
Temporal and Wash-Out Studies Identify Medicines for Malaria Venture Pathogen Box Compounds with Fast-Acting Activity against Both Trypanosoma cruzi and Trypanosoma brucei

 ,
, 
Abstract
1. Introduction
2. Methods
2.1. Growth and Maintenance of Parasites and Host Mammalian Cell Lines
2.2. Compounds
2.3. T.b. brucei 72 h REDOX-Based Viability Assay
2.4. T. cruzi 48 h Image-Based Assay
2.5. Pathogen Box IC50 Values and Parasite Selectivity
2.6. T.b. brucei Temporal Viability Assays
2.7. T. cruzi Temporal Image-Based Assays
2.8. T.b. brucei Subspecies Assays
2.9. HEK293 Cytotoxicity Assay
2.10. HEPG2 Cytotoxicity Assay
2.11. Compound Activity against T. cruzi Recombinant CYP51 Enzyme
3. Results
3.1. Confirming Activity of Pathogen Box Solid Compound Stocks against T.b. brucei and T. cruzi
3.2. Time-Kill: Compounds Active against Both T. cruzi and T.b. brucei
3.3. Fast-Acting Compound Activity against T.b. rhodesiense and T.b. gambiense
3.4. Physicochemical Properties of Fast-Acting Compounds
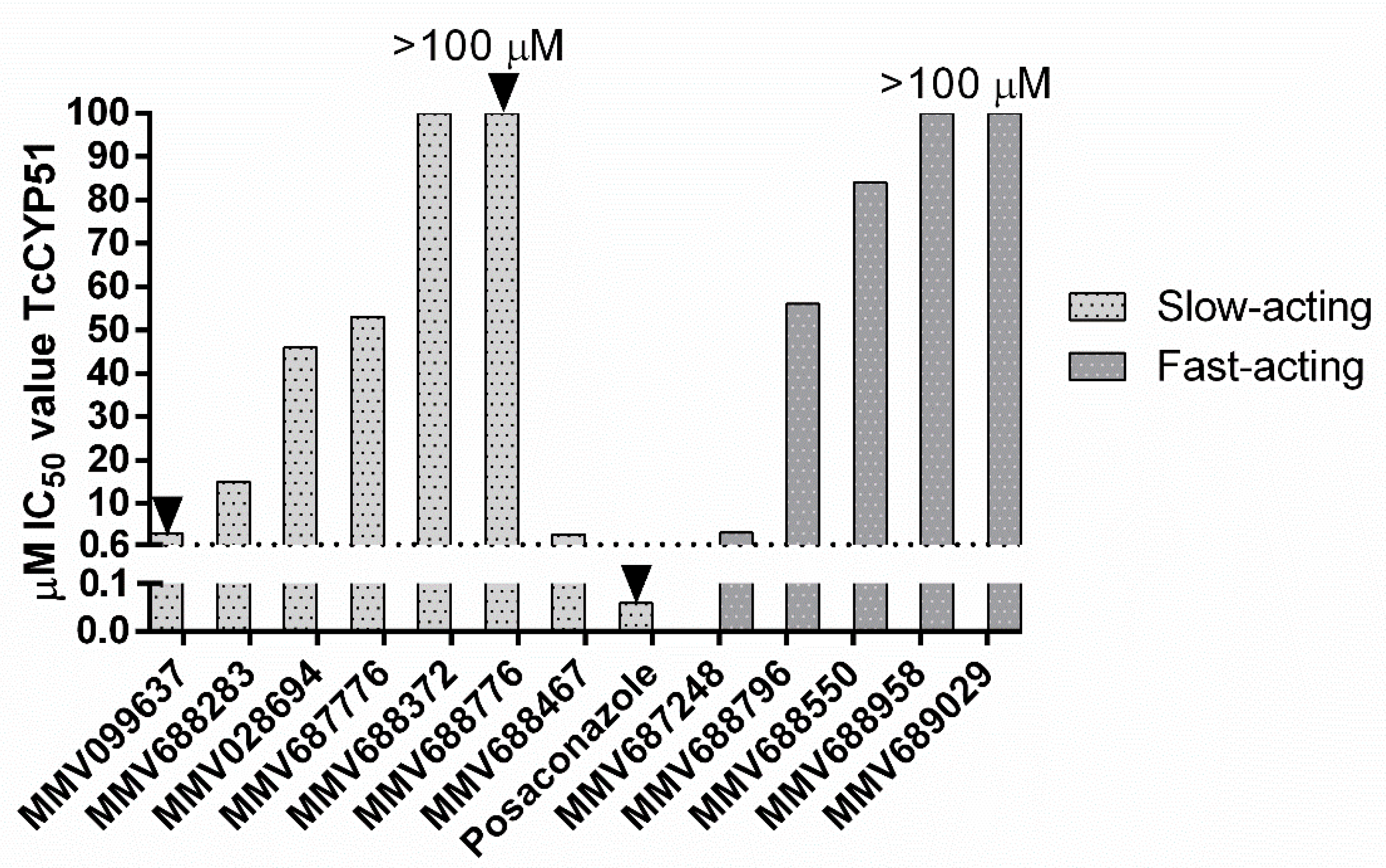
3.5. TcCYP51 Biochemical Activity
3.6. Compounds Recommended for Further Investigation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Neglected Tropical Diseases. Available online: https://www.who.int/health-topics/neglected-tropical-diseases (accessed on 27 May 2022).
