Abstract
Weathered crust elution-deposited rare earth ores represent a significant source of medium and heavy rare earth elements. Their mineralization mechanism fundamentally involves the adsorption and enrichment of rare earth ions on the surfaces of secondary minerals within the weathering crust. To elucidate the role of nano-sized iron oxide–clay mineral composites in the migration and enrichment of rare earth elements (REEs) in weathered crusts, α-Fe2O3, oolitic hematite, Fe2O3-kaolinite, and FeOOH-kaolinite composites were selected as adsorbents. Using La3+, Ce3+, and Y3+ as target ions, their adsorption performance and fractionation mechanisms were systematically investigated through batch adsorption experiments combined with characterization techniques including Langmuir/Freundlich model fitting, Zeta potential, XPS, and FT-IR. The results demonstrate that the adsorption of REEs on iron oxides and their composites follows the Langmuir model (R2 > 0.96), indicating monolayer homogeneous adsorption. The FeOOH-kaolinite composite exhibited the highest adsorption capacity (0.6531 mmol/g for Ce). Amorphous iron oxide–clay mineral composites showed superior complexation adsorption and enrichment capabilities due to their higher oxygen vacancy concentration (Oβ/Oα = 3.86 for FeOOH-kaolinite), whereas crystalline iron oxide–clay mineral composites contributed significantly to REE fractionation and enrichment. Among the influencing factors, elevated temperature (20–60 °C) favored the adsorption process, with an activation energy > 4.2 kJ/mol indicating chemical adsorption dominance. Within the pH range of 3.0–7.0, adsorption capacity increased with rising pH, with Ce being more sensitive to ionic strength and pH variations. The adsorption mechanism was further elucidated through post-adsorption XPS, FT-IR in the low-wavenumber region, and desorption studies. This study confirms that iron oxide–clay mineral composites regulate the enrichment and fractionation of REEs in weathered crusts through the interplay of their surface properties and environmental factors, providing a scientific basis for refining the metallogenic theory and guiding the exploration and development of weathered crust elution-deposited rare earth ore resources.
1. Introduction
Rare earth elements (REEs) are indispensable strategic resources for high-tech industries [1]. Weathered crust elution-deposited REE ores, characterized by complete fractionation patterns and heavy REE enrichment, are formed primarily through the liberation of REEs from parent rocks and their subsequent adsorption onto secondary minerals [2,3,4,5,6]. Understanding these adsorption mechanisms is fundamental for advancing ore genesis theories and optimizing resource exploitation.
Current research on REE migration and enrichment in weathering crusts indicates that under weathering conditions, readily weathered REE-bearing minerals decompose, and REEs undergo activation and leaching, ultimately enriching mainly in ionic forms on secondary minerals such as clay minerals and iron-manganese oxides. Clay minerals, as the principal clay-sized components of weathering crusts, are widely recognized as important hosts for REEs [6,7,8]. While clay minerals (e.g., kaolinite, halloysite) adsorb REEs mainly via non-selective ion exchange limited by their cation exchange capacity (CEC) [8,9,10], iron-manganese oxides play a pivotal role due to their high surface hydroxyl densities and large specific surface areas [11,12]. These oxides enable efficient REE capture through surface complexation and drive significant fractionation, such as the oxidative scavenging of Ce [13,14,15,16,17,18].
Recent studies highlight that REE enrichment is further regulated by iron oxide–clay mineral composites (e.g., kaolinite-hematite, halloysite-goethite associations) [19,20]. These composites, formed via cementation or surface coating, exhibit distinct physicochemical properties and enhanced adsorption capacities compared to individual minerals [21,22,23]. Their formation and reactivity are strictly controlled by environmental conditions like pH and redox potential, which influence the crystallinity and distribution of iron oxides within the weathering crust [24,25,26].
However, several key issues regarding the mechanisms of iron oxide–clay mineral composites in REE enrichment remain unresolved. The quantitative differences in adsorption selectivity among composites with varying iron oxide crystallinities are poorly established. The regulatory mechanisms of environmental factors (e.g., pH, ionic strength) on composite-mediated adsorption require deeper investigation. The specific contribution of these composites to REE fractionation remains unclear. To address these issues, this study selects typical iron oxides (α-Fe2O3, FeOOH) and their kaolinite composites as research subjects to systematically conduct adsorption experiments on REE ions. By integrating modern analytical techniques with adsorption experiments, it focuses on investigating the influence of factors such as iron oxide crystallinity, surface characteristics, and their composite effects with clay minerals on REE adsorption capacity and fractionation behavior. The aim is to reveal the mechanism of iron oxide–clay mineral composites in the REE enrichment and ore-forming process at the microscopic level, providing an important scientific basis for improving the metallogenic theory of weathered crust elution-deposited rare earth ores and offering guidance for the prospecting, evaluation, and sustainable development and utilization of REE resources.
2. Materials and Methods
2.1. Adsorbents and Reagents
The adsorbents used in this study comprised two iron oxide pure minerals (α-Fe2O3 and oolitic hematite) and two laboratory-simulated iron oxide-kaolinite composites (Fe2O3-kaolinite and FeOOH-kaolinite). The iron oxide pure minerals were sourced from actual mines and exhibited a high degree of crystallinity. The composites were synthesized according to a method described in the literature [14]. The adsorbates were solutions of La3+, Ce3+, and Y3+, prepared by dissolving their corresponding rare earth nitrates in deionized water. The rare earth nitrate salts—La(NO3)3·6H2O, Ce(NO3)3·6H2O, and Y(NO3)3·6H2O—were purchased from Aladdin Reagent (analytical grade, purity ≥ 99.9%).
2.2. Batch Adsorption Experiments
Batch adsorption experiments were conducted under isothermal conditions at 25 °C. Precisely 50 mL of rare earth ion solutions with varying initial concentrations (3–40 mmol/L) were transferred into conical flasks. Then, 1.0 g of adsorbent was added to each flask, resulting in a liquid-to-solid ratio of 50:1. The mixtures were shaken in a constant-temperature incubator shaker at a constant speed for 10 h to ensure adsorption equilibrium was reached. After the reaction, solid–liquid separation was achieved using a high-speed centrifuge operated at 9000 r/min for 5 min. The supernatant was collected for subsequent determination of the equilibrium rare earth ion concentration. All batch adsorption experiments were conducted in triplicate. The mean values are reported, with standard deviations consistently below 5%. The pH of the solutions was adjusted using 0.1 M HNO3 and 0.1 M NaOH, with continuous monitoring using a pH meter.
To gain deeper insight into the adsorption mechanism, the experimental data were fitted using both the Langmuir and Freundlich adsorption isotherm models. This analysis aimed to elucidate the nature of the interaction between the rare earth ions and the surface sites of the adsorbents.
