
Targeting the Kynureninase–HDAC6–Complement Axis as a Novel Therapeutic Strategy in Glioblastoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
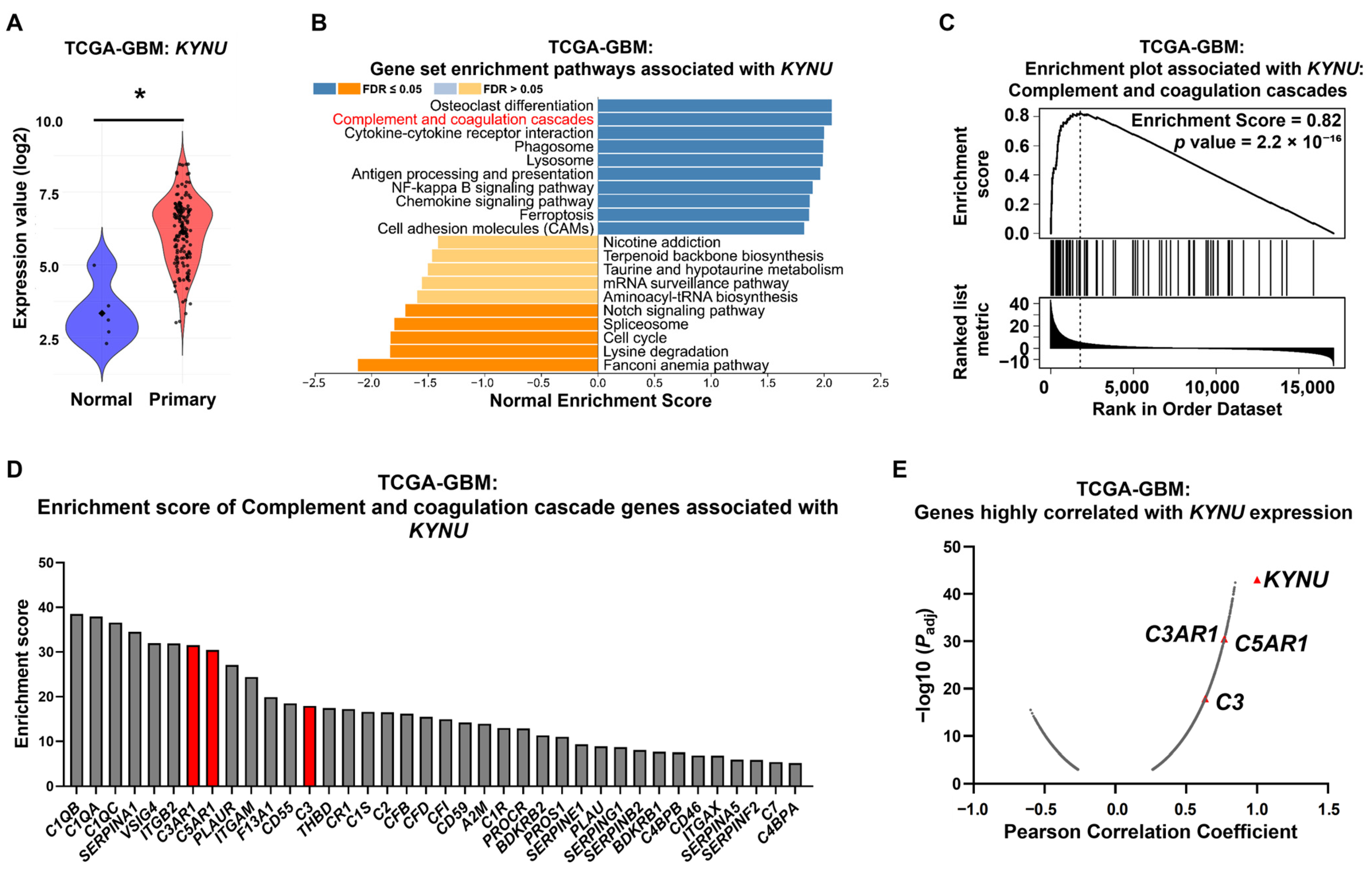
2.1. KYNU Overexpression Is Strongly Correlated with Genes Related to the Complement Cascade in GBM Patients
2.2. C3, C3AR1, and C5AR1 Along with KYNU Are Overexpressed in Selected GBM Cell Lines
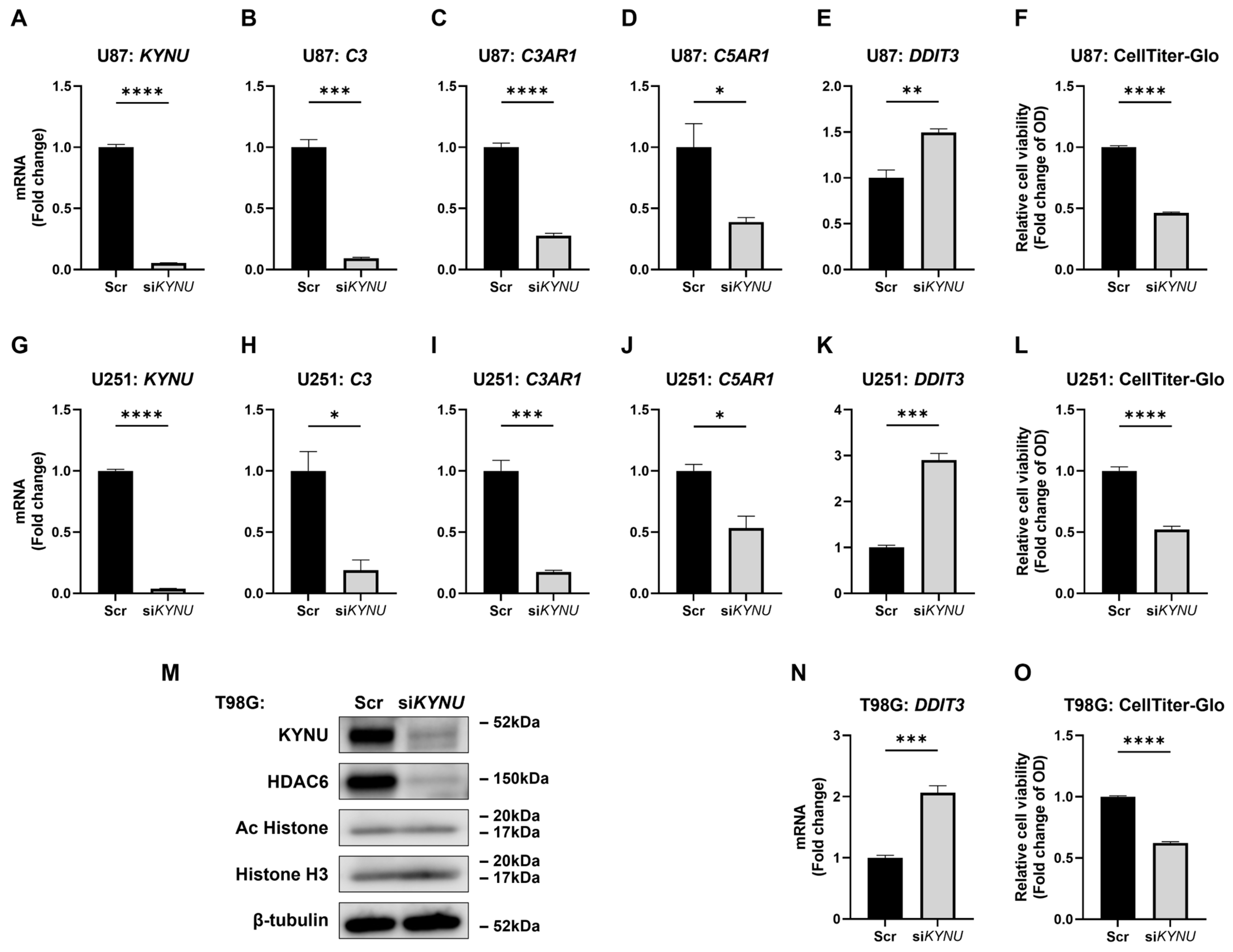
2.3. KYNU Is an Upstream Regulator of C3, C3AR1, and C5AR1
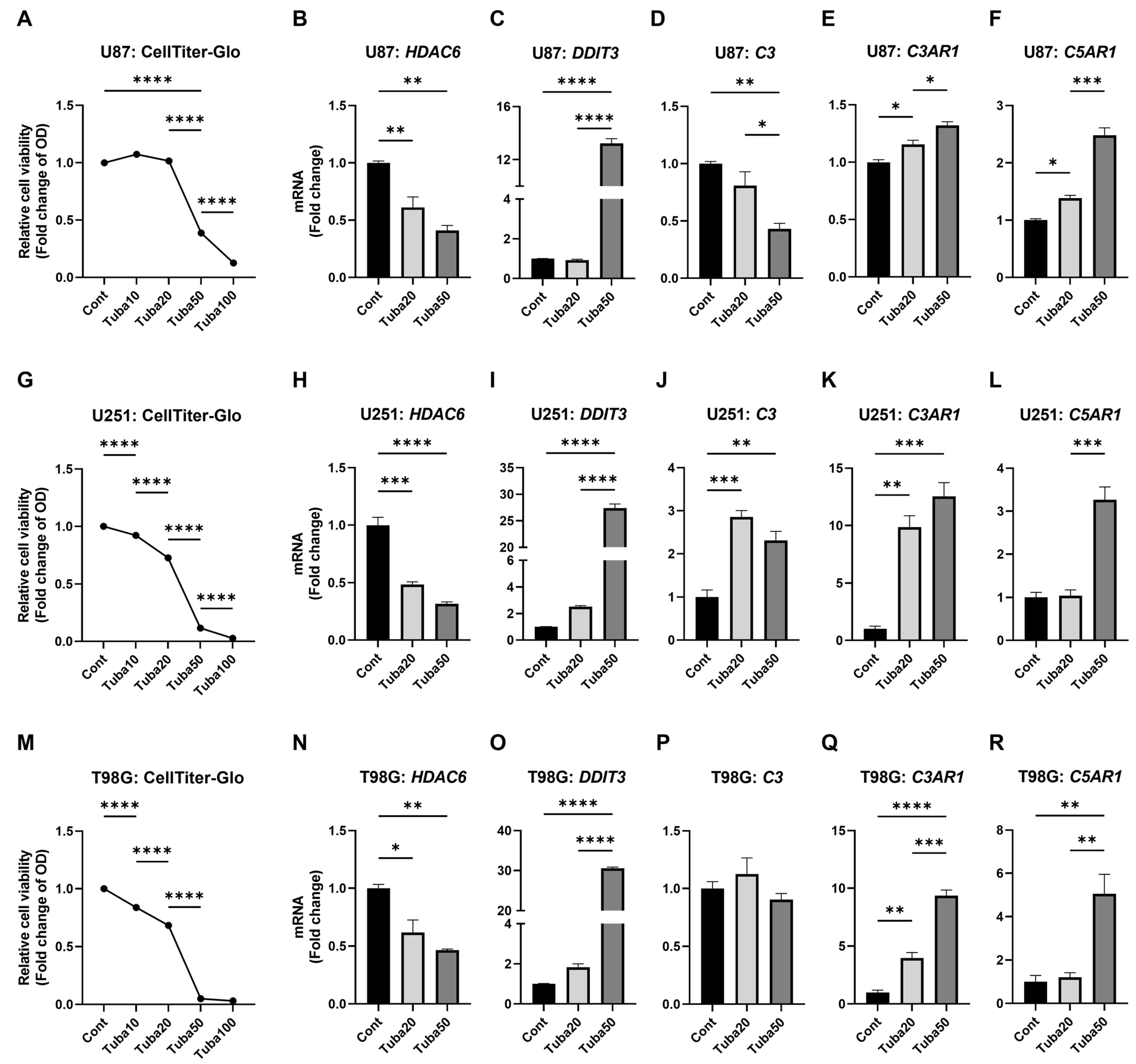
2.4. HDAC6 Inhibition Upregulates Some Complement Components
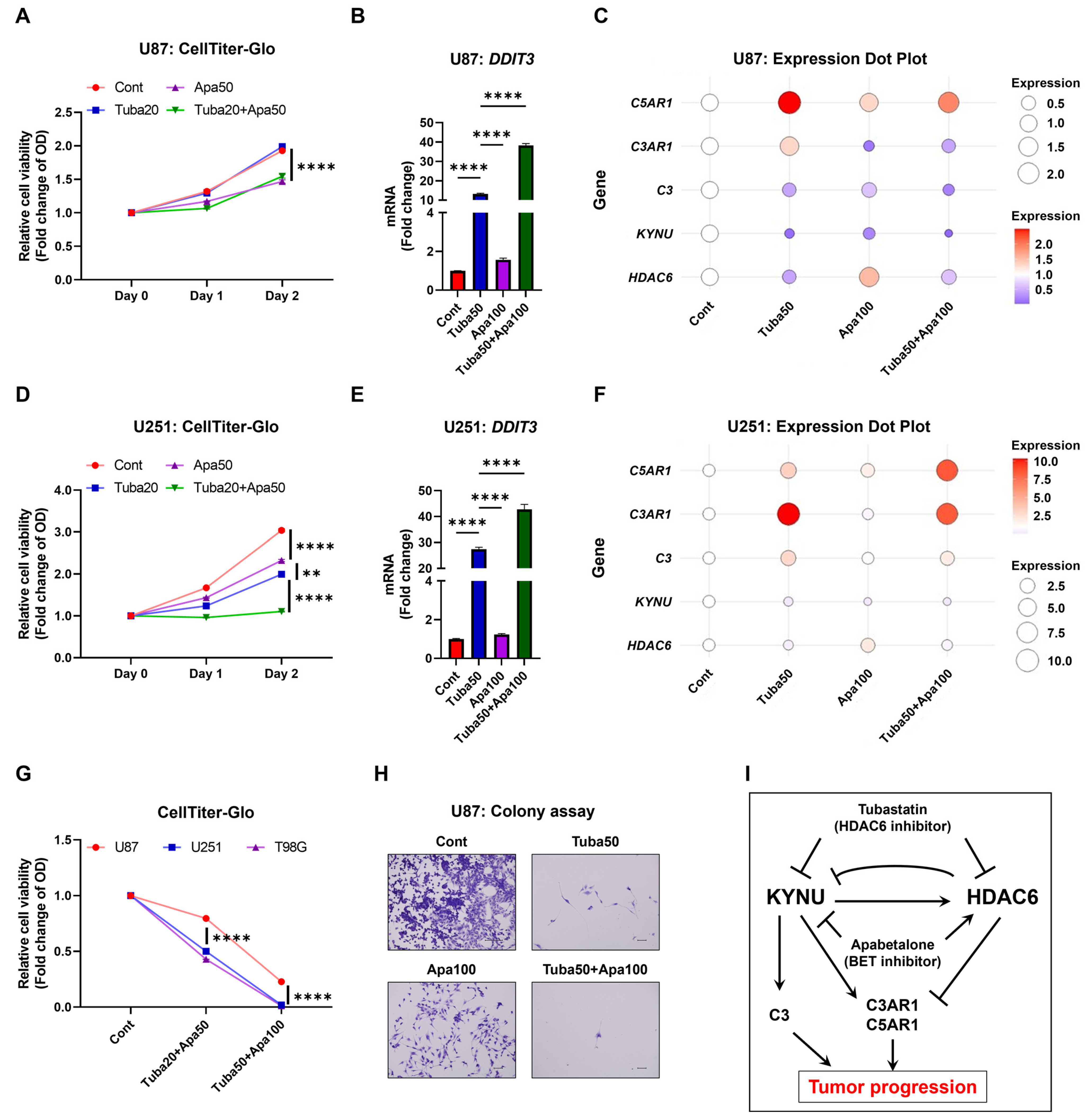
2.5. KYNU Inhibition Attenuates HDAC6-Mediated Dysregulation of Complement Genes
2.6. Inhibition of KYNU-HDAC6-Mediated Complement Activation Reduces GBM Cell Viability
3. Discussion
4. Materials and Methods
4.1. Public Dataset Acquisition and Data Processing
4.2. Cell Lines, Culture, and Reagents for Major Interventions
4.3. Gene Silencing Using siRNA Transfection
4.4. Total RNA Extraction, cDNA Synthesis, and qRT PCR
4.5. SDS-PAGE and Western Blotting
4.6. Cell Viability Assay
4.7. Colony Formation Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: A practical update on what neurosurgeons need to know—A minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma Multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Tenner, A.J.; Mollnes, T.E.; Levy, F.O. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol. Rev. 2021, 73, 792–827. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef]
