammatory response and tissue integration [18]. Thus, optimizing the morphology of CP granules is essential to enhance their potential functionality in bone repair [19].
In this study, we focus on the controlled synthesis and detailed physicochemical characterization of Mg-WH granules produced via a dissolution–precipitation method. The aim was to systematically explore how variations in precursor, reaction solution compositions, and reaction time influence the phase evolution, morphology, and surface properties of the synthesized granules. While these characteristics are relevant to potential bone regeneration applications, this work does not include biological assessments, and the biomedical suitability of the materials remains a prospective direction for future re-search.
2. Materials and Methods
2.1. Materials
Calcium sulfate dihydrate (CSD, CaSO4·2H2O, 99.0%, Sigma-Aldrich, St. Louis, MO, USA), diammonium hydrogen phosphate ((NH4)2HPO4, ≥99%; Chempur, Karlsruhe, Germany), magnesium hydrogen phosphate trihydrate (MgHPO4·3H2O, 99%; Alfa Aesar, Heysham, UK), magnesium acetate tetrahydrate (Mg(CH3COO)2·4H2O, 98%, Roth, Newport Beach, CA, USA), sodium dihydrogen phosphate (NaH2PO4, 99%, Merck, Rahway, NJ, USA), and disodium hydrogen phosphate (Na2HPO4, 98%, Merck).
2.2. Characterization of the Granules
2.2.1. X-Ray Diffraction (XRD) Analysis
The samples obtained were subjected to XRD analysis using a Rigaku MiniFlex II diffractometer (Tokyo, Japan) equipped with CuKα radiation (λ = 1.5418 Å) in a 2θ range of 10–50° (70°) using a step scan mode with a step size of 0.02° and a scan speed of 2°/min. A quality analysis of the synthesized products was conducted using XRD, which enabled the characterization of the phase composition through a comparative analysis of the obtained diffraction patterns with reference standards from the International Centre for Diffraction Data database. The Mg-WH sample underwent Le Bail structure refinement with FullProf software(Version 19.04.24). In order to accurately determine the crystallite size of the prepared single-phase materials, standard lanthanum hexaborate (LaB6) measurements were performed to account for instrumental broadening. This is essential to eliminate its contribution from the width of the experimental diffraction peak [20].
2.2.2. Fourier-Transform Infrared Spectroscopy (FTIR)
FTIR spectra of the synthesized samples were recorded using an FTIR spectrometer equipped with an attenuated total reflectance (ATR) accessory (Alpha, Bruker, Bremen, Germany). Spectra were collected in the range of 1350–450 cm−1 at a spectral resolution of 4 cm−1, averaging 25 scans.
2.2.3. Scanning Electron Microscopy (SEM)
The microstructure of the synthesized samples was examined by SEM using an SU-70 scanning electron microscope (Hitachi, Tokyo, Japan). Images were collected at an accelerating voltage of 10 kV.
2.2.4. N2 Adsorption–Desorption Analyses
The specific surface area (SBET) of the synthesized materials was determined by nitrogen (N2) adsorption–desorption analysis using the Brunauer–Emmett–Teller (BET) method on a TriStar II surface area analyzer (Micromeritics, Norcross, GA, USA). Prior to measurement, the samples were degassed at 120 °C for 2 h. The total pore volume (Vp) was calculated based on the quantity of N2 adsorbed at a relative pressure (p/p0) close to 1. The micropore volume (Vμ) was estimated using the T-plot method.
2.3. Synthesis of Granules
Mg-WH granules were synthesized through a dissolution–precipitation reaction, as illustrated schematically in Figure 1. Calcium sulfate (CS, CaSO4) was selected as the starting material, which was prepared by heating CSD powder at 170 °C for 5 h. Initially, three types of precursor granules with different chemical compositions were synthesized: monophasic (R1) and mixed (R2, and R3) granules. Monophasic precursor granules R1 were prepared by thoroughly mixing 20.0 g of CS with 12 mL of deionized water. Mixed granules R2 were prepared by homogenizing 20.0 g of CS with 8.0 g of (NH4)2HPO4, and then mixing with 12 mL of deionized water. Mixed granules R3 were prepared by homogenizing 20.0 g of CS with 8.0 g of (NH4)2HPO4 and 4.0 g of MgHPO4·3H2O, then mixing with 12 mL of deionized water. The R1–R3 samples were kept at room temperature for a week to allow the solidification process to occur. Then, solidified samples were crushed in an agate mortar and sieved to obtain granules with a size of approximately 200–355 µm.