- World Health Organisation. Chagas Disease (American trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 27 May 2022).
- Castro, J.A.; de Mecca, M.M.; Bartel, L.C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Sales Junior, P.A.; Molina, I.; Fonseca Murta, S.M.; Sanchez-Montalva, A.; Salvador, F.; Correa-Oliveira, R.; Carneiro, C.M. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Drugs for Neglected Diseases Initiative. Fexinidazole for T.b. rhodesiense. Available online: https://dndi.org/research-development/portfolio/fexinidazole-tb-rhodesiense/ (accessed on 27 May 2022).
- Efficacy and Safety of Fexinidazole in Patients with Human African Trypanosomiasis (HAT) Due to Trypanosoma brucei rhodesiense. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03974178 (accessed on 27 May 2022).
- World Health Organization. Control and surveillance of African trypanosomiasis. In Report of a WHO Expert Committee; World Health Organisation Technical Report Series: Geneva, Switzerland, 1998; Volume 881, pp. 1–114. [Google Scholar]
- Arrowsmith, J. Trial Watch Phase III and submission failures: 2007–2010. Nat. Rev. Drug Discov. 2011, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- MMV About the Pathogen Box. Available online: https://www.mmv.org/mmv-open/pathogen-box/about-pathogen-box (accessed on 27 May 2022).
- Hunter, P. Tropical diseases and the poor: Neglected tropical diseases are a public health problem for developing and developed countries alike. EMBO Rep. 2014, 15, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Maccesi, M.; Aguiar, P.H.N.; Pasche, V.; Padilla, M.; Suzuki, B.M.; Montefusco, S.; Abagyan, R.; Keiser, J.; Mourão, M.M.; Caffrey, C.R. Multi-center screening of the Pathogen Box collection for schistosomiasis drug discovery. Parasit Vectors 2019, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, A.B.; Tuvshintulga, B.; Guswanto, A.; Tayebwa, D.S.; Rizk, M.A.; Gantuya, S.; Batiha, G.E.-S.; Beshbishy, A.M.; Sivakumar, T.; Yokoyama, N.; et al. Screening the Medicines for Malaria Venture Pathogen Box against piroplasm parasites. Int. J. Parasitol. Drugs Drug Resist. 2019, 10, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Vila, T.; Lopez-Ribot, J.L. Screening the Pathogen Box for Identification of Candida albicans Biofilm Inhibitors. Antimicrob. Agents Chemother. 2017, 61, e02006-16. [Google Scholar] [CrossRef] [PubMed]
- Machicado, C.; Soto, M.P.; Timoteo, O.; Vaisberg, A.; Pajuelo, M.; Ortiz, P.; Marcos, L.A. Screening the Pathogen Box for Identification of New Chemical Agents with Anti-Fasciola hepatica Activity. Antimicrob. Agents Chemother. 2019, 63, e02373-18. [Google Scholar] [CrossRef]
- Duffy, S.; Sykes, M.L.; Jones, A.J.; Shelper, T.B.; Simpson, M.; Lang, R.; Poulsen, S.-A.; Sleebs, B.E.; Avery, V.M. Screening the Medicines for Malaria Venture Pathogen Box across Multiple Pathogens Reclassifies Starting Points for Open-Source Drug Discovery. Antimicrob. Agents Chemother. 2017, 61, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Veale, C.G.L.; Hoppe, H.C. Screening of the Pathogen Box reveals new starting points for anti-trypanosomal drug discovery. MedChemComm 2018, 9, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Human African Trypanosomiasis (Sleeping Sickness). Available online: https://www.who.int/health-topics/human-african-trypanosomiasis (accessed on 27 May 2022).