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116. [Google Scholar] [CrossRef]
- Rosberg, R.; Smolag, K.I.; Sjölund, J.; Johansson, E.; Bergelin, C.; Wahldén, J.; Pantazopoulou, V.; Ceberg, C.; Pietras, K.; Blom, A.M.; et al. Hypoxia-induced complement component 3 promotes aggressive tumor growth in the glioblastoma microenvironment. JCI Insight 2024, 9, e179854. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, X.; Zhang, S.; Shu, K. Targeting the Complement Pathway in Malignant Glioma Microenvironments. Front. Cell Dev. Biol. 2021, 9, 657472. [Google Scholar] [CrossRef]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massagué, J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 2017, 168, 1101–1113.e13. [Google Scholar] [CrossRef]
- Bouwens, T.; Trouw, L.; Veerhuis, R.; Dirven, C.; Lamfers, M.; Al-Khawaja, H. Complement activation in Glioblastoma Multiforme pathophysiology: Evidence from serum levels and presence of complement activation products in tumor tissue. J. Neuroimmunol. 2015, 278, 271–276. [Google Scholar] [CrossRef]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic targeting of glioblastoma. Front. Oncol. (OCT) 2018, 8, 448. [Google Scholar] [CrossRef]
- Yang, H.; Wei, L.; Xun, Y.; Yang, A.; You, H. BRD4: An emerging prospective therapeutic target in glioma. Mol. Ther. Oncolytics 2021, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Yu, M.; Chen, J. BRD4: New hope in the battle against glioblastoma. Pharmacol. Res. 2023, 191, 106767. [Google Scholar] [CrossRef] [PubMed]