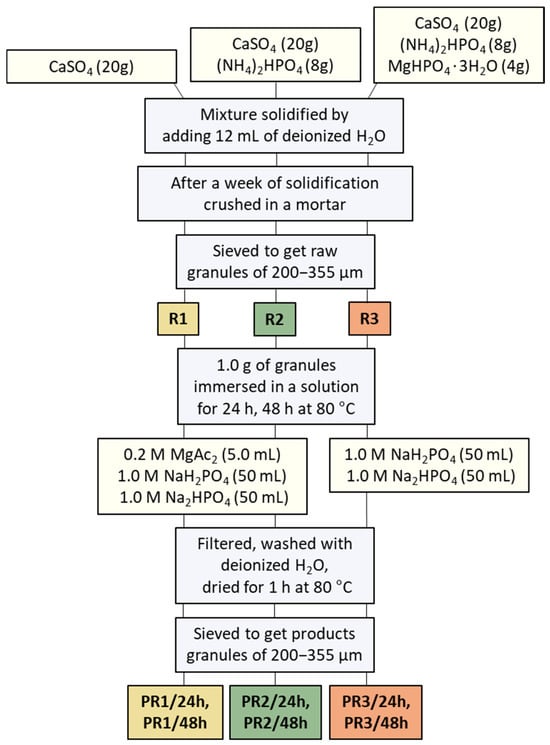
Figure 1.
The following schematic diagram illustrates the preparation process of the raw R1–R3 granules, which are later converted into the final product, PR1/24h, PR1/48h, PR2/24h, PR2/48h, PR3/24h, and PR3/48h granules.
The synthesis of the Mg-WH product granules (see Figure 1) from R1 and R2 precursor granules was carried out as follows: 1 g of each sample was mixed with a 0.20 M magnesium acetate (Mg(CH3COO)2; Mg(Ac)2) solution (0.20 g Mg(CH3COO)2·4H2O dissolved in 5.0 mL of water). Then, 50 mL of 1.0 M NaH2PO4 and 50 mL of 1.0 M Na2HPO4 solutions were added sequentially. The mixture was placed in a capped bottle and kept in a drying oven at 80 °C for 24 h and 48 h. The synthesis of the Mg-WH from R3 granules followed the same procedure, except that the addition of the Mg(Ac)2 solution was omitted. After 24 h and 48 h, all synthesized samples were filtered, washed several times with deionized water, and dried in the oven at 80 °C for 1 h. The samples were then sieved to collect granules of approximately 200–355 µm in size. Granules synthesized from R1, R2, and R3 granules for 28 h and 48 h were named PR1/24h, PR2/24h, PR3/24h, PR1/48h, PR2/48h, and PR3/48h granules, respectively.
3. Results and Discussion
Figure 2a–c shows the XRD patterns of synthesized R1–R3, PR1/24h–PR3/24h, and PR1/48h–PR3/48h granules. Figure 2a illustrates that the XRD results reveal that the synthesis of the R1–R3 granules predominantly led to the formation of CSD as the major crystalline phase. This observation indicates that the CS powder utilized as a precursor underwent a hydration reaction during synthesis in an aqueous environment, resulting in its transformation into the dihydrate form of calcium sulfate. This hydration-induced phase transformation facilitated the transition from a loose powder to a consolidated solid through crystal growth and interlocking, which was subsequently processed into granules [21].
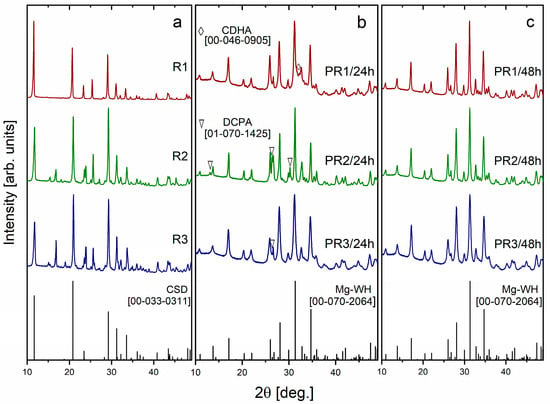
Figure 2.
XRD patterns of R1–R3 (a), PR1/24h–PR3/24h (b), and PR1/48h–PR3/48h (c) granules synthesized via a dissolution–precipitation reaction.