2.3. Material Characterization
A suite of characterization techniques was employed to systematically investigate the physicochemical properties of the adsorbents and elucidate the underlying adsorption mechanisms.
The micro-morphology and surface elemental distribution of the samples were observed using field-emission scanning electron microscopy (FESEM; GeminiSEM 300, Carl Zeiss, Oberkochen, Germany). The specific surface area and pore-size distribution were determined via nitrogen adsorption–desorption measurements on a Brunauer–Emmett–Teller (BET) surface area analyzer (ASAP 2460, Micromeritics, Norcross, GA, USA). Surface charge characteristics under different pH conditions were evaluated with a Zeta potential analyzer (Zetasizer Ultra, Malvern Panalytical, UK). The chemical states of surface elements were analyzed by X-ray photoelectron spectroscopy (XPS; ESCALAB XI+, Thermo Fisher Scientific, Waltham, MA, USA), while functional-group variations were examined by Fourier-transform infrared spectroscopy (FT-IR; Nicolet 6700, Thermo Fisher Scientific, USA) in transmission mode using KBr pellets (spectral range 400–4000 cm−1).
Additional instrumental analyses included: X-ray diffraction (XRD; D8 ADVANCE, Bruker, Karlsruhe, Germany) for phase identification; Laser particle-size analysis (S3500 series, Microtrac, York, PA, USA) for particle-size distribution.
Solution pH was adjusted with 0.1 M HNO3 or NaOH and monitored with a pH meter (Mettler Toledo, Greifensee, Switzerland). All adsorption experiments were conducted in a constant-temperature incubator shaker (SHZ-82A) with temperature control accuracy of ±0.3 °C.
3. Results and Discussion
3.1. Morphology and Adsorption Performance of Iron Oxides and Their Clay Mineral Composites
3.1.1. Microstructure of Iron Oxides and Their Composites
In this study, kaolinite, a primary clay mineral, served as a substrate for loading iron oxides with different structures to simulate their composites for investigating rare earth element (REE) adsorption. Figure 1 presents the field emission scanning electron microscopy (FESEM) images of pure α-Fe2O3 and various iron oxide-kaolinite composites.
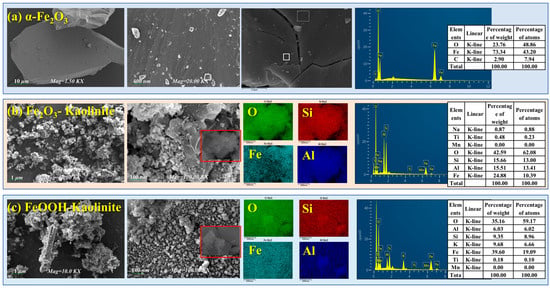
Figure 1.
FESEM and EDS spectra of iron oxides and their composites: (a) α-Fe2O3, (b) Fe2O3-Kaolinite, and (c) FeOOH-Kaolinite.
As shown in Figure 1, compared to the smooth euhedral platy layers and dense cryptocrystalline massive structure of pure α-Fe2O3, the FESEM images of the synthesized iron oxide-kaolinite composites clearly reveal the presence of granular particles with varied shapes and sizes on the layered kaolinite surface. The particle sizes range approximately from 50 to 100 nm, resulting from the accumulation of iron oxide nanoparticles formed during the crystallization process. Figure 1c shows that the FeOOH-kaolinite composite exhibits distinct rod-shaped particles with relatively larger sizes, which is a typical morphology of FeOOH. The iron oxides primarily combine with kaolinite, forming a “vermiform structure.” Unlike the FeOOH-kaolinite composite, where nanoparticles concentrate on the external surface, the surface particles of the Fe2O3-kaolinite composite are mainly located within the interlayer micro-environment, predominantly adhering closely to the kaolinite surface as spherical microcrystals. Energy-dispersive X-ray spectroscopy (EDS) analysis indicates surface elemental compositions of approximately 60.0 wt% O, 6.0–15.5 wt% Al, and 9.3–15.7 wt% Si, likely related to the parent composition of kaolinite. Additionally, the Fe content on the kaolinite surface ranges from 24.8 wt% to 39.6 wt%. These results further confirm the successful loading of iron oxide nanoparticles with different morphologies on the kaolinite surface, providing additional reactive sites.
3.1.2. BET Analysis of Iron Oxides and Their Clay Mineral Composites
The N2 adsorption–desorption isotherms of the iron oxides and their composites were investigated to determine their specific surface areas and total pore volumes. All samples were degassed at 150 °C for 12 h before analysis. The corresponding BET analysis results are shown in Figure 2 and Table 1.
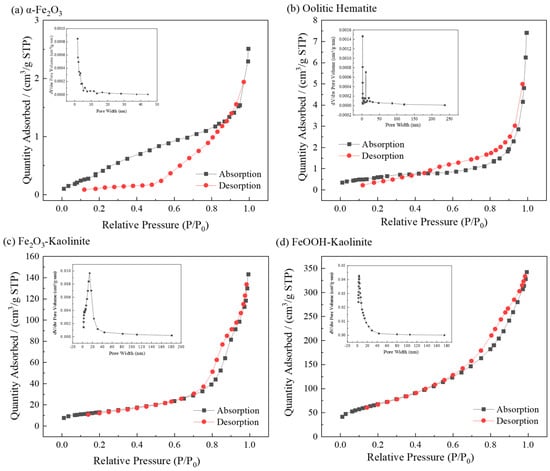
Figure 2.
Nitrogen adsorption and desorption isotherms and pore size distribution of iron oxides and their composites: (a) α-Fe2O3, (b) Oolitic hematite, (c) Fe2O3-Kaolinite, and (d) FeOOH-Kaolinite.

Table 1.
BET parameters of iron oxides and their composites.
The isotherms of α-Fe2O3 and oolitic hematite exhibit features consistent with Type II behavior according to the IUPAC classification, indicating non-porous or macroporous materials [27]. However, the extremely low specific surface areas (≈2.2 m2/g) place these measurements near the instrument’s detection limit, which may lead to less distinct hysteresis loops. Alternatively, the isotherms can also be interpreted as Type IVa with H2b hysteresis, suggesting some mesoporosity, which is supported by the pore-size distributions showing significant contributions in the 2–50 nm range.
The Fe2O3-Kaolinite composite shows a more defined Type IVa isotherm with H2b hysteresis, indicating the development of a mesoporous structure upon composite formation. The FeOOH-Kaolinite composite exhibits a clear Type IVa isotherm with a pronounced H2b hysteresis loop, characteristic of well-developed mesoporosity and interconnected pore networks. BET surface areas follow the order: α-Fe2O3 ≈ oolitic hematite (≈2.2 m2/g) < Fe2O3-Kaolinite (47.1 m2/g) < FeOOH-Kaolinite (244.5 m2/g), with pore volumes showing a corresponding trend. Pore sizes are predominantly in the mesoporous range (2–50 nm), with most below 20 nm. Generally, adsorbents with larger surface areas and denser pore structures exhibit superior adsorption capacities.