- Dhulkifle, H.; Diab, M.I.; Algonaiah, M.; Korashy, H.M.; Maayah, Z.H. Apabetalone (RVX-208): A Potential Epigenetic Therapy for the Treatment of Cardiovascular, Renal, Neurological, Viral, and Cancer Disorders. ACS Pharmacol. Transl. Sci. 2024, 7, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Schwartz, G.G.; Nicholls, S.J.; Khan, A.; Halliday, C.; Toth, P.P.; Sweeney, M.; Johansson, J.O.; Wong, N.C.; Kulikowski, E.; et al. Cognitive Effects of the BET Protein Inhibitor Apabetalone: A Prespecified Montreal Cognitive Assessment Analysis Nested in the BETonMACE Randomized Controlled Trial. J. Alzheimer's Dis. 2021, 83, 1703–1715. [Google Scholar] [CrossRef]
- Wasiak, S.; Gilham, D.; Tsujikawa, L.M.; Halliday, C.; Calosing, C.; Jahagirdar, R.; Johansson, J.; Sweeney, M.; Wong, N.C.; Kulikowski, E. Downregulation of the Complement Cascade In Vitro, in Mice and in Patients with Cardiovascular Disease by the BET Protein Inhibitor Apabetalone (RVX-208). J. Cardiovasc. Transl. Res. 2017, 10, 337–347. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Saenz-Antoñanzas, A.; Mikelez-Alonso, I.; Carrasco-Garcia, E.; Tellaetxe-Abete, M.; Lawrie, C.H.; Sampron, N.; Cortajarena, A.L.; Matheu, A. Characterization of a new small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis. 2020, 11, 417. [Google Scholar] [CrossRef]