The XRD patterns of the reaction products, obtained after the R1–R3 precursor granules were immersed in a solution containing Mg2+, H2PO42−, HPO42−, and PO43− ions for a period of 24 h, are shown in Figure 2b. These include PR1/24h, PR2/24h, and PR3/24h, which are derived from the monophasic R1 granules and the mixed R2 and R3 granules, respectively, along with the standard Mg-WH diffractogram [ICDD #00-070-2064]. The XRD patterns of PR1/24h, PR2/24h, and PR3/24h granules revealed the formation of biphasic materials, with PR1/24h consisting of Mg-WH and calcium deficient hydroxyapatite (CDHA, Ca10−x(HPO4)x(PO4)6−x(OH)2−x, where 0 < x < 1) phases, while PR2/24h and PR3/24h were composed of Mg-WH and dicalcium phosphate anhydrous (DCPA, CaHPO4) phases. Notably, the amount of DCPA in the PR2/24h sample was significantly higher compared to PR3/24h. The formation of CDHA phase is commonly observed at higher pH values, as Mg-WH is thermodynamically more stable than apatite under moderately low pH conditions. In this study, however, all syntheses were performed under nearly identical pH conditions, suggesting that the appearance of CDHA is influenced by variations in the Ca/Mg ratio within the reaction solution, as previously reported in [22]. The presence of the DCPA phase in the PR2/24h and PR3/24h samples can be ascribed to its function as a transient intermediate in the formation pathway of Mg-WH. It is known that DCPA undergoes a gradual conversion into Mg-WH over time, a transformation that is promoted by magnesium-rich conditions and a moderately acidic pH. This observation is consistent with previous findings [6] and underscores the pivotal role of reaction time, precursor composition, and solution composition on the resulting phase.
In contrast, the XRD patterns of the products synthesized for 48 h (PR1/48h, PR2/48h, and PR3/48h granules) demonstrated the formation of single-phase materials, as no diffraction peaks corresponding to CDHA, DCPA, or any other secondary phases were observed (see Figure 2c). The broad and low-intensity diffraction peaks indicate the formation of nanocrystallites with moderate crystallinity [23]. The reflection positions identified in the XRD analysis were subjected to careful indexing and were found to correspond with the standard diffractogram for rhombohedral Mg-WH [00-070-2064]. The diffraction peaks observed at 2θ values of 10.92°, 13.72°, 17.14°, 28.02°, 31.28°, and 34.70° can be attributed to the (0 1 2), (1 0 4), (1 1 0), (214), (0 2 10), and (2 2 0) crystallographic planes of the Mg-WH phase, respectively. Additionally, smaller peaks align with the reference data too. A 48 h synthesis duration was sufficient for the complete phase transformation of the raw granules into Mg-WH ones. The resulting products exhibited consistent composition, irrespective of whether the starting granules contained magnesium salts or were synthesized using Mg-containing or Mg-free solutions.
In order to ascertain the structural parameters of the synthesized single-phase PR1/48h–PR3/48h samples, a Le Bail refinement of the XRD data was conducted. The XRD patterns, together with the fitting curves derived from the Le Bail refinement, are displayed in Figure 3. The Le Bail refinement plots demonstrate an excellent correlation between the observed XRD patterns (red circles) and the calculated patterns (black lines), which correspond to the allowed Bragg reflections (green ticks), while the discrepancy between the experimental and refined data is shown by the blue line. This confirms the successful formation of the Mg-WH phase with a rhombohedral structure and space group R3c. The structural parameters of the Mg-WH phase synthesized from various starting compositions of raw granules during the 48 h synthesis process are presented in Table 1. The resulting products exhibited consistent structural characteristics, both within the set of samples and when compared to previously published data [24].
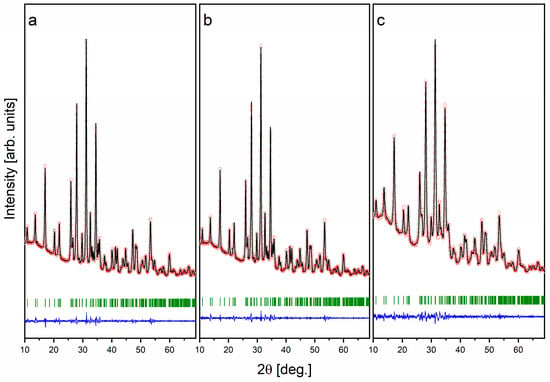
Figure 3.
XRD patterns with the fitting curve by Le Bail refinement of the synthesized PR1/48h (a), PR2/48h (b), and PR3/48h (c) granules.

Table 1.
Structural, R-values, χ2, and microstructural data elucidated from XRD for PR1/48h, PR2/48h, and PR3/48h samples.