- Zrein, M.; Granjon, E.; Gueyffier, L.; Caillaudeau, J.; Liehl, P.; Pottel, H.; Cardoso, C.S.; Oliveira, C.D.L.; De Oliveira, L.C.; Lee, T.-H.; et al. A novel antibody surrogate biomarker to monitor parasite persistence in Trypanosoma cruzi-infected patients. PLoS Negl. Trop. Dis. 2018, 12, e0006226. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, E.; Konar, N. Translational challenges of animal models in Chagas disease drug development: A review. Drug. Des. Dev. Ther. 2015, 9, 4807–4823. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Cucunubá, Z.; Flórez, C.; Olivera, M.; Valencia, C.; Zambrano, P.; León, C.; Ramírez, J.D. Molecular Diagnosis of Chagas Disease in Colombia: Parasitic Loads and Discrete Typing Units in Patients from Acute and Chronic Phases. PLoS Negl. Trop. Dis. 2016, 10, e0004997. [Google Scholar] [CrossRef]
- Ward, A.I.; Lewis, M.D.; Khan, A.A.; McCann, C.J.; Francisco, A.F.; Jayawardhana, S.; Taylor, M.C.; Kelly, J.M. In Vivo Analysis of Trypanosoma cruzi Persistence Foci a Single-Cell Resolution. Mbio 2020, 11, e01242-20. [Google Scholar] [CrossRef]
- Sykes, M.L.; Avery, V.M. 3-pyridyl inhibitors with novel activity against Trypanosoma cruzi reveal in vitro profiles can aid prediction of putative cytochrome P450 inhibition. Sci. Rep. 2018, 8, 4901. [Google Scholar] [CrossRef]
- Sykes, M.L.; Baell, J.; Kaiser, M.; Chatelain, E.; Moawad, S.R.; Ganame, D.; Ioset, J.-R.; Avery, V.M. Identification of compounds with anti-proliferative activity against Trypanosoma brucei brucei strain 427 by a whole cell viability based HTS campaign. PLoS Negl. Trop. Dis. 2012, 6, e1896. [Google Scholar] [CrossRef]
- Chatelain, E. Chagas disease drug discovery: Toward a new era. J. Biomol. Screen. 2015, 20, 22–35. [Google Scholar] [CrossRef]
- Cal, M.; Ioset, J.R.; Fugi, M.A.; Maser, P.; Kaiser, M. Assessing anti-T. cruzi candidates in vitro for sterile cidality. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 165–170. [Google Scholar] [CrossRef]
- MacLean, L.M.; Thomas, J.; Lewis, M.D.; Cotillo, I.; Gray, D.W.; De Rycker, M. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl. Trop. Dis. 2018, 12, e0006612. [Google Scholar] [CrossRef]
- Sykes, M.L.; Avery, V.M. Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol. Drug. 2015, 5, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; i Prat, J.G.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; Bangher, M.D.C.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. Benznidazole and Posaconazole in Eliminating Parasites in Asymptomatic T. Cruzi Carriers: The STOP-CHAGAS Trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef]
- Molina, I.; Salvador, F.; Sanchez-Montalva, A. The use of posaconazole against Chagas disease. Curr. Opin. Infect. Dis. 2015, 28, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.B.; Giardini, M.A.; Kim, H.; Franco, C.H.; Araujo, A.M.; Schenkman, S.; Chatelain, E.; Freitas, L.H. Nitroheterocyclic compounds are more efficacious than CYP, 51 inhibitors against Trypanosoma cruzi: Implications for Chagas disease drug discovery and development. Sci. Rep. 2014, 4, 4703. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.; Brand, S.; Voice, M.; Caballero, I.; Calvo, D.; Read, K.D. Development of a Fluorescence-based Trypanosoma cruzi CYP51 Inhibition Assay for Effective Compound Triaging in Drug Discovery Programmes for Chagas Disease. PLoS Neglect. Trop. D 2015, 9, e0004014. [Google Scholar] [CrossRef]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef]