- Yang, W.-B.; Wu, A.-C.; Hsu, T.-I.; Liou, J.-P.; Lo, W.-L.; Chang, K.-Y.; Chen, P.-Y.; Kikkawa, U.; Yang, S.-T.; Kao, T.-J.; et al. Histone deacetylase 6 acts upstream of DNA damage response activation to support the survival of glioblastoma cells. Cell Death Dis. 2021, 12, 884. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-B.; Hsu, C.-C.; Hsu, T.-I.; Liou, J.-P.; Chang, K.-Y.; Chen, P.-Y.; Yang, S.-T.; Wang, J.-Y.; Yeh, S.-H.; Chen, R.-M.; et al. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro-Oncol. 2020, 22, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234. [Google Scholar] [CrossRef]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef]
- Jin, J.; Meng, T.; Yu, Y.; Wu, S.; Jiao, C.-C.; Song, S.; Li, Y.-X.; Zhang, Y.; Zhao, Y.-Y.; Li, X.; et al. Human HDAC6 senses valine abundancy to regulate DNA damage. Nature 2024, 637, 215–223. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2020, 6, 2. [Google Scholar] [CrossRef]
- Kesarwani, P.; Prabhu, A.; Kant, S.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Wilson, G.D.; Miller, C.R.; Chinnaiyan, P. Tryptophan metabolism contributes to radiation-induced immune checkpoint reactivation in glioblastoma. Clin. Cancer Res. 2018, 24, 3632–3643. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef]
- Widodo, S.S.; Dinevska, M.; Furst, L.M.; Stylli, S.S.; Mantamadiotis, T. IL-10 in glioma. Br. J. Cancer 2021, 125, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xu, Y.; Liu, J.; Wu, S.; Zhang, R.; Cao, H.; Sun, J. MAP3K8 Is a Prognostic Biomarker and Correlated With Immune Response in Glioma. Front. Mol. Biosci. 