3.1.3. XRD Analysis of Iron Oxides and Their Composites
The crystal structures of hematite, hematite-kaolinite composite, and goethite-kaolinite composite were analyzed using X-ray diffraction (XRD) under the following conditions: copper target (Cu-Kα radiation), scanning angle range of 10° to 90°, and scanning speed of 2.4°/min. The XRD patterns of the adsorbents are presented in Figure 3.
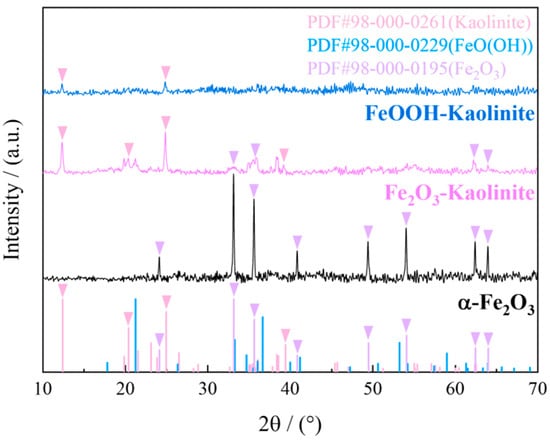
Figure 3.
XRD spectrum of iron oxides and their composites.
As shown in Figure 3, the XRD pattern of hematite exhibits distinct characteristic peaks at 28.5°, 29.5°, and 30.5°, which correspond well to the standard diffraction card (PDF#98-000-0195) of α-Fe2O3, confirming its high crystallinity. Similarly, the hematite-kaolinite composite displays prominent peaks at 32.5°, 33.5°, and 34.5°, indicating the presence of crystalline α-Fe2O3 phases along with the kaolinite substrate (PDF#98-000-0261). In contrast, the XRD pattern of the goethite-kaolinite composite shows only a broad and weak hump in the range of 37.5–39.5°, without any sharp diffraction peaks. This diffuse scattering feature is characteristic of amorphous or poorly crystalline iron oxides, suggesting that the iron oxide phase in the FeOOH-kaolinite composite exists predominantly in an amorphous state. The absence of well-defined peaks further supports the low crystallinity of the FeOOH component, which aligns with its expected structural nature as a hydrous iron oxide with high surface reactivity.
3.1.4. Particle Size Distribution Analysis of Iron Oxides and Their Composites
Laser particle size analysis of iron oxides and its kaolinite composites (Figure 4) revealed a distinct bimodal particle-size distribution, indicating the coexistence of micrometer-sized clay aggregates and surface-attached iron-oxide nanoparticles. The D50 values were 175.82 μm for pure hematite, 60.06 μm for the hematite-kaolinite composite, and 50.88 μm for the goethite-kaolinite composite. The micron-scale peaks correspond to the overall size of clay-based aggregates carrying nanoscale iron-oxide coatings, rather than the intrinsic size of the oxides themselves. This macro-scale measurement complements the nanoscale morphology observed by SEM, which clearly shows iron-oxide nanoparticles adhering to clay surfaces. The apparent discrepancy between the two techniques reflects their different observational scales—SEM resolves surface nanostructures, while laser diffraction captures the larger aggregated architectures. Thus, the composites exhibit a hierarchical structure: nanoscale iron-oxide particles are anchored onto clay surfaces, while the bulk material remains a macroscopic physical mixture. This configuration combines the structural integrity of clay minerals with the high surface reactivity of iron oxides, contributing to their enhanced adsorption capacity for rare-earth ions.
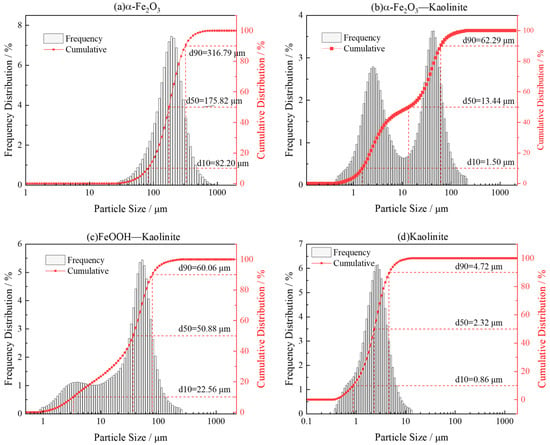
Figure 4.
Particle size distribution analysis of iron oxides and their composites: (a) α-Fe2O3, (b) α-Fe2O3-Kaolinite, (c) FeOOH-Kaolinite, and (d) Kaolinite.
3.2. Static Adsorption of Rare Earth Ions on Iron Oxides and Their Composites
3.2.1. Adsorption Isotherm and Kinetic Models
The Langmuir and Freundlich isotherm models are two of the most commonly used mathematical models in adsorption studies. The Langmuir model assumes homogeneous distribution of adsorption sites with no interactions between adsorbed molecules, and it describes monolayer adsorption equilibrium. The Freundlich model represents monolayer adsorption on heterogeneous surfaces [28]. Adsorption kinetic models are employed to analyze the rate-controlling steps of the adsorption process. Commonly used models include the pseudo-first-order, pseudo-second-order, intra-particle diffusion, and Elovich models. In this study, the above isotherm and kinetic models were applied to fit the adsorption behavior of rare earth ions on iron oxide–clay mineral composites, in order to identify the most appropriate model for describing the adsorption process.
3.2.2. Adsorption Isothermal Model for the Adsorption of Rare Earth Ions by Iron Oxides and Their Composites
To investigate the adsorption isotherm model for the interaction of rare earth ions with the surface of each adsorbent, static adsorption experiments were carried out using different iron oxides and their composites as adsorbents for different initial concentrations of rare earth ion solutions. Two adsorption isotherm models were fitted to the adsorption experimental data. The results are shown in Figure 5 and Table 2.
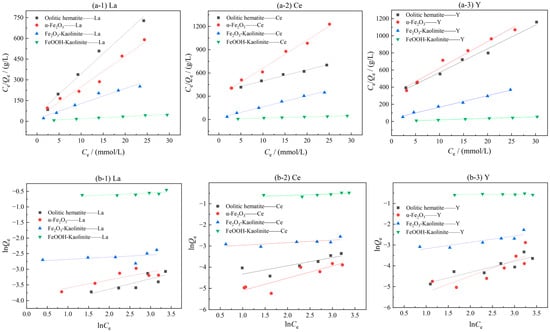
Figure 5.
Isothermal model fitting of iron oxidation and its complex adsorption of rare earth ions: (a-1–a-3) Langmuir model; (b-1–b-3) Freundlich model.