- Passos-Silva, D.G.; Rajão, M.A.; de Aguiar, P.H.N.; Vieira-Da-Rocha, J.P.; Machado, C.R.; Furtado, C. Overview of DNA Repair in Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major. J. Nucleic Acids 2010, 2010, 840768. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; A Akl, E.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef]
- Watson, J.A.; Strub-Wourgraft, N.; Tarral, A.; Ribeiro, I.; Tarning, J.; White, N.J. Pharmacokinetic-Pharmacodynamic Assessment of the Hepatic and Bone Marrow Toxicities of the New Trypanoside Fexinidazole. Antimicrob. Agents Chemother. 2019, 63, e02515-18. [Google Scholar] [CrossRef]
- Azzam, M.E.; Algranati, I.D. Mechanism of Puromycin Action—Fate of Ribosomes after Release of Nascent Protein Chains from Polysomes. Proc. Natl. Acad. Sci. USA 1973, 70, 3866–3869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Bray, M.A.; Cal, M.; Trunz, B.B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef]
- Brand, S.; Ko, E.J.; Viayna, E.; Thompson, S.; Spinks, D.; Thomas, M.; Sandberg, L.; Francisco, A.F.; Jayawardhana, S.; Smith, V.C.; et al. Discovery and Optimization of 5-Amino-1,2,3-triazole-4-carboxamide Series against Trypanosoma cruzi. J. Med. Chem. 2017, 60, 7284–7299. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug. Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Nare, B.; Wring, S.; Bacchi, C.; Beaudet, B.; Bowling, T.; Brun, R.; Chen, D.; Ding, C.; Freund, Y.; Gaukel, E.; et al. Discovery of Novel Orally Bioavailable Oxaborole 6-Carboxamides That Demonstrate Cure in a Murine Model of Late-Stage Central Nervous System African Trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 4379–4388. [Google Scholar] [CrossRef]
- Bustamante, J.M.; Craft, J.M.; Crowe, B.D.; Ketchie, S.A.; Tarleton, R.L. New, combined, and reduced dosing treatment protocols cure Trypanosoma cruzi infection in mice. J. Infect. Dis. 2014, 209, 150–162. [Google Scholar] [CrossRef]
- Svensen, N.; Wyllie, S.; Gray, D.W.; De Rycker, M. Live-imaging rate-of-kill compound profiling for Chagas disease drug discovery with a new automated high-content assay. PLoS Negl. Trop. Dis. 2021, 15, e0009870. [Google Scholar] [CrossRef]
- Wall, R.J.; Rico, E.; Lukac, I.; Zuccotto, F.; Elg, S.; Gilbert, I.H.; Freund, Y.; Alley, M.R.K.; Field, M.C.; Wyllie, S.; et al. Clinical and veterinary trypanocidal benzoxaboroles target CPSF. Proc. Natl. Acad. Sci. USA 2018, 115, 9616–9621. [Google Scholar] [CrossRef]
- Russell, S.; Rahmani, R.; Jones, A.J.; Newson, H.L.; Neilde, K.; Cotillo, I.; Khajouei, M.R.; Ferrins, L.; Qureishi, S.; Nguyen, N.; et al. Hit-to-Lead Optimization of a Novel Class of Potent, Broad-Spectrum Trypanosomacides. J. Med. Chem. 2016, 59, 9686–9720. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valdez, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. eLife 2018, 7, e34039. [Google Scholar] [CrossRef] [PubMed]
- Veale, C.G.L. Unpacking the Pathogen Box-An Open Source Tool for Fighting Neglected Tropical Disease. ChemMedChem 2019, 14, 386–453. [Google Scholar] [CrossRef] [PubMed]
- MMV Pathogen Box Supporting Information. Available online: https://www.mmv.org/mmv-open/pathogen-box/pathogen-box-supporting-information (accessed on 27 May 2022).