2021, 8, 779290. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.-D.; Li, Q.-Z.; Zhang, X.-J.; Li, W.-P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Bornes, K.E.; Moody, M.A.; Huckaba, T.M.; Benz, M.C.; McConnell, E.C.; Foroozesh, M.; Barnes, V.H.; Collins-Burow, B.M.; Burow, M.E.; Watt, T.J.; et al. Lysine deacetylase inhibitors have low selectivity in cells and exhibit predominantly off-target effects. FEBS Open Bio 2025, 15, 94–107. [Google Scholar] [CrossRef]
- Saito, S.; Zhuang, Y.; Shan, B.; Danchuk, S.; Luo, F.; Korfei, M.; Guenther, A.; Lasky, J.A.; Ding, Q. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFβ-PI3K-Akt pathway. PLoS ONE 2017, 12, e0186615. [Google Scholar] [CrossRef]
- Liljedahl, E.; Konradsson, E.; Gustafsson, E.; Jonsson, K.F.; Olofsson, J.K.; Osther, K.; Ceberg, C.; Redebrandt, H.N. Combined anti-C1-INH and radiotherapy against glioblastoma. BMC Cancer 2023, 23, 106. [Google Scholar] [CrossRef]
- Lim, E.-J.; Kim, S.; Oh, Y.; Suh, Y.; Kaushik, N.; Lee, J.-H.; Lee, H.-J.; Kim, M.-J.; Park, M.-J.; Kim, R.-K.; et al. Crosstalk between GBM cells and mesenchymal stemlike cells promotes the invasiveness of GBM through the C5a/p38/ZEB1 axis. Neuro-Oncol. 2020, 22, 1452–1462. [Google Scholar] [CrossRef]
- Beach, C.; MacLean, D.; Majorova, D.; Melemenidis, S.; Nambiar, D.K.; Kim, R.K.; Valbuena, G.N.; Guglietta, S.; Krieg, C.; Darvish-Damavandi, M.; et al. Improving radiotherapy in immunosuppressive microenvironments by targeting complement receptor C5aR. J. J. Clin. Investig. 