Table 2.
Adsorption isothermal model parameters.
As can be seen from Figure 5 and Table 2, the adsorption process of rare earth ions on iron oxide and its clay mineral complex is more consistent with the Langmuir adsorption isotherm model than the Freundlich adsorption isotherm model, and the fitting coefficients all reach above 0.96, indicating that the adsorption process of rare earth ions on iron oxide and its clay mineral complex is consistent with monolayer adsorption, and the adsorption capacity depends on the number of active adsorption sites on the iron oxide and its clay mineral complex. The parameter 1/n in the Freundlich adsorption isotherm model represents the inhomogeneity or adsorption intensity of the adsorbent surface, and when the value of 1/n is in the range of 0–1, it indicates that the adsorption process is more consistent with monolayer adsorption [29].
3.3. Influence of Various Factors on the Adsorption of Rare Earth Ions by Iron Oxides and Their Composites
3.3.1. Effect of Initial Concentration on the Adsorption of Rare Earth Ions by Iron Oxides and Their Composites
To investigate the effect of initial rare earth ion concentration on adsorption performance, the adsorption capacities of iron oxides and their composites were measured in solutions with varying initial concentrations (3–30 mmol/L) under conditions of a liquid-to-solid ratio of 50:1 and room temperature oscillation for 10 h (Figure 6). The results indicate that the initial concentration is a critical parameter governing adsorption behavior. In the low-concentration range, the equilibrium adsorption capacities of all adsorbents for La3+, Ce3+, and Y3+ increased with rising initial concentration. Beyond a certain threshold, the growth in adsorption capacity plateaued, indicating the near-saturation of available adsorption sites.
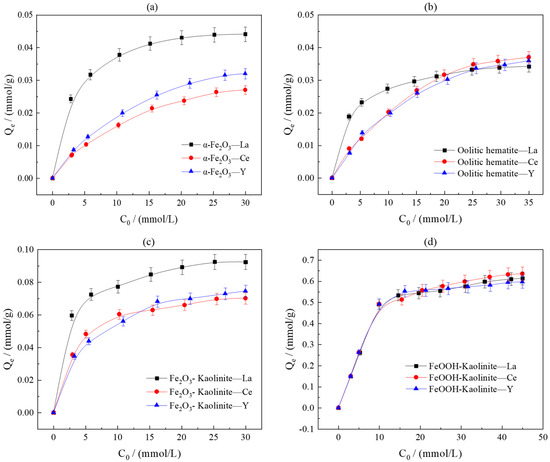
Figure 6.
Effect of initial concentration on adsorption of rare earth ions by iron oxides and their composites: (a) α-Fe2O3, (b) Oolitic hematite, (c) Fe2O3-Kaolinite, and (d) FeOOH-Kaolinite.
Detailed data analysis revealed that pure α-Fe2O3 exhibited a higher adsorption capacity for La3+ than oolitic hematite, suggesting that crystallinity and specific surface area are key structural parameters influencing adsorption capability. The adsorption capacities of Fe2O3-kaolinite and FeOOH-kaolinite composites were significantly higher than those of the pure minerals, with the FeOOH-kaolinite composite performing optimally. This confirms the synergistic enhancement between iron oxides and clay minerals in the adsorption process.
The saturated adsorption capacities for the three rare earth ions followed the order La3+ > Y3+ > Ce3+. The introduction of clay minerals did not alter this trend, indicating their limited influence on rare earth fractionation. However, differences in crystal structure led to distinct adsorption characteristics between the two types of composites: the FeOOH-kaolinite composite (low crystallinity) demonstrated superior overall adsorption capacity due to its higher specific surface area and abundant surface sites, whereas the Fe2O3-kaolinite composite (high crystallinity) exhibited greater adsorption disparity among the three rare earth ions, reflecting its stronger selectivity for rare earth fractionation.
3.3.2. Effect of pH and Ionic Strength on the Adsorption of Rare Earth Ions by Iron Oxides and Their Composites
In order to investigate the effects of solution pH and ionic strength on the adsorption effect of iron oxides and their composites, NaNO3 at 0.10 mol/L and 0.01 mol/L were used as two ionic strength background solutions of the solutions, and the initial concentrations of rare earth solutions were both 5 mmol/L. Under the conditions of liquid-solid ratio of 50:1 and room temperature of 25 °C, the adsorption performance of iron oxides and their composites were investigated under different pH conditions. The experimental results are shown in Figure 7.
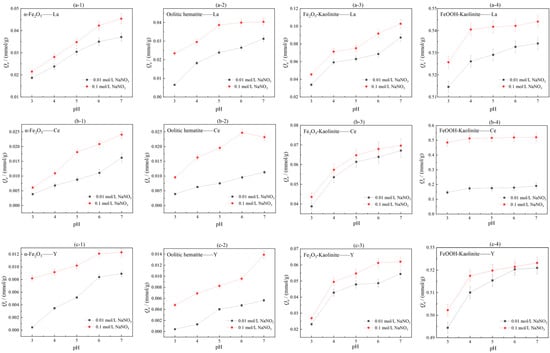
Figure 7.
Influence of ionic strength and pH on adsorption of rare earth ions by iron oxides and their composites: (a-1–a-4) La, (b-1–b-4) Ce, (c-1–c-4) Y.
As can be seen from Figure 7, the adsorption of rare earth ions by iron oxides and their composites increases with increasing pH in the range of pH = 3–7. This is due to the presence of a large amount of H+ in the acidic environment, which favors the surface protonation of iron oxides, and the free H+ will compete with the rare earth ions in solution for adsorption, leading to a decrease in the proportion of negatively charged groups that can complex with rare earths. This leads to a decrease in the adsorption of rare earth ions. In addition, iron oxides dissolve into the solution at low pH, leading to a decrease in adsorption capacity. As the pH increases, the H+ concentration in the solution gradually decreases, and the deprotonation of the surface of iron oxides and their clay mineral composites results in a weaker competitive adsorption and a significant increase in the adsorption of rare earth ions. The degree of adsorption/isostructural substitution of rare earth ions on iron oxides and their composites is also related to the pH environment of the solution. At pH = 5, iron elements are transformed into akaganéite (β-FeOOH) and α-Fe2O3 by dissolution-recrystallization and aggregation-recrystallization, respectively, and the structural substitution of REEs decreases during the transformation process, and the concentration of rare earth ions in the liquid phase solution increases; at pH 7, hydrated iron oxide particles aggregate and effectively enrichment of rare earths [16]. Under the same pH conditions, the adsorption amount Q1 when 0.10 mol/L NaNO3 was used as the solution ionic background was greater than the adsorption amount Q2 when 0.01 mol/L NaNO3 was used as the solution ionic background, i.e., Q1 > Q2. The experimental results indicate that the adsorption of rare earth ions by iron oxides increases when the ionic strength of the solution increases. Comparing the adsorption effects of the three rare earth ions on iron oxides and their composites in two ionic strength solution environments, it can be seen that the change in ionic strength has the most significant effect on the adsorption effect of Ce3+, indicating that the element Ce is more sensitive to the ionic strength and pH conditions in the solution environment, and the adsorption complexation of rare earth ions on the surface of iron oxides and their composites is not only dependent on their charge and radius, but also influenced by the rare earth ion The adsorption complexation of rare earth ions on the surface of iron oxides and their composites is not only dependent on their charges and radii, but also controlled by the changes of electronic configuration and the type of complexing ligands. Complexation of REEs by iron oxides is selective for the adsorption of HREEs, and rare earth ions are adsorbed on the surface of iron oxides mainly through inner circle complexation [30].
