Genome-Wide DNA Methylation and LncRNA-Associated DNA Methylation in Metformin-Treated and -Untreated Diabetes

 ,
, 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of the Study Population
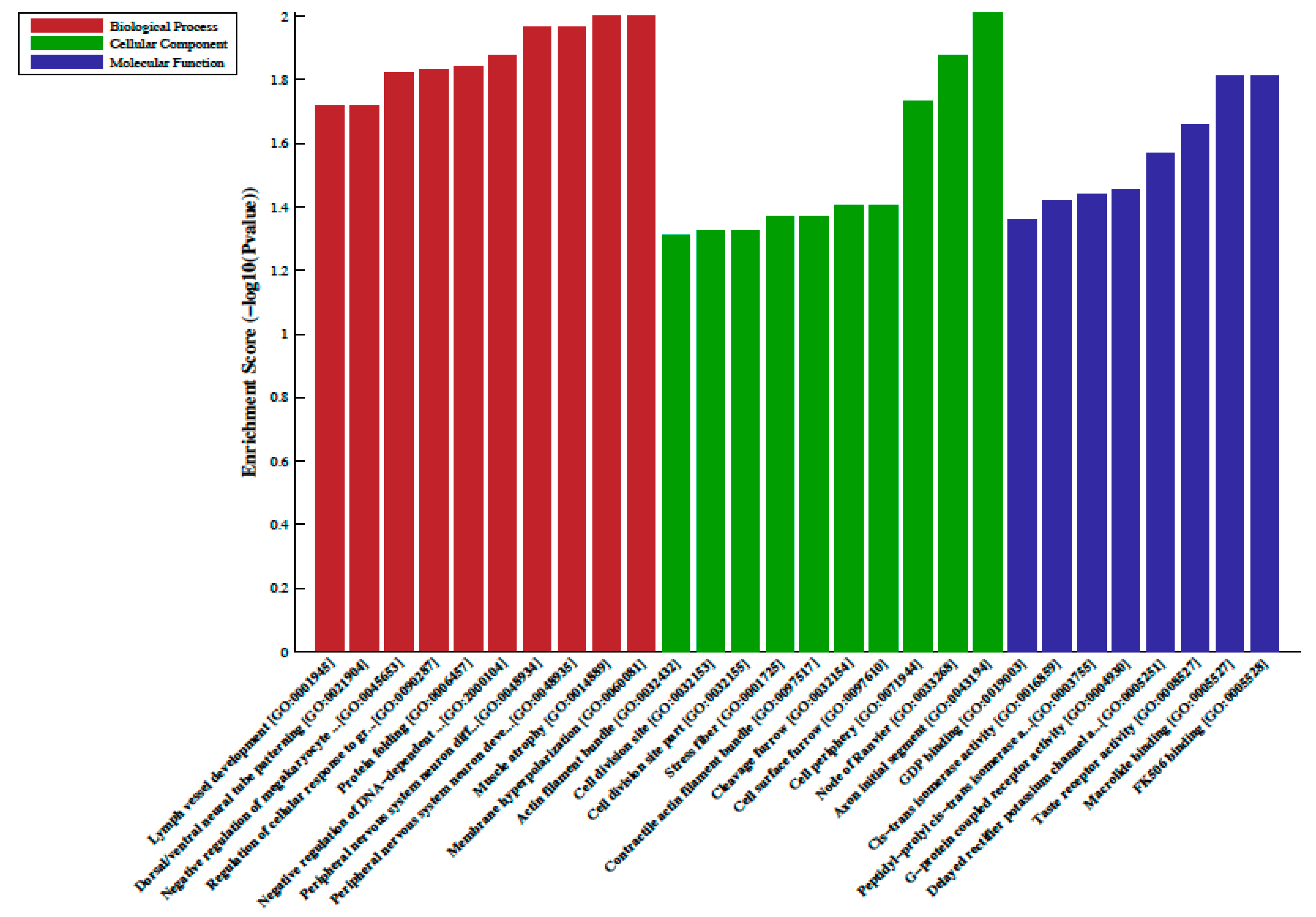
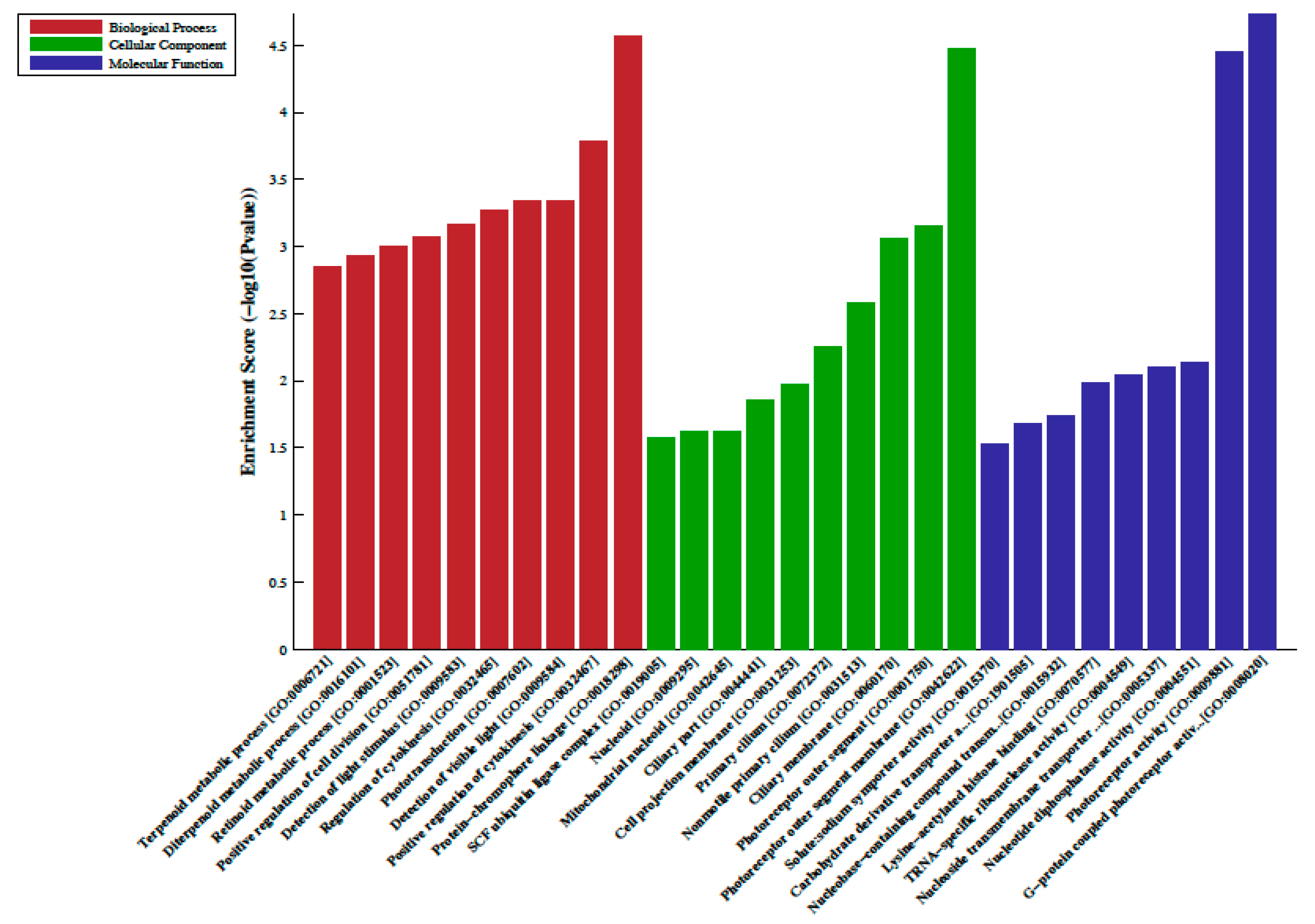
2.2. Differentially Methylated Regions, LncRNA-Associated DNA Methylation, Gene Ontology (GO) and Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Ethical Approval of the Study
4.2. Study Procedures
4.3. Genome-Wide DNA Methylation Sequencing
4.4. MeDIP-Seq Data Analysis
4.5. Gene Ontology (GO) and KEGG Pathway Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deaton, A.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Hoarau-Vechot, J.; Fakhro, K.; Rafii, A.; Abi Khalil, C. Epigenetics and Cardiovascular Disease in Diabetes. Curr. Diab. Rep. 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pinney, S.E. DNA methylation and its role in the pathogenesis of diabetes. Pediatr. Diabetes 2017, 18, 167–177. [Google Scholar] [CrossRef]
- Muka, T.; Nano, J.; Voortman, T.; Braun, K.V.E.; Ligthart, S.; Stranges, S.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; et al. The role of global and regional DNA methylation and histone modifications in glycemic traits and type 2 diabetes: A systematic review. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 553–566. [Google Scholar] [CrossRef]
- Ronn, T.; Ling, C. DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics 2015, 7, 451–460. [Google Scholar] [CrossRef]
- Li, Y.; Xu, K.; Xu, K.; Chen, S.; Cao, Y.; Zhan, H. Roles of Identified Long Noncoding RNA in Diabetic Nephropathy. J. Diabetes Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, X.; Wu, Z.; Hu, D.; Jia, J.; Guo, J.; Tang, T.; Yao, J.; Liu, H.; Tang, H. Long noncoding RNA LINC00467 promotes glioma progression through inhibiting p53 expression via binding to DNMT1. J. Cancer 2020, 11, 2935–2944. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Chen, S.; Zhao, S.; Huang, J.; Zhou, J.; Xu, Y. Identification of Potential Therapeutic Targets and Pathways of Liraglutide Against Type 2 Diabetes Mellitus (T2DM) Based on Long Non-Coding RNA (lncRNA) Sequencing. Med. Sci. Monit. 2020, 26, e922210. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, J.; Hu, X.; Chen, L. Dysregulated expression of long noncoding RNAs serves as diagnostic biomarkers of type 2 diabetes mellitus. Endocrine 2019, 65, 494–503. [Google Scholar] [CrossRef]
- Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum. Genomics 2018, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; DiStefano, J.K. Long noncoding RNAs as diagnostic and therapeutic targets in type 2 diabetes and related complications. Genes 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Priya, G.; Kalra, S. Metformin in the management of diabetes during pregnancy and lactation. Drugs Context 2018, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-term pancreatic beta cell exposure to high levels of glucose but not palmitate induces DNA methylation within the insulin gene promoter and represses transcriptional activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef]
- García-Calzón, S.; Perfilyev, A.; Männistö, V.; de Mello, V.D.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Diabetes medication associates with DNA methylation of metformin transporter genes in the human liver. Clin. Epigenetics 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Elbere, I.; Silamikelis, I.; Ustinova, M.; Kalnina, I.; Zaharenko, L.; Peculis, R.; Konrade, I.; Ciuculete, D.M.; Zhukovsky, C.; Gudra, D.; et al. Significantly altered peripheral blood cell DNA methylation profile as a result of immediate effect of metformin use in healthy individuals. Clin. Epigenetics 2018, 10, 156. [Google Scholar] [CrossRef]
- Banerjee, P.; Surendran, H.; Chowdhury, D.R.; Prabhakar, K.; Pal, R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J. Mol. Med. 2016, 94, 1397–1409. [Google Scholar] [CrossRef]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.; Li, S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Sun, K.; Meng, Z.; Chen, L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Haitina, T.; Lindblom, J.; Renström, T.; Fredriksson, R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006, 88, 779–790. [Google Scholar] [CrossRef]
- Rodríguez-Mulero, S.; Errasti-Murugarren, E.; Ballarín, J.; Felipe, A.; Doucet, A.; Casado, F.J.; Pastor-Anglada, M. Expression of concentrative nucleoside transporters SLC28 (CNT1, CNT2, and CNT3) along the rat nephron: Effect of diabetes. Kidney Int. 2005, 68, 665–672. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta—Mol. Cell Res. 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Guo, K.; Elzinga, S.; Eid, S.; Figueroa-Romero, C.; Hinder, L.M.; Pacut, C.; Feldman, E.L.; Hur, J. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics 2019, 14, 766–779. [Google Scholar] [CrossRef]