- Parmentier, Y.; Pothier, C.; Hewitt, N.; Vincent, L.; Caradec, F.; Liu, J.; Lin, F.; Trancart, M.-M.; Guillet, F.; Bouaita, B.; et al. Direct and quantitative evaluation of the major human CYP contribution (fmCYP) to drug clearance using the in vitro Silensomes (TM) model. Xenobiotica 2019, 49, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, Y.; Yang, G.; Byun, S.; Rao, G.; Shoen, C.; Yang, H.; Gulati, A.; Crick, D.C.; Cynamon, M.; et al. Oxa, Thia, Heterocycle, and Carborane Analogues of SQ109: Bacterial and Protozoal Cell Growth Inhibitors. ACS Infect. Dis. 2015, 1, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Santos, P.; Li, K.; Lameira, L.; de Carvalho, T.M.U.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a New Drug Lead for Chagas Disease. Antimicrob. Agents Chemother. 2015, 59, 1950–1961. [Google Scholar] [CrossRef]
- Nyagwange, J.; Awino, E.; Tijhaar, E.; Svitek, N.; Pelle, R.; Nene, V. Leveraging the Medicines for Malaria Venture malaria and pathogen boxes to discover chemical inhibitors of East Coast fever. Int. J. Parasitol. Drug 2019, 9, 80–86. [Google Scholar] [CrossRef]
- Vanveller, B.; Aronoff, M.R.; Raines, R.T. A Divalent Protecting Group for Benzoxaboroles. RSC Adv. 2013, 44, 21331–21334. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nocentini, A.; Cadoni, R.; Dumy, P.; Supuran, C.T.; Winum, J.Y. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J. Enzym. Inhib. Med. Chem. 2017, 33, 286–289. [Google Scholar] [CrossRef]
- Cheng, X.; Hochlowski, J.; Tang, H.; Hepp, D.; Beckner, C.; Kantor, S.; Schmitt, R. Studies on repository compound stability in DMSO under various conditions. J. Biomol. Screen. 2003, 8, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L.; Carter, G.T. In vitro solubility assays in drug discovery. Curr. Drug Metab. 2008, 9, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Taliani, S.; Pugliesi, I.; Barresi, E.; Salerno, S.; Marchand, C.; Agama, K.; Simorini, F.; La Motta, C.; Marini, A.M.; Di Leva, F.S.; et al. Phenylpyrazolo[1,5-a]quinazolin-5(4H)-one: A suitable scaffold for the development of noncamptothecin topoisomerase I (Top1) inhibitors. J. Med. Chem. 2013, 56, 7458–7462. [Google Scholar] [CrossRef][Green Version]
- Costa-Silva, H.M.; Resende, B.C.; Umaki, A.C.S.; Prado, W.; da Silva, M.S.; Virgílio, S.; Macedo, A.M.; Pena, S.D.J.; Tahara, E.B.; Tosi, L.R.O.; et al. DNA Topoisomerase 3alpha Is Involved in Homologous Recombination Repair and Replication Stress Response in Trypanosoma cruzi. Front. Cell Dev. Biol. 2021, 9, 633195w. [Google Scholar] [CrossRef]


{kind=link}
| Compound | IC50 (T.b. brucei) (Emax %) | Chemical Class | IC50 (T. cruzi) (Emax %) | IC50 HEK 2 (HEP) 1 | SI (T.b. brucei) HEK (HEP) | SI (T. cruzi) 3T3 3 | SI (T. cruzi) HEK (HEP) |
|---|---|---|---|---|---|---|---|
| MMV 688372 | 0.014 ± 0.0024 (99) | substituted 2-phenylimidazopyridine | 0.13 ± 0.0097 (99) | 1.8 ± 0.0502 (3.6 ± 0.54) | 129 (257) | >561 | 14 (28) |
| MMV 688550 | 0.025 ± 0.00083 (99) | imidazo [1,2] purine | 0.53 ± 0.110 (100) | NA (NA) | >3160 (>3160) | >138 | >149 (>149) |
| MMV 652003 | 0.092 ± 0.0027 (100) | benzoxaborole | 0.15 ± 0.004 (100) | 6.8 ± 0.204 (50%) | 74 (>862) | >487 | 45 (>548) |
| MMV 688797 | 0.097 ± 0.0021 (99) | 2-aryl oxazole | 0.51 ± 0.11 (100) | NA (NA) | >814 (>814) | >143 | >154 (>154) |
| MMV 688795 | 0.13 ± 0.052 (99) | 2-aryl oxazole | 0.58 ± 0.0503 (100) | NA (NA) | >608 (>608) | >126 | >136 (>136) |