3.3.3. Effect of Temperature on the Adsorption of Rare Earth Ions by Iron Oxides and Their Composites
In order to explore the effect of reaction temperature on the adsorption effect of iron oxides and their composites, the experiments were carried out at 20 °C, 30 °C, 40 °C, 50 °C and 60 °C with constant temperature oscillation at the reaction temperatures of 15 mmol/L for both rare earth solutions, with a liquid-solid ratio of 50:1 and a reaction time of 10 h. The experimental results are shown in Figure 8.
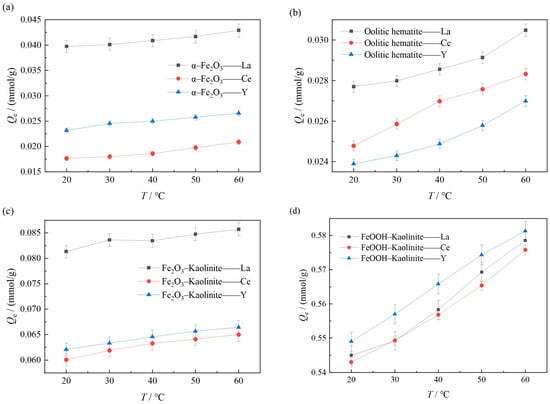
Figure 8.
Influence of temperature on adsorption of rare earth ions by iron oxides and their composites: (a) α-Fe2O3, (b) Oolitic hematite, (c) Fe2O3-Kaolinite, and (d) FeOOH-Kaolinite.
As can be seen from Figure 8, the adsorption of rare earth ions on iron oxides and their composites increases with the increase in the reaction temperature, and the experimental results indicate that increasing the temperature is favorable for the adsorption reaction. On the one hand, this is due to the fact that increasing the temperature induces a swelling effect inside the adsorbent [31], which leads to an increase in the adsorption amount due to the expansion of the adsorbent molecules, and on the other hand, increasing the temperature helps rare earth ions to overcome the adsorption energy barrier, which is more favorable to the adsorption of rare earth ions on iron oxides and their composites.
3.3.4. Effect of Temperature on Adsorption Kinetics and Thermodynamic Analysis
To investigate the influence of temperature on adsorption kinetics, systematic studies on the dynamic adsorption processes of La3+ onto α-Fe2O3, Fe2O3-Kaolinite, and FeOOH-Kaolinite were conducted over a temperature range of 20 °C to 60 °C.
As can be seen from Figure 9, the kinetic curves at various temperatures consistently showed that the adsorption capacity increased gradually with time and reached equilibrium at approximately 20 min. As the temperature increased from 20 °C to 60 °C, the equilibrium adsorption capacities of La3+ onto all three adsorbents exhibited a notable upward trend, indicating that elevated temperature favored the adsorption process. The kinetic data were fitted using pseudo-first-order, pseudo-second-order, intra-particle diffusion, and elovich models (Figure 10 and Table 3). The results demonstrated that the pseudo-second-order model yielded the highest correlation coefficients (R2 > 0.99), suggesting that this model provided a more accurate description of the overall adsorption process and that the adsorption rate was governed by a chemisorption mechanism.
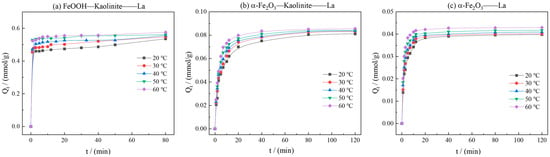
Figure 9.
Effects of reaction temperature on kinetics curve of adsorption of rare earth ions by iron oxides and their composites: (a) FeOOH-Kaolinite, (b) α-Fe2O3-Kaolinite, and (c) α-Fe2O3.
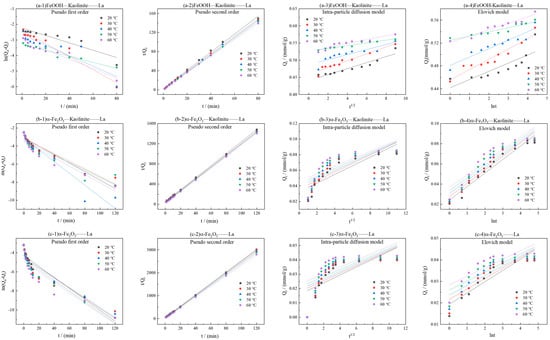
Figure 10.
Kinetic model fitting for adsorption of La3+ by iron oxides and their composites: (a-1–a-4) FeOOH-Kaolinite, (b-1–b-4) α-Fe2O3-Kaolinite, and (c-1–c-4) α-Fe2O3.
Table 3.
Kinetic parameters of adsorption of La3+ by iron oxides and their composites.
The rate constants (k2) derived from the pseudo-second-order model increased significantly with rising temperature. Linear fitting of ln k2 versus 1/T based on the Arrhenius equation (Figure 11 and Table 4) allowed calculation of the apparent activation energies (Ea) for La3+ adsorption onto FeOOH-Kaolinite, Fe2O3-Kaolinite, and α-Fe2O3, which were determined to be 18.35 kJ/mol, 12.59 kJ/mol, and 15.62 kJ/mol, respectively. All Ea values exceeded 4.2 kJ/mol, further confirming the dominance of chemical adsorption in the process.
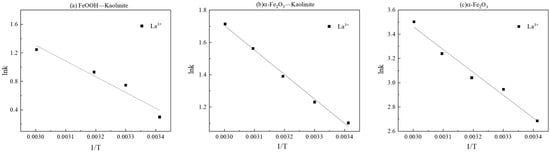
Figure 11.
Linear fitting of lnk-1/T: (a) FeOOH-Kaolinite, (b) α-Fe2O3-Kaolinite, and (c) α-Fe2O3.
Table 4.
Fitting results and activation energy results of La3+ by iron oxides and their composites.