2023, 133, e168277. [Google Scholar] [CrossRef]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Perepechaeva, M.L.; Grishanova, A.Y. The Role of Aryl Hydrocarbon Receptor (AhR) in Brain Tumors. Int. J. Mol. Sci. 2020, 21, 2863. [Google Scholar] [CrossRef] [PubMed]
- Guastella, A.R.; Michelhaugh, S.K.; Klinger, N.V.; Fadel, H.A.; Kiousis, S.; Ali-Fehmi, R.; Kupsky, W.J.; Juhász, C.; Mittal, S. Investigation of the aryl hydrocarbon receptor and the intrinsic tumoral component of the kynurenine pathway of tryptophan metabolism in primary brain tumors. J. Neuro-Oncol. 2018, 139, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Sato, S.; Hasan, A.U.; Obara, M.; Kondo, Y.; Taira, E. Long-term consumption of moderate amounts of sucrose-sweetened drinks disrupts intestinal barrier function by impairing goblet cell differentiation. Cell Tissue Res. 2025, 400, 273–285. [Google Scholar] [CrossRef]
- Hasan, A.U.; Serada, S.; Sato, S.; Obara, M.; Hirata, S.; Nagase, Y.; Kondo, Y.; Taira, E. KDM4B Histone Demethylase Inhibition Attenuates Tumorigenicity of Malignant Melanoma Cells by Overriding the p53-Mediated Tumor Suppressor Pathway. J. Cell. Biochem. 2024, 126, e30643. [Google Scholar] [CrossRef]
- Obara, M.; Sato, S.; Takahashi, K.; Kondo, Y.; Hirose, M.; Nata, K.; Taira, E. Expression of cell adhesion molecule, Gicerin/CD146 during the formation of heart and in the cardiac hypertrophy. Mol. Cell. Biochem. 