| MMV 688796 | 0.12 ± 0.023 (99) | 2,4-substituted furan | 1.2 ± 0.4 (100) | NA (NA) | >658 (>658) | >61 | >66 (>66) |
| MMV 689028 | 0.12 ± 0.030 (99) | benzyl piperazine | 0.70 ± 0.13 (100) | NA (NA) | >658 (>658) | >104 | >113 (>113) |
| MMV 688958 | 0.14 ± 0.028 (99) | 2-aryl oxazole | 0.98 ± 0.11 (100) | NA (NA) | >564 (>564) | >75 | >81 (>81) |
| MMV 688776 | 0.79 ± 0.050 (99) | pyrazoloquinazoline | 3.1 ± 0.73 (87) 5 | NA (NA) | >100 (>100) | >24 | >25 (>25) |
| MMV 688467 | 0.31 ± 0.0069 (99) | butyl sulfanilamide | 2.3 ± 0.46 (98) | 102% (23 ± 3.8) | >254 (74) | >32 | >34 (10) |
| MMV 689029 | 0.42 ± 0.053 (99) | benzyl piperazine | 1.2 ± 0.067 (100) | NA (NA) | >188 (>188) | >61 | >66 (>66) |
| MMV 687248 | 0.51 ± 0.045 (99) | 3,5-disubstituted pyridine | 1.5 ± 0.13 (99) | NA (19.6 ± 0.083) | >155 (38) | >49 | 53 (13) |
| MMV 099637 | 2.13 ± 0.064 (99) | pyrazolopyridine | 2.7 ± 0.41 (70) 5 | NA (NA) | >37 (>37) | >27 | >29 (>29) |
| MMV 028694 | 0.83 ± 0.057 (100) | 2,4-disubstituted pyrimidine | 9.4 ± 1.8 (98) | 9.7 ± 0.050 (NT) | 12 | >7.8 | 1.0 (NT) |
| MMV 687776 | 4.3 ± 1.3 (99) | benzoxaborole | 1.6 ± 0.38 (100) | 13 ± 1.6 (89%) | 3.0 (>18) | >46 | 8.1 (>50) |
| MMV 688283 | 7.6 ± 0.27 (99) | 4-amino quinoline | 6.6 ± 1.2 (100) | 100% (109%) | >10 (>10) | >11 | >12 (>12) |
| MMV 006901 | 4.9 ± 0.0085 (99) | 2,4-aminoquinoline | 1.2 ± 0.11 (100) | 18 ± 0.81 (9.8 ± 1.5) | 3.7 (1.8) | >60 4 | 15 (7.1) |
| MMV 22029 | 1.5 ± 0.059 (99) | biaryl sulfonamide | 8.2 ± 0.69 (100) | 8.1 ± 0.054 (NT) | 5.6 | 1.2 | 1.0 (NT) |
| MMV 688514 | 1.50 ± 0.027 (99) | ND | 4.9 ± 0.16 (100) | 9.1 ± 0.46 (NT) | 6.0 | 2.7 | 1.9 (NT) |
| MMV 22478 | 1.5 ± 0.18 (98) | pyrazolo[1,5-a]pyrimidine | 5.9 ± 1.3 (100) | 12 ± 0.39 (NT) | 8.0 | 1.04 | 2.03 (NT) |
| MMV 21013 | 2.2 ± 0.088 (100) | 2-pyridyl-4-aminopyrimidine | 1.8 ± 0.49 (100) | 4.0 ± 0.22 (106%) | 1.8 (>36) | >41 4 | 2.2 (NT) |
| MMV 687706 | 2.2 ± 0.0028 (99) | 1-[3-(4-phenoxyphenyl)-1H-pyrazol-5-yl]piperazine | 11 ± 0.205 (99) | 16.2 ± 5.2 (NT) | 7.4 | >6.6 | 1.5 (NT) |
| Pentamidine | 0.0031 ± 0.0001 (100) | aromatic diamidine | NT | NA at 0.67 µM (NT) | >216 | NT | NT |
| Diminazene aceturate | 0.037 ± 0.00041 (100) | phenylhydrazine | NT | NA at 38 µM (NT) | >1027 | NT | NT |
| Amphotericin B | 1.5 ± 0.088 (100) | polyene macrolide | NT | NA at 4.0 µM (NT) | >2.6 | NT | NT |
| Posaconazole | NT | triazole | 0.0055 ± 0.0000041 (77) 5 | NT (NA at 1 µM) | NT | >333 | NT |
| Benznidazole | NT | nitroheterocyclic | 3.4 ± 0.71 (100) | NT (NA at 127 µM) | NT | >37 | NT |
| Nifurtimox | NT | nitroheterocyclic | 0.85 ± 0.048 (100) | NT (74% at 127 µM) | NT | >149 | NT |
| Puromycin | 0.063 ± 0.00041 (100) | peptidyl nucleoside | 5.6 ± 0.011 (100) | 0.73 ± 0.27 (0.25 ± 0.015) | 12 (4.0) | 1.2 | 0.13 (0.044) |
| Compound | IC50 24 h T. cruzi (Emax) | IC50 72 h T. cruzi (Emax) | IC50 Wash Off (Emax, Fold Change 1) | IC50 24 h T.b. brucei (Emax) | IC50 48 h T.b. brucei (Emax) | Set 2 | Speed of Action 3 |
|---|---|---|---|---|---|---|---|
| MMV 688550 | 0.90 ± 0.17 (80) | 0.44 ± 0.089 (100) | 0.31 ± 0.067 (100; 0.58) | 0.022 ± 0.014 (100) | 0.021 ± 0.00053 (99) | Kinetoplastid | FA Cidal |
| MMV 652003 | 1.28 ± 0.56 (93) | 0.28 ± 0.18 (100) | 0.51 ± 0.080 (99; 3.4) | 0.45 ± 0.067 (100) | 0.11 ± 0.012 (100) | Kinetoplastid | FA Static4 |