3.4. The Adsorption Mechanism of Iron Oxides and Their Composites on REEs
3.4.1. Zeta Potential Analysis
The variation of surface potentials of iron oxides and their composites under different pH conditions can be obtained by zeta potential analysis. The changes of the surface zeta potential of iron oxides and their composites before and after adsorption were investigated in the range of pH 2~6, and the results are shown in Figure 12.
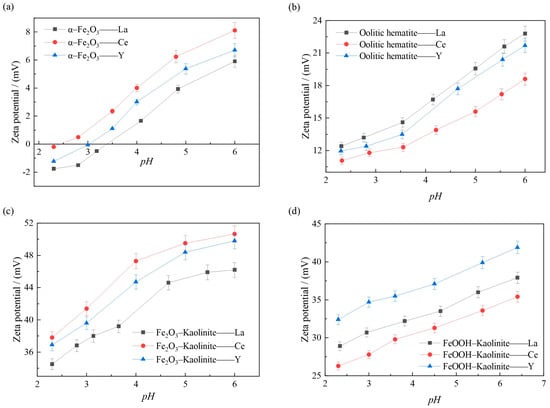
Figure 12.
Effect of pH on the surface Zeta potential of adsorption of rare earth ions by iron oxides and their complex: (a) α-Fe2O3, (b) Oolitic hematite, (c) Fe2O3-Kaolinite, and (d) FeOOH-Kaolinite.
As can be seen from Figure 12, during the adsorption process, the ζ-potential on the surface of the iron oxide and its composites increases as the pH of the solution increases, which is due to the fact that the magnitude of the ζ-potential depends on the difference between the amount of negative charge carried on the surface of the iron oxide and its complex particles and the amount of positive charge carried by the cations within the adsorbed layer, i.e., the effective charge. The higher the ζ-potential, the greater the amount of cations adsorbed in the diffusion layer. It shows that when cations are present in the solution, cations are adsorbed on the surface of the particles, making the potential increase, and the higher ζ potential indicates that more cations are adsorbed on the surface of the particles. As can be seen from Figure 12, the order of the magnitude of the adsorption of different rare earth ions on the surface of the iron oxide and its complex at 15 mmol/L in the same pH environment is consistent with the order of the magnitude of the ζ potential on the surface of the iron oxide and its complex, further indicating that the adsorption of rare earth ions by the iron oxide and its complex increases with increasing pH, and the more cations adsorbed on the surface of the particles, the surface ζ potential the higher the surface ζ potential.
3.4.2. XPS Analysis
To investigate the surface chemical states and elemental composition of the iron oxide-based adsorbents before and after La3+ adsorption, X-ray photoelectron spectroscopy (XPS) was performed. The Fe 2p and O 1s spectra are shown in Figure 13 and Figure 14, and the corresponding peak assignments are summarized in Table 5.
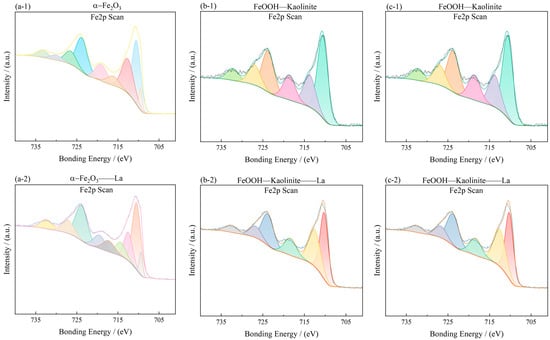
Figure 13.
Fe2p spectra of iron oxides and their complexes before and after La3+ adsorption: (a-1,a-2) α-Fe2O3, (b-1,b-2) Fe2O3-Kaolinite, and (c-1,c-2) FeOOH-Kaolinite.
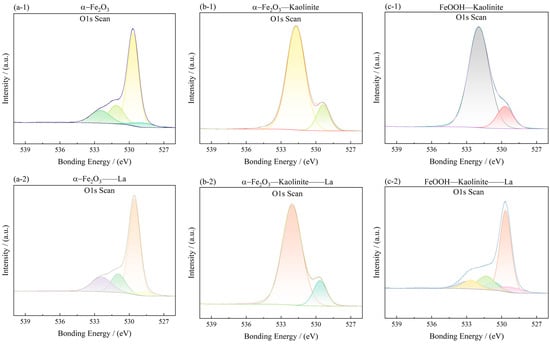
Figure 14.
O1s spectra of iron oxides and their complexes before and after La3+ adsorption: (a-1,a-2) α-Fe2O3, (b-1,b-2) Fe2O3-Kaolinite, and (c-1,c-2) FeOOH-Kaolinite.
Table 5.
Surface oxygen vacancy XPS results of iron oxides and their composites.
In the Fe 2p spectra (Figure 13), the characteristic Fe 2p3/2 and Fe 2p1/2 peaks were observed at approximately 710.45 eV and 724.19 eV for α-Fe2O3, 711.33 eV and 725.01 eV for Fe2O3-Kaolinite, and 711.45 eV and 724.94 eV for FeOOH-Kaolinite, respectively. These binding energies are consistent with Fe(III) in oxide/hydroxide phases. After La3+ adsorption, the Fe 2p peaks of all samples exhibited a slight positive shift (≈0.2–0.5 eV), indicating an increase in the oxidation state of surface Fe species due to electron transfer toward the adsorbed La3+ ions.
The O 1s spectra (Figure 14) were deconvoluted into two main components: a lower-energy peak at 529.60–530.13 eV, attributed to lattice oxygen (Oα, i.e., O2− in the oxide lattice), and a higher-energy peak at 531.15–531.34 eV, assigned to surface-adsorbed oxygen (Oβ) associated with hydroxyl groups, water, and/or oxygen vacancies. The area ratio Oβ/Oα, which reflects the relative abundance of surface reactive oxygen species, was calculated for each sample (Table 5). The values follow the order: FeOOH-Kaolinite (3.86) > α-Fe2O3 (3.68) > Fe2O3-Kaolinite (0.28). This trend indicates that the FeOOH-Kaolinite composite possesses the highest concentration of surface oxygen vacancies, which are known to act as active sites for metal-ion coordination.
After La3+ adsorption, the O 1s spectra showed a noticeable increase in the Oβ component, especially for the FeOOH-Kaolinite composite, confirming the participation of surface hydroxyl groups in the adsorption process. The enhanced Oβ signal, together with the slight shift of Fe 2p peaks, supports the formation of Fe-O-La surface complexes through inner-sphere coordination. These results provide direct spectroscopic evidence that the adsorption of La3+ onto iron oxide-kaolinite composites is driven by chemical interaction rather than purely physical forces, consistent with the kinetic and thermodynamic analyses presented earlier.