2021, 476, 2021–2028. [Google Scholar] [CrossRef]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Noma, T.; Igarashi, J.; Tsuboi, K.; Tokuda, M.; Nishiyama, A.; et al. GPR120 in adipocytes has differential roles in the production of pro-inflammatory adipocytokines. Biochem. Biophys. Res. Commun. 2017, 486, 76–82. [Google Scholar] [CrossRef]
- Hasan, A.U.; Obara, M.; Sato, S.; Kondo, Y.; Taira, E. CD146/MCAM links doxorubicin-induced epigenetic dysregulation to the impaired fatty acid transportation in H9c2 cardiomyoblasts. Biochem. Biophys. Res. Commun. 2024, 693, 149370. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, A.U.; Sato, S.; Obara, M.; Kondo, Y.; Taira, E. Targeting the Kynureninase–HDAC6–Complement Axis as a Novel Therapeutic Strategy in Glioblastoma. Epigenomes 2025, 9, 27. https://doi.org/10.3390/epigenomes9030027
Hasan AU, Sato S, Obara M, Kondo Y, Taira E. Targeting the Kynureninase–HDAC6–Complement Axis as a Novel Therapeutic Strategy in Glioblastoma. Epigenomes. 2025; 9(3):27. https://doi.org/10.3390/epigenomes9030027
Chicago/Turabian StyleHasan, Arif Ul, Sachiko Sato, Mami Obara, Yukiko Kondo, and Eiichi Taira. 2025. "Targeting the Kynureninase–HDAC6–Complement Axis as a Novel Therapeutic Strategy in Glioblastoma" Epigenomes 9, no. 3: 27. https://doi.org/10.3390/epigenomes9030027
APA StyleHasan, A. U., Sato, S., Obara, M., Kondo, Y., & Taira, E. (2025). Targeting the Kynureninase–HDAC6–Complement Axis as a Novel Therapeutic Strategy in Glioblastoma. Epigenomes, 9(3), 27. https://doi.org/10.3390/epigenomes9030027