| MMV 688797 | 0.89 ± 0.135 (86) | 0.43 ± 0.042 (99) | 0.38 ± 0.056 (99; 1.3) | 0.15 ± 0.029 (99) | 0.10 ± 0.0062 (99) | Kinetoplastid | FA Cidal |
| MMV 688795 | 1.3 ± 0.12 (89) | 0.62 ± 0.05 (99) | 0.38 ± 0.056 (99; 0.74) | 0.29 ± 0.0047 (99) | 0.106 ± 0.020 (99) | Kinetoplastid | FA Cidal |
| MMV 688796 | 2.4 ± 1.3 (91) | 1.02 ± 0.057 (100) | 1.1 ± 0.48 (98; 1.1) | 0.24 ± 0.053 (82) | 0.097 ± 0.0048 (100) | Kinetoplastid | FA Cidal |
| MMV 689028 | 1.1 ± 0.43 (91) | 0.52 ± 0.13 (100) | 0.39 ± 0.020 (99; 0.86) | 0.72 ± 0.38 (78) | 0.11 ± 0.0032 (99) | Kinetoplastid | FA Cidal |
| MMV 688958 | 1.74 ± 0.18 (94) | 0.601 ± 0.093 (100) | 0.601 ± 0.093 (100; 0.73) | 0.201 ± 0.028 (78) | 0.14 ± 0.017 (99) | Kinetoplastid | FA Cidal |
| MMV 689029 | 3.7 ± 0.26 (90) | 1.3 ± 0.28 (99) | 0.88 ± 0.056 (98; 0.73) | 0.98 ± 0.34 (89) | 0.40 ± 0.023 (100) | Kinetoplastid | FA Cidal |
| MMV 687248 | 4.1 ± 1.3 (100) | 3.5 ± 0.16 (100) | 2.0 ± 0.039 (100; 1.3) | 0.93 ± 0.047 (87) | 0.2 ± 0.032 (99) | Tuberculosis | FA Cidal |
| MMV 688372 | 1.6 ± 0.064 (75) | 0.12 ± 0.052 (100) | 0.13 ± 0.0029 (100; 0.86) | 0.034 ± 0.0020 (83) | 0.013 ± 0.00032 (100) | Kinetoplastid | SA (TC) Cidal |
| MMV 688467 | 4.4 ± 0.73 (75) | 2.5 ± 1.4 (99) | 2.10 ± 0.44 (100; 0.91) | 1.2 ± 0.35 (88) | 0.46 ± 0.018 (100) | Kinetoplastid | SA (TC) Cidal |
| MMV 028694 | 58% at 73 µM | 16 ± 1.70 (100) | 12 ± 1.8 (98; 1.2) | 0.70 ± 0.19 (87) | 0.71 ± 0.13 (100) | Malaria | SA (TC) Cidal |
| MMV 687776 | 94% at 73.3 µM | 1.3 ± 0.30 (99) | 1.6 ± 0.064 (100; 1.0) | 5.35 ± 0.103 (89) | 4.04 ± 1.78 (99) | Lymphatic filiariasis | SA (TC) Cidal |
| MMV 688283 | 58% at 73 µM | 7.9 ± 0.51 (96) | 7.6 ± 1.5 (100; 1.2) | 7.8 ± 0.32 (94) | 6.2 ± 0.45 (96) | Kinetoplastid | SA (TC) Cidal |
| MMV 688776 | 82% at 49 73 µM | 2.43 ± 0.023 (820) | 3.5 ± 0.101 (87; 1.1) | 2.3 ± 0.17 (60) | 0.86 ± 0.14 (99) | Kinetoplastid | SA (TC, TBB) Not efficacious (TC) 4 |
| MMV 099637 | NA at 73 µM | 2.41 ± 0.070 (65) | 3.3 ± 0.045 (92; 1.2) | 2.6 ± 0.11 (50) | 2.2 ± 0.13 (99) | Kinetoplastid | SA (TC, TBB) Not efficacious (TC) 4 |
| Pentamidine | NT | NT | NT | 0.012 ± 0.0021 (99) | 0.0002 ± 0.0000021 (100) | - | FA Cidal |
| Diminazene aceturate | NT | NT | NT | 0.21 ± 0.058 (99) | 0.070 ± 0.027 (100) | - | FA Cidal |
| Amphotericin B | NT | NT | NT | 0.88 ± 0.163 (100) | 0.26 ± 0.0016 (100) | - | FA Cidal |
| Posaconazole | NA at 1 µM | 0.0035 ± 0.000065 (97) 4 | 0.0018 ± 0.000078 (100; 0.36) | NT | NT | - | SA Not efficacious (TC) 4 |
| Benznidazole | 4.7 ± 1.4 (100) | 5.6 ± 0.0021 (100) | 1.2 ± 0.11 (100; 0.29) | NT | NT | - | FA Cidal |
| Nifurtimox | 1.03 ± 0.076 (100) | 0.85 ± 0.088 (100) | 0.35 ± 0.014 (100; 0.60) | NT | NT | - | FA Cidal |
| Puromycin | 3.5 ± 0.44 (98101) | 2.55 ± 0.47 (100) | 2.8 ± 0.13 (100; 0.50) | 0.096 ± 0.013 (100) | 0.097 ± 0.00042 (100) | - | FA Cidal |
| Compound | Chemical Structure | Chemical Class | IC50 T. bR1 1 | IC50 T. Bg 2 STIB930 | IC50 T.b. brucei 3 |
|---|---|---|---|---|---|
| MMV 688550 |  | imidazo [1,2] purine | 0.080 ± 0.0014 | <0.001 | 0.025 ± 0.00083 |
| MMV 652003 1 |  | benzoxaborole | NT | NT | 0.092 ± 0.0027 |
| MMV 688797 2 |  | 2-aryl oxazole | NT | NT | 0.097 ± 0.0021 |
| MMV 688795 |  | 2-aryl oxazole | 0.079 ± 0.00070 | 0.63 ± 0.0014 | 0.13 ± 0.052 |
| MMV 688958 |  | 2-aryl oxazole | 0.067 ± 0.0028 | 0.65 ± 0.050 | 0.14 ± 0.028 |
| MMV 688796 |  | 2,4-substituted furan | 0.046 ± 0.031 | 0.16 ± 0.011 | 0.12 ± 0.023 |