- Fernyhough, P. Mitochondrial Dysfunction in Diabetic Neuropathy: A Series of Unfortunate Metabolic Events. Curr. Diab. Rep. 2015, 15, 24–27. [Google Scholar] [CrossRef]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Lu, J.; Brooks, M.M.; Albert, S.; Althouse, A.D.; Escobedo, J.; Green, J.; Palumbo, P.; Perkins, B.A.; Whitehouse, F.; et al. Impact of glycemic control strategies ontheprogressionofdiabeticperipheral neuropathy in the bypass angioplasty revascularization investigation 2 diabetes (BARI 2D) Cohort. Diabetes Care 2013, 36, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Lin, J.D. Long Noncoding RNAs: A New Regulatory Code in Metabolic Control. Trends Biochem. Sci. 2015, 40, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.H.; Kaur, S.; Brorsson, C.A.; Pociot, F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE 2014, 9, e105723. [Google Scholar] [CrossRef] [PubMed]
- Pengyu, Z.; Yan, Y.; Xiying, F.; Maoguang, Y.; Mo, L.; Yan, C.; Hong, S.; Lijuan, W.; Xiujuan, Z.; Hanqing, C. The Differential Expression of Long Noncoding RNAs in Type 2 Diabetes Mellitus and Latent Autoimmune Diabetes in Adults. Int. J. Endocrinol. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kim, Y.; Caramori, M.L.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Walker, P.C.; Mauer, M. Diabetic nephropathy is associated with gene expression levels of oxidative phosphorylation and related pathways. Diabetes 2006, 55, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Farré, P.; Jones, M.J.; Meaney, M.J.; Emberly, E.; Turecki, G.; Kobor, M.S. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 2015, 8, 1–17. [Google Scholar] [CrossRef]
- Crujeiras, A.B.; Diaz-Lagares, A.; Sandoval, J.; Milagro, F.I.; Navas-Carretero, S.; Carreira, M.C.; Gomez, A.; Hervas, D.; Monteiro, M.P.; Casanueva, F.F.; et al. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: A genome-wide analysis from non-obese and obese patients. Sci. Rep. 2017, 7, 41903. [Google Scholar] [CrossRef]