3.4.3. Fourier Infrared Spectroscopy
Fourier transform infrared spectroscopy analyzers are mainly used for qualitative and quantitative analysis of samples and can provide more useful information about the interaction between iron oxides and their composites with rare earth ions. Metal hydroxyl groups are usually present on the surface of many metal oxides, which are the most abundant and active adsorption sites for adsorbates and can be detected by IR spectroscopy. The changes of iron oxides and their composites before and after adsorption were analyzed and determined using a Nicolet 6700 Fourier transform infrared spectrometer (Waltham, MA, USA), and the results are shown in Figure 15.
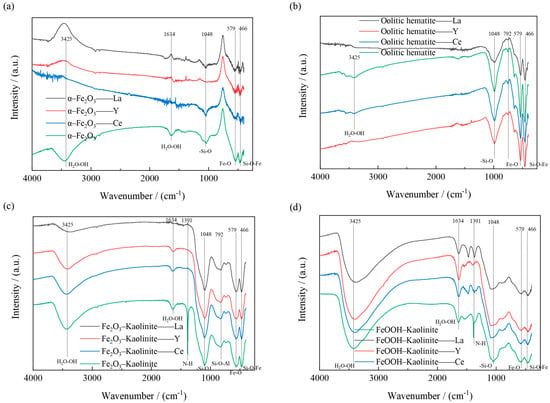
Figure 15.
FT-IR spectra of iron oxides and their composites before and after adsorption: (a) α-Fe2O3, (b) Oolitic hematite, (c) Fe2O3-Kaolinite, and (d) FeOOH-Kaolinite.
The FT-IR spectra of the iron oxides and their composites before and after adsorption are shown in Figure 14. As can be seen from Figure 14, two significant peaks are found at 1391 and 579 cm−1. The former is the -NO3 stretching mode, indicating the presence of excess positively charged iron aggregates outside the interlayer space of the composite; the latter is the vibrational mode of Fe-O The bonding positions at 1048, 792 and 466 cm−1 are caused by the deformation vibrations of the -Si-O, Si-O-Al and Si-O-Fe structures. The bonds near 3425 cm−1 and 1634 cm−1 are caused by the -OH stretching and deformation of water. All the above results indicate the presence of iron oxides on the kaolinite surface.
4. Conclusions
This study systematically elucidates the adsorption behavior and mechanisms of rare earth ions (La3+, Ce3+, Y3+) onto iron oxides and their kaolinite composites. The main conclusions are as follows:
The adsorption isotherms of all four adsorbents for the three REEs were better described by the Langmuir model (R2 > 0.96), indicating monolayer homogeneous adsorption where the adsorption capacity depends on the number of surface active sites. The adsorption kinetics followed the pseudo-second-order model (R2 > 0.99). The calculated activation energies (Ea > 12.59 kJ/mol) all exceeded 4.2 kJ/mol, confirming that the process is dominated by chemical adsorption.
Compared to pure iron oxides (α-Fe2O3, BET surface area ≈ 2.2 m2/g), the adsorption capacities of the Fe2O3-kaolinite (47.1 m2/g) and FeOOH-kaolinite (244.5 m2/g) composites were significantly higher. Among them, the amorphous/poorly crystalline FeOOH-kaolinite composite exhibited the highest saturated adsorption capacity (0.6531 mmol/g for Ce3+). XPS analysis revealed that it possesses the highest surface oxygen vacancy concentration (Oβ/Oα = 3.86), which is the key reason for its superior complexation adsorption and enrichment capability.
The highly crystalline α-Fe2O3-kaolinite composite showed a greater disparity in adsorption amounts for the three REEs (La > Y > Ce), indicating stronger fractionation selectivity. In contrast, the amorphous FeOOH-kaolinite composite, with its high specific surface area and high density of active sites, demonstrated superior overall enrichment capacity. This suggests that within weathered crusts, crystalline iron oxide–clay composites may be the key phase controlling REE fractionation, while amorphous composites dominate the overall REE enrichment intensity.
Environmental factors exert a regular controlling influence on adsorption behavior. Within the pH range of 3.0–7.0, the adsorption amount increased with rising pH, attributable to surface deprotonation reducing competitive adsorption from H+. The adsorption behavior of Ce3+ was the most sensitive to changes in solution ionic strength and pH, likely due to its variable valence characteristics and more complex surface complexation chemistry.
Author Contributions
Conceptualization, R.C. and Z.Z.; methodology, Z.C.; software, W.G. and H.W.; validation, H.W., X.D. and D.L. (Defeng Liu); formal analysis, D.L. (Defeng Liu); investigation, W.G. and X.D.; resources, Z.C. and H.W.; data curation, D.L. (Dan Li); writing—original draft preparation, D.L. (Dan Li); writing—review and editing, Z.Z. and W.G.; visualization, Z.C. and X.D.; supervision, Z.Z. and D.L. (Defeng Liu); project administration, Z.Z. and R.C.; funding acquisition, Z.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (52222405, 92475206, 52304293, 92462303) and the Natural Science Foundation Innovation Group Project of Hubei Province (2023AFA044).
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding authors.