The granules synthesized by a dissolution–precipitation reaction were subjected to analysis by FTIR spectroscopy. Figure 4a–c illustrates the FTIR spectra of the materials obtained under varying conditions, with a particular emphasis on the 1350–400 cm−1 region to elucidate the distinctive functional groups present in the synthesized granules. The FTIR spectra of the R1–R3 raw granules (see Figure 4a) display absorption bands centered at 1110 cm−1, 669 cm−1, and 598 cm−1, which are attributed to the stretching and bending modes of the sulfate (SO42−) ion [25]. A weak absorption band centered at 869 cm−1 is attributed to the stretching vibration of hydrogen phosphate (P–O(H)) groups, originating from the precursor salts (NH4)2HPO4 and MgHPO4·3H2O.
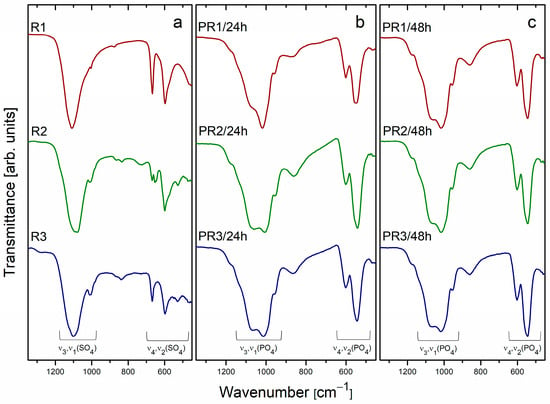
Figure 4.
FTIR spectra of R1–R3 (a), PR1/24h–PR3/24h (b), and PR1/48h–PR3/48h (c) granules synthesized via a dissolution–precipitation reaction.
The FTIR spectra of the granules PR1/24h-PR3/24h (Figure 4b) display the presence of absorption bands corresponding to PO43− and HPO42− functional groups. The absence of SO42−-related absorption bands suggests that the sulphate-containing precursors were completely dissolved and subsequently transformed into CP materials during the 24 h synthesis process. These findings are corroborated by the XRD analysis, which identified specific CP phases: the PR1/24h sample exhibited the presence of CDHA and Mg-WH; the PR2/24h and PR3/24h samples displayed the presence of DCPA and Mg-WH phases (see Figure 2b). The observed absorption bands at 1180 cm−1, 1065 cm−1, 1011 cm−1, and 954 cm−1 are attributed to the P–O and P–O(H) stretching vibrations associated with the PO43− and HPO42− groups, which are integral to the crystal structures of the CDHA, DCPA, and Mg-WH phases, are present to different extents in the synthesized granules. The differences in the intensity of the absorption band at 1065 cm−1 between the PR1/24h and PR2/24h samples provide further insight into the phase composition. In the case of PR1/24h, which comprises CDHA in conjunction with the Mg-WH phase, the increased intensity of the 1065 cm−1 band can be attributed to the absence of a notable contribution from CDHA, given that this phase does not exhibit a pronounced absorption at this wavenumber [26]. In contrast, the 1065 cm−1 band is prominent in the PR2/24h sample and barely noticeable in the PR3/24h sample, where it corresponds to the P–O stretching vibration characteristic of the DCPA phase, resulting in a smaller intensity for this absorption band [27]. Furthermore, the PR1/24h and PR2/24h samples display a relatively broad absorption band with a maximum at 863 cm−1, which is attributed to the stretching vibration of the P–O(H) bond. The broadness of this band indicates the coexistence of two distinct crystallographic environments for hydrogen phosphate ions, which are likely the result of the presence of two different calcium phosphate phases in the samples. At lower wavenumbers, the absorption bands at 601 cm−1, 545 cm−1, and 465 cm−1 are primarily associated with the Mg-WH phase and correspond to the bending modes of P–O and O–P–O bonds [5]. In the case of the PR1/24h sample, the peak at 545 cm−1 displays a slight degree of broadening due to the presence of CDHA, which exhibits an absorption band in the vicinity of 560 cm−1. The FTIR spectra of PR1/48h–PR3/48h granules (Figure 4c) display a series of absorption bands at 1180 cm−1, 1065 cm−1, 1011 cm−1, 954 cm−1, 865 cm−1, and within the 600–460 cm−1 range. These bands are ascribed to the P–O and P–O(H) stretching and deformation vibrations of the PO43− and HPO42− groups, which are distinctive of the crystal structure of the Mg-WH phase.