| MMV 689028 |  | benzyl piperazine | 0.28 ± 0.011 | 0.048 ± 0.0011 | 0.12 ± 0.030 |
| MMV 689029 |  | benzyl piperazine | 0.63 ± 0.11 | 0.40 ± 0.12 | 0.42 ± 0.053 |
| MMV 687248 |  | 3,5-disubstituted pyridine | 1.2 ± 0.54 | 0.24 ± 0.022 | 0.51 ± 0.039 |
| Melarsoprol |  | substituted aniline | 0.015 ± 0.0040 | 0.011 ± 0.017 | 0.0031 ± 0.00019 |
| Pentamidine |  | aromatic diamidine | NT | 0.0030 ± 0.0020 | NT |
| Compound | Mwt | LogP 1 | Pathogen Box Set | Commercially Available |
|---|---|---|---|---|
| MMV 688550 | 437.49 | 4.6 | Kinetoplastid | No |
| MMV 652003 | 321.06 | NC | Kinetoplastid | No |
| MMV 688797 | 331.38 | 3 | Kinetoplastid | Yes |
| MMV 688795 | 297.39 | 3.6 | Kinetoplastid | No |
| MMV 688796 | 354.38 | 2.5 | Kinetoplastid | Yes |
| MMV 689028 | 474.57 | 3.3 | Kinetoplastid | No |
| MMV 688958 | 308.40 | 3.9 | Kinetoplastid | Yes |
| MMV 689029 | 449.62 | 2.9 | Kinetoplastid | No |
| MMV 687248 | 320.32 | 3.3 | Tuberculosis | No |
| Melarsoprol | 398.341 | 1.3 | Not in set (control) | Yes |
| Pentamidine | 340.42 | 2.3 | Not in set (control) | Yes |
| Compound/s | Chemical Class | Speed of Action | Potential Liability | Recommendation | References | New Class |
|---|---|---|---|---|---|---|
| MMV652003 | benzoxaborole | Fast | Static action 1 | Identify target/s (T. cruzi) | [4,42,44,45,46,47] | No |
| MMV688797 MMV688958 MMV688795 | 2-aryl oxazole | Fast | Metabolic Stability 2 | Identify target/s Improve PK 2 | [24,48] | No |
| MMV688796 | 2,4-substituted furan | Fast | - | In vivo studies | - | Yes |
| MMV688550 | imidazo [1,2] purine | Fast | DMPK 3 | In vivo studies 4 Improve PK 3 | - | Yes |
| MMV689028 MMV689029 | benzyl piperazine | Fast | DMPK 3 | In vivo studies 4 Improve PK 3 | - | Yes |
| MMV687248 | 3,5-disubstituted pyridine | Fast | Selectivity T. cruzi 1 | Improve moderate selectivity (SAR) | - | Yes |
| MMV688372 | substituted 2-phenylimidazopyridine | Slow | Selectivity T. cruzi 1 | Improve moderate selectivity (SAR) | - | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sykes, M.L.; Kennedy, E.K.; Read, K.D.; Kaiser, M.; Avery, V.M. Temporal and Wash-Out Studies Identify Medicines for Malaria Venture Pathogen Box Compounds with Fast-Acting Activity against Both Trypanosoma cruzi and Trypanosoma brucei. Microorganisms 2022, 10, 1287. https://doi.org/10.3390/microorganisms10071287
Sykes ML, Kennedy EK, Read KD, Kaiser M, Avery VM. Temporal and Wash-Out Studies Identify Medicines for Malaria Venture Pathogen Box Compounds with Fast-Acting Activity against Both Trypanosoma cruzi and Trypanosoma brucei. Microorganisms. 2022; 10(7):1287. https://doi.org/10.3390/microorganisms10071287
Chicago/Turabian StyleSykes, Melissa L., Emily K. Kennedy, Kevin D. Read, Marcel Kaiser, and Vicky M. Avery. 2022. "Temporal and Wash-Out Studies Identify Medicines for Malaria Venture Pathogen Box Compounds with Fast-Acting Activity against Both Trypanosoma cruzi and Trypanosoma brucei" Microorganisms 10, no. 7: 1287. https://doi.org/10.3390/microorganisms10071287
APA StyleSykes, M. L., Kennedy, E. K., Read, K. D., Kaiser, M., & Avery, V. M. (2022). Temporal and Wash-Out Studies Identify Medicines for Malaria Venture Pathogen Box Compounds with Fast-Acting Activity against Both Trypanosoma cruzi and Trypanosoma brucei. Microorganisms, 10(7), 1287. https://doi.org/10.3390/microorganisms10071287