Acknowledgments
Thanks for the great effort by the editors and reviewers.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gu, B.J.; Long, Z.Q.; Huang, X.W.; Li, H.W. Present status and prospect of RE compound industry in China. Chin. J. Rare Met. 2003, 391–394. [Google Scholar] [CrossRef]
- Chi, R.A.; Tian, J. Review of weathered crust rare earth ore. J. Chin. Soc. Rare Earths 2007, 06, 641–650. [Google Scholar]
- García, M.V.R.; Krzemień, A.; Del Campo, M.Á.M.; Álvarez, M.M.; Gent, M.R. Rare earth elements mining investment: It is not all about China. Resour. Policy 2017, 53, 66–76. [Google Scholar] [CrossRef]
- Fan, H.R.; Niu, H.C.; Li, X.C.; Yang, K.F.; Yang, Z.F.; Wang, Q.W. The types, ore genesis and resource perspective of endogenic REE deposits in China. Chin. Sci. Bull. 2020, 65, 3778–3793. [Google Scholar] [CrossRef]
- He, Y.; Cheng, L.; Li, Y.; Ran, D.J.; Wei, Q.S. The mineralization mechanism of the ion adsorption type rare earths ore and prospecting marks. Chin. Rare Earths 2015, 36, 98–103. [Google Scholar]
- Li, Y.; Zhao, W.W.; Zhou, M.F. Nature of parent rocks, mineralization styles and ore genesis of regolith-hosted REE deposits in South China: An integrated genetic model. J. Asian Earth Sci. 2017, 148, 65–95. [Google Scholar] [CrossRef]
- Borst, A.M.; Smith, M.P.; Finch, A.A.; Estrade, G.; Villanova-De-Benavent, C.; Nason, P.; Marquis, E.; Horsburgh, N.J.; Goodenough, K.M.; Xu, C. Adsorption of rare earth elements in regolith-hosted clay deposits. Nat. Commun. 2020, 11, 4386. [Google Scholar] [CrossRef]
- Yang, M.J.; Liang, X.L.; Ma, L.Y.; Huang, J.; He, H.P.; Zhu, J.X. Adsorption of REEs on kaolinite and halloysite: A link to the REE distribution on clays in the weathering crust of granite. Chem. Geol. 2019, 525, 210–217. [Google Scholar] [CrossRef]
- Li, M.Y.H.; Zhou, M.F. The role of clay minerals in formation of the regolith-hosted heavy rare earth element deposits. Am. Miner. 2020, 105, 92–108. [Google Scholar] [CrossRef]
- Sanematsu, K.; Kon, Y.; Imai, A.; Watanabe, K.; Watanabe, Y. Geochemical and mineralogical characteristics of ion-adsorption type REE mineralization in Phuket, Thailand. Miner. Depos. 2013, 48, 437–451. [Google Scholar] [CrossRef]
- Braun, J.J.; Riotte, J.; Battacharya, S.; Violette, A.; Oliva, P.; Prunier, J.; Maréchal, J.C.; Ruiz, L.; Audry, S.; Subramanian, S. REY-Th-U dynamics in the critical zone: Combined influence of reactive bedrock accessory minerals, authigenic phases, and hydrological sorting (Mule Hole Watershed, South India). Geochem. Geophys. Geosyst. 2018, 19, 1611–1635. [Google Scholar] [CrossRef]
- Li, Z.B.; Liu, L.W.; Chen, J.; Teng, H.H. Cellular dissolution at hypha-and spore-mineral interfaces revealing unrecognized mechanisms and scales of fungal weathering. Geology 2016, 44, 319–322. [Google Scholar] [CrossRef]
- Yu, C.; Drake, H.; Mathurin, F.A.; Åström, M.E. Cerium sequestration and accumulation in fractured crystalline bedrock: The role of Mn-Fe (hydr-) oxides and clay minerals. Geochim. Cosmochim. Acta 2017, 199, 370–389. [Google Scholar] [CrossRef]
- Ni, Z. Transformation Performance and Potential Mechanisms of Polycyclic Aromatic Hydrocarbons on Iron Oxide/Clay Composite. Master’s Thesis, Northwest A&F University, Xianyang, China, 2022. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Ohta, A.; Kagi, H.; Nomura, M.; Tsuno, H.; Kawabe, I. Coordination study of rare earth elements on Fe oxyhydroxide and Mn dioxides: Part I. Influence of a multi-electron excitation on EXAFS analyses of La, Pr, Nd, and Sm. Am. Miner. 2009, 94, 467–475. [Google Scholar] [CrossRef]
- Ichimura, K.; Sanematsu, K.; Kon, Y.; Takagi, T.; Murakami, T. REE redistributions during granite weathering: Implications for Ce anomaly as a proxy for paleoredox states. Am. Miner. 2020, 105, 848–859. [Google Scholar] [CrossRef]
- Janots, E.; Bernier, F.; Brunet, F.; Muñoz, M.; Trcera, N.; Berger, A.; Lanson, M. Ce(III) and Ce(IV) (re)distribution and fractionation in a laterite profile from Madagascar: Insights from in situ XANES spectroscopy at the Ce LIII-edge. Geochim. Cosmochim. Acta 2015, 153, 134–148. [Google Scholar] [CrossRef]
- Tan, W.F.; Zhou, S.Z.; Liu, F.; Feng, X.H.; Li, X.Y. Advancement in the study on interactions between iron-aluminum (hydro-) oxides and clay minerals in soil. Soils 2007, 39, 726–730. [Google Scholar]
- Duiker, S.W.; Rhoton, F.E.; Torrent, J.; Smeck, N.E.; Lal, R. Iron (hydr) oxide crystallinity effects on soil aggregation. Soil. Sci. Soc. Am. J. 2003, 67, 606–611. [Google Scholar] [CrossRef]
- Spadini, L.; Manceau, A.; Schindler, P.W.; Charlet, L. Structure and stability of Cd2+ surface complexes on ferric oxides: 1. Results from EXAFS spectroscopy. J. Colloid Interface Sci. 1994, 168, 73–86. [Google Scholar] [CrossRef]
- Liang, J.; Li, X.M.; Yu, Z.G.; Zeng, G.M.; Luo, Y.; Jiang, L.B.; Yang, Z.X.; Qian, Y.Y.; Wu, H.P. Amorphous MnO2 modified biochar derived from aerobically composted swine manure for adsorption of Pb (II) and Cd (II). ACS Sustain. Chem. Eng. 2017, 5, 5049–5058. [Google Scholar] [CrossRef]
- Horváth, Z.; Varga, B.; Mindszenty, A. Micromorphological and chemical complexities of a lateritic profile from basalt (Jos Plateau, Central Nigeria). Chem. Geol. 2000, 170, 81–93. [Google Scholar] [CrossRef]
- Yang, M.J.; Liang, X.L.; He, H.P.; Arai, Y.J. Distribution and fractionation of REEs during ferrihydrite transformation. In Proceedings of the Thirtieth Annual Goldschmidt Conference 2020, Online, 21–26 June 2020. [Google Scholar]
- Vaniman, D.T.; Chipera, S.J.; Bish, D.L.; Duff, M.C.; Hunter, D.B. Crystal chemistry of clay-Mn oxide associations in soils, fractures, and matrix of the Bandelier Tuff, Pajarito Mesa, New Mexico. Geochim. Cosmochim. Acta 2002, 66, 1349–1374. [Google Scholar] [CrossRef]
- Wu, P.Q.; Zhou, J.W.; Huang, J.; Lin, X.J.; Liang, X.L. Enrichment and fractionation of rare earth elements in ion-adsorption rare earth elements deposits: Constraints of iron oxide-clay mineral composites. Geochimica 2022, 51, 271–282. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Bolanz, R.M.; Kiefer, S.; Göttlicher, J.; Steininger, R. Hematite (α-Fe2O3)—A potential Ce4+ carrier in red mud. Sci. Total Environ. 2018, 622, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, F.; Chi, R.; Feng, J.; Ding, Y.; Liu, Q. Preparation of modified montmorillonite and its application to rare earth adsorption. Minerals 2019, 9, 747. [Google Scholar] [CrossRef]
- Koeppenkastrop, D.; Carlo, E. Sorption of rare-earth elements from seawater onto synthetic mineral particles: An experimental approach. Chem. Geol. 1992, 95, 251–263. [Google Scholar] [CrossRef]
- Deng, Y.F. An attempt to search for concealed leaching adsorption rare earth minerals in granite weathering crusts using soil pH values. Earth Environ. 1985, 2, 74–78. [Google Scholar]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.