Figure 5, Figure 6 and Figure 7 present the SEM micrographs of the synthesized R1–R3 raw granules, the PR1/24h–PR3/24h granules, and the PR1/48h–PR3/48h granules, respectively. An inspection of the surface of approximately 200–400 µm rough-edged raw granules (see Figure 5a,d,g; surface views in Figure 5b,e,h) discloses the presence of rod-like crystals, which are indicative of calcium sulfate dihydrate. These crystals are accompanied by randomly oriented plate-shaped crystals, as observed in the higher-magnification images (see Figure 5c,f,i). The surface morphology of the irregularly shaped PR1/24h to PR3/24h granules (Figure 6a,d,g; surface views in Figure 6b,e,h) shows notable variation, influenced by both the type of raw granules used and possible differences in local reaction environments during synthesis.

Figure 5.
SEM images of raw granules R1 (a–c), R2 (d–f), and R3 (g–i).

Figure 6.
SEM images of PR1/24h (a–c), PR2/24h (d–f), and PR3/24h (g–i) granules synthesized via a dissolution–precipitation reaction.

Figure 7.
SEM images of PR1/48h (a–c), PR2/48h (d–f), and PR3/48h (g–i) granules synthesized via a dissolution–precipitation reaction.
A thorough inspection of the surface of the PR1/48h–PR3/48h granules (see Figure 7a,d,g; surface views in Figure 7b,e,h) of 200–450 µm granules reveals that they are composed of interlocked spherical and rod-like particles, which are composed of fine crystals. The higher magnification SEM micrograph provides a detailed analysis of the granules, revealing their composition as a stack of characteristic Mg-WH rhombohedral crystals (see Figure 7c,f) with an average size of approximately 150–200 nm [5]. As illustrated in Figure 7i, the PR3/48h specimen displays a well-developed porous microstructure, which is composed of an assembly of plate-like crystals with an approximate diameter of 200 nm. As illustrated in Figure 7i, the PR3/48h specimen displays a well-developed porous microstructure, which is composed of an assembly of plate-like crystals with an approximate diameter of 200 nm, similar to the interconnected porous structures reported in sintered WH granules, which have been shown to facilitate fluid transport and nutrient exchange essential for bone regeneration [13]. It is worth noting that the granule size range of approximately 200–450 µm is particularly suitable for use as bone graft substitutes. Previous studies have demonstrated that hydroxyapatite-based granules within the 212–300 µm size range promote superior bone formation in vivo compared to larger granules, which tend to limit osteogenesis [28].
An examination of the textural characteristics of samples PR1/24h, PR2/24h, PR3/24h, PR1/48h, PR2/48h, and PR3/48h, as determined by N2 adsorption–desorption measurements, reveals significant differences in surface area and porosity characteristics (see Table 2). Among the 24 h samples, PR1/24h, consisting of a biphasic mixture of Mg-WH and CDHA, exhibits the highest BET surface area (SBET, 35 m2/g), external surface area (Sext) of 30 m2/g, and total pore volume (Vp, 0.13 cm3/g), indicating a highly porous structure with significant external surface exposure. In contrast, PR2/24h, composed mainly of Mg-WH and a significant amount of DCPA, shows a lower SBET (4 m2/g), Sext (3 m2/g), and Vp (0.010 cm3/g), reflecting a denser microstructure. PR3/24h, containing Mg-WH with a smaller amount of DCPA, exhibits intermediate properties, with a relatively high SBET (30 m2/g), Sext (27 m2/g), and moderate Vp (0.069 cm3/g). Overall, PR1/24h demonstrates the greatest combination of surface area and pore volume among the 24 h samples, suggesting superior surface accessibility and porosity.
Table 2.
Data derived from N2 adsorption–desorption measurements: SBET, Vμ, and Vp of all synthesized product samples.
For the 48 h samples, PR3/48h exhibits a substantially higher BET surface area (38 m2/g) and total pore volume (0.077 cm3/g) compared to PR1/48h (9 m2/g, 0.025 cm3/g) and PR2/48h (11 m2/g, 0.027 cm3/g). All these samples composed solely of the Mg-WH phase but were derived from three chemically distinct precursor granules and reaction solution chemistries. Specifically, the R3 granules incorporated MgHPO4·3H2O directly into the precursor, providing an intrinsic magnesium source. Unlike the syntheses of PR1 and PR2, the PR3 samples did not require the addition of external magnesium acetate solution. The in situ availability of magnesium likely facilitated a more homogeneous and controlled precipitation process, leading to the formation of finer particles, smaller crystallites (see Table 1) and consequently a larger surface area of PR3/48h sample.
The higher surface area of PR3/48h is expected to substantially enhance its bioactivity, as materials with greater surface areas provide expanded sites for ion exchange, faster dissolution kinetics, and increased protein adsorption which all are known as critical factors for bone regeneration [29]. Conversely, the lower surface areas of PR1/48h and PR2/48h suggest denser, less permeable structures with reduced surface exposure, which may limit their bioactivity and hinder effective tissue integration [30].
Micropore analysis further supports these differences. The external surface areas (Sext) of PR1/48h, PR2/48h, and PR3/48h were 7 m2/g, 8 m2/g, and 35 m2/g, respectively, closely matching their total surface areas and indicating that the surfaces are predominantly external. Micropores account for a mere 12%, 17%, and 8%, respectively, of the total surface area for these specimens, indicating the mesoporous nature of the materials [31]. This assertion is further substantiated by the uniformity in micropore volumes across all samples (Vμ ≈ 0.0011–0.0012 cm3/g), thereby confirming the predominance of meso- to macroporous characteristics in the material [32].
The significantly higher surface area and pore volume of PR3/48h, attributed to its finer particle morphology, likely provide a more open and biologically favorable structure. This is consistent with previous findings that sintered WH granules with macroporous structures enhance bioactivity and support bone tissue integration due to improved fluid and nutrient transport [13]. Studies on porous calcium phosphate ceramics have shown that elevated surface area and porosity significantly enhance protein adsorption, osteoblastic differentiation, and ionic interactions essential for new bone formation [28,33]. Therefore, PR3/48h may offer superior bioactivity compared to the other samples. In contrast, the denser microstructures and underdeveloped mesoporosity of PR1/48h and PR2/48h may negatively impact their performance in surface-driven biological processes [30].
4. Conclusions
This study successfully demonstrated a systematic approach to synthesizing magnesium whitlockite (Mg-WH) granules with tailored phase compositions, morphologies, and surface properties through a controlled dissolution–precipitation process. By varying the precursor compositions, reaction solution chemistries, and synthesis durations, it was possible to direct the phase evolution from biphasic systems comprising Mg-WH with calcium-deficient hydroxyapatite or dicalcium phosphate anhydrous to single-phase Mg-WH granules. XRD and FTIR analyses confirmed the progressive transformation of the initial calcium sulfate-based precursors into Mg-WH, with complete phase purity achieved after 48 h of synthesis. Notably, the incorporation of magnesium directly into the precursor granules led to the development of finer particles with significantly higher surface areas and porosity compared to those synthesized from precursors requiring external magnesium sources. SEM observations revealed that the final Mg-WH granules exhibited favorable size and microstructures, composed of interlocked spherical and rod-like particles formed by nanoscale Mg-WH rhombohedral crystals. Additionally, N2 adsorption–desorption analyses underscored that the surface area and porosity of the granules could be finely tuned by adjusting the synthesis parameters. Although the materials demonstrate promising characteristics for potential bone regeneration applications, further biological assessments are necessary to validate their biomedical suitability, which remains an important direction for future research.
Author Contributions
Conceptualization, I.G. and R.R.; methodology, I.G.; software, R.R.; validation, R.R., G.L., and A.E.; formal analysis, R.R.; investigation, R.R.; resources, G.L.; data curation, R.R. and G.L.; writing—original draft preparation, R.R.; writing—review and editing, I.G. and A.E.; visualization, R.R.; supervision, I.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Andrius Pakalniskis (Vilnius University) is acknowledged for assistance with SEM measurements and helpful discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BET | Brunauer–Emmett–Teller |
| CDHA | Calcium-deficient hydroxyapatite |
| CP | Calcium phosphate |
| CS | Calcium sulfate |
| CSD | Calcium sulfate dihydrate |
| DCPA | Dicalcium phosphate anhydrous |
| FTIR | Fourier transform infrared spectroscopy |
| Mg(Ac)2 | Magnesium acetate |
| Mg-WH | Magnesium whitlockite |
| SEM | Scanning electron microscopy |
References
- Bussell, M.B. Improving bone health: Addressing the burden through an integrated approach. Aging Clin. Exp. Res. 2021, 33, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Mastnak, T.; Maver, U.; Finšgar, M. Addressing the Needs of the Rapidly Aging Society through the Development of Multifunctional Bioactive Coatings for Orthopedic Applications. Int. J. Mol. Sci. 2022, 23, 2786. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, O.; Yanovska, A.; Kosinov, O.; Maksymov, D.; Moskalenko, R.; Ramanavicius, A.; Pogorielov, M. Synthetic Calcium–Phosphate Materials for Bone Grafting. Polymers 2023, 15, 3822. [Google Scholar] [CrossRef] [PubMed]
- Sinusaite, L.; Kareiva, A.; Zarkov, A. Thermally Induced Crystallization and Phase Evolution of Amorphous Calcium Phosphate Substituted with Divalent Cations Having Different Sizes. Cryst. Growth Des. 2021, 21, 1242–1248. [Google Scholar] [CrossRef]
- Kizalaite, A.; Klimavicius, V.; Balevicius, V.; Niaura, G.; Salak, A.N.; Yang, J.-C.; Cho, S.H.; Goto, T.; Sekino, T.; Zarkov, A. Dissolution–precipitation synthesis and thermal stability of magnesium whitlockite. CrystEngComm 2023, 25, 4370–4379. [Google Scholar] [CrossRef]
- Raiseliene, R.; Linkaite, G.; Zarkov, A.; Kareiva, A.; Grigoraviciute, I. Large-Scale Green Synthesis of Magnesium Whitlockite from Environmentally Benign Precursor. Materials 2024, 17, 788. [Google Scholar] [CrossRef]
- Ishikawa, K. Bone Substitute Fabrication Based on Dissolution-Precipitation Reactions. Materials 2010, 3, 1138–1155. [Google Scholar] [CrossRef]
- Jamilludin, M.A.; Hayashi, K.; Yusuf, Y.; Ishikawa, K. Low-crystalline magnesium-doped carbonate apatite/β-tricalcium phosphate granules from sea urchin spine. J. Am. Ceram. Soc. 2025, 108, e20498. [Google Scholar] [CrossRef]
- Ginebra, M.P.; Espanol, M.; Maazouz, Y.; Bergez, V.; Pastorino, D. Bioceramics and bone healing. EFORT Open Rev. 2018, 3, 173–183. [Google Scholar] [CrossRef]
- Fuchs, A.; Kreczy, D.; Brückner, T.; Gbureck, U.; Stahlhut, P.; Bengel, M.; Hoess, A.; Nies, B.; Bator, J.; Klammert, U.; et al. Bone regeneration capacity of newly developed spherical magnesium phosphate cement granules. Clin. Oral Investig. 2022, 26, 2619–2633. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Yang, H.; Zhao, Y.; Guo, J.; Yin, X.; Ma, T.; Liu, X.; Li, L. Magnesium-Based Whitlockite Bone Mineral Promotes Neural and Osteogenic Activities. ACS Biomater. Sci. Eng. 2020, 6, 5785–5796. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.Z.; Bin Zheng, G.; Jang, H.L.; Lee, K.M.; Lee, J.H. Whitlockite Promotes Bone Healing in Rabbit Ilium Defect Model. J. Med. Biol. Eng. 2019, 39, 944–951. [Google Scholar] [CrossRef]
- Lee, W.B.; Wang, C.; Lee, J.H.; Jeong, K.J.; Jang, Y.S.; Park, J.Y.; Ryu, M.H.; Kim, U.K.; Lee, J.; Hwang, D.S. Whitlockite Granules on Bone Regeneration in Defect of Rat Calvaria. ACS Appl. Bio Mater. 2020, 3, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Qi, C.; Chen, Y.X.; Zhu, Y.J.; Sun, T.W.; Chen, F.; Zhang, C.Q. Comparative study of porous hydroxyapatite/chitosan and whitlockite/chitosan scaffolds for bone regeneration in calvarial defects. Int. J. Nanomed. 2017, 12, 2673–2687. [Google Scholar] [CrossRef]
- Maximiano, L.V.; Correa, L.B.; Gomes-da-Silva, N.C.; da Costa, L.S.; Da Silva, M.G.P.; Chaves, A.V.; Franco, M.L.; Fechine, P.B.A.; de Menezes, A.S.; Santos-Oliveira, R.; et al. Magnesium whitlockite nanoparticles: Hydrothermal synthesis, anti-inflammatory and anti-cancer potential. Colloids Surf. B Biointerfaces 2024, 239, 113931. [Google Scholar] [CrossRef]
- Jia, X.; Luo, J.; Li, K.; Wang, C.; Li, Z.; Wang, M.; Jiang, Z.; Veiko, V.P.; Duan, J. Ultrafast Laser Welding of Transparent Materials: From Principles to Applications. Int. J. Extrem. Manuf. 2025, 7, 032001. [Google Scholar] [CrossRef]
- Yuan, H.; Fernandes, H.; Habibovic, P.; de Boer, J.; Barradas, A.M.C.; de Ruiter, A.; Walsh, W.R.; van Blitterswijk, C.A.; de Bruijn, J.D. Osteoinductive ceramics as a synthetic alternative to autologous bone grafting. Proc. Natl. Acad. Sci. USA 2010, 107, 13614–13619. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Q.; Wu, Y.; Feng, C.; Yang, X.; Wang, L.; Xiao, Y.; Zhang, K.; Zhu, X.; Liu, L.; et al. Enhanced bone regenerative properties of calcium phosphate ceramic granules in rabbit posterolateral spinal fusion through a reduction of grain size. Bioact. Mater. 2022, 11, 90–106. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, L.; Chen, L.; Geng, M.; Xing, Z.; Chen, S.; Zeng, Y.; Zhou, J.; Sun, K.; Yang, X.; et al. Core–Shell Structured Porous Calcium Phosphate Bioceramic Spheres for Enhanced Bone Regeneration. ACS Appl. Mater. Interfaces 2022, 14, 47491–47506. [Google Scholar] [CrossRef]
- Rodriguez-Carvajal, J.; Roisnel, T. Line broadening analysis using FullProf: Determination of microstructural properties. Mater. Sci. Forum 2004, 443–444, 123–126. [Google Scholar] [CrossRef]
- Nomura, S.; Tsuru, K.; Matsuya, S.; Takahashi, I.; Kunio, K. Fabrication of Spherical Carbonate Apatite Using Calcium Sulfate as a Precursor by W/O Emulsion Method. KEM 2012, 529–530, 78–81. [Google Scholar] [CrossRef]
- Angelova, N.; Koleva, S.; Kostadinov, M.; Yordanov, G. Preparation, characterization and protein adsorption properties of nanostructured magnesium whitlockite. J. Mater. Sci. 2022, 57, 21571–21582. [Google Scholar] [CrossRef]
- Holder, C.F.; Schaak, R.E. Tutorial on Powder X-ray Diffraction for Characterizing Nanoscale Materials. ACS Nano 2019, 13, 7359–7365. [Google Scholar] [CrossRef]
- Konishi, T.; Watanabe, S. Hydrothermal transformation of calcium hydrogen phosphate dihydrate into magnesium whitlockite. Phosphorus Res. Bull. 2021, 37, 21–25. [Google Scholar] [CrossRef]
- Wu, X.Q.; Sun, F.; Yan, W.; Chen, L.; Wang, S.M.; Liang, S.W. Fingerprinting of Mineral Medicine Natrii Sulfas by Fourier Transform Infrared Spectroscopy. Spectroscopy 2021, 36, 38–43. [Google Scholar]
- Karalkeviciene, R.; Raudonyte-Svirbutaviciene, E.; Zarkov, A.; Yang, J.C.; Popov, A.I.; Kareiva, A. Solvothermal Synthesis of Calcium Hydroxyapatite via Hydrolysis of Alpha-Tricalcium Phosphate in the Presence of Different Organic Additives. Crystals 2023, 13, 265. [Google Scholar] [CrossRef]
- Djošić, M.S.; Mišković-Stanković, V.B.; Kačarević-Popović, Z.M.; Jokić, B.M.; Bibić, N.; Mitrić, M.; Milonjić, S.K.; Jančić-Heinemann, R.; Stojanović, J. Electrochemical synthesis of nanosized monetite powder and its electrophoretic deposition on titanium. Colloids Surf. A Physicochem. Eng. Asp. 2009, 341, 110–117. [Google Scholar] [CrossRef]
- Fischer, E.M.; Layrolle, P.; van Blitterswijk, C.A.; de Bruijn, J.D. Bone Formation by Mesenchymal Progenitor Cells Cultured on Dense and Microporous Hydroxyapatite Particles. Tissue Eng. 2003, 9, 1179–1188. [Google Scholar] [CrossRef]
- Lee, D.S.H.; Pai, Y.; Chang, S.; Kim, D.H. Microstructure, physical properties, and bone regeneration effect of the nano-sized β-tricalcium phosphate granules. Mater. Sci. Eng. C 2016, 58, 971–976. [Google Scholar] [CrossRef]
- Dehkord, E.S.; De Carvalho, B.; Ernst, M.; Albert, A.; Lambert, F.; Geris, L. Influence of physicochemical characteristics of calcium phosphate-based biomaterials in cranio-maxillofacial bone regeneration: A systematic literature review and meta-analysis of preclinical models. Mater. Today Bio 2024, 26, 101100. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, C.; Hong, Y.; Zhang, X. A Review of Protein Adsorption on Bioceramics. Interface Focus 2012, 2, 259–277. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).