- Porta, M.; Curletto, G.; Cipullo, D.; De la Longrais, R.R.; Trento, M.; Passera, P.; Taulaigo, A.V.; Di Miceli, S.; Cenci, A.; Dalmasso, P.; et al. Estimating the Delay Between Onset and Diagnosis of Type 2 Diabetes From the Time Course of Retinopathy Prevalence. Diabetes Care 2014, 37, 1668–1674. [Google Scholar] [CrossRef]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Gräf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A Bayesian deconvolution strategy for immunoprecipitation- based DNA methylome analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristics | Screen-Detected Diabetes Mellitus n = 12 | Known Diabetes Mellitus n = 12 | p-Value |
|---|---|---|---|
| Mean ± SD | Mean ± SD | ||
| Age (years) | 54.8 ± 7.5 | 53.2 ± 9.6 | 0.658 |
| Body mass index (kg/m2) | 33.5 ± 8.9 | 29.4 ± 5.0 | 0.174 |
| Waist circumference (cm) | 101.3 ± 19.7 | 91.7 ± 10.5 | 0.150 |
| Hip circumference (cm) | 109.4 ± 16.6 | 103.3 ± 11.9 | 0.311 |
| Waist hip ratio | 0.92 ± 0.07 | 0.89 ± 0.05 | 0.195 |
| Systolic blood pressure (mmHg) | 142.9 ± 32.9 | 136.3 ± 25.9 | 0.587 |
| Diastolic blood pressure (mmHg) | 94.2 ± 22.1 | 83.8 ± 11.5 | 0.165 |
| Fasting plasma glucose (mmol/L) | 9.1 ± 3.6 | 11.0 ± 5.8 | 0.352 |
| Post 2-h plasma glucose (mmol/L) | 16.5 ± 4.71 | - | - |
| HbA1c (%) | 7.97 ± 2.58 | 9.33 ± 3.04 | 0.254 |
| Fasting serum insulin (mIU/L) | 15.3 ± 10.6 | 11.3 ± 7.6 | 0.316 |
| Triglycerides (mmol/L) | 2.18 ± 1.35 | 2.02 ± 0.96 | 0.760 |
| Total cholesterol (mmol/L) | 6.36 ± 0.85 | 6.10 ± 1.47 | 0.607 |
| Low-density lipoprotein-cholesterol (mmol/L) | 4.17 ± 0.84 | 4.08 ± 1.22 | 0.831 |
| High-density lipoprotein-cholesterol (mmol/L) | 1.38 ± 0.58 | 1.23 ± 0.35 | 0.448 |
| Ultrasensitive C-reactive protein (mg/L) | 11.1 ± 12.3 | 14.4 ± 12.5 | 0.531 |
| Serum cotinine (ng/mL) | 127.4 ± 149.5 | 120.7 ± 179.7 | 0.921 |
| Gamma-glutamyl transferase (IU/L) | 64.5 ± 57.4 | 43.7 ± 27.2 | 0.287 |
| Hypermethylated DMRs | |||||
|---|---|---|---|---|---|
| Gene Name | Genomic Coordinates | DMR Length | log2FC | p-Value | q-Value |
| XAGE1E | chrX:52260741-52261020 | 279 | 1.66 | <0.001 | 0.001 |
| XAGE1B | chrX:52260741-52261020 | 279 | 1.66 | <0.001 | 0.001 |
| KIAA1467 | chr12:13198981-13199200 | 219 | 1.62 | <0.001 | 0.001 |
| ASB2 | chr14:94442921-94443160 | 239 | 1.6 | <0.001 | 0.001 |
| GABPA | chr21:27105221-27105420 | 199 | 1.56 | <0.001 | 0.004 |
| ZNF346 | chr5:176448161-176448360 | 199 | 1.47 | <0.001 | 0.004 |
| FKBP8 | chr19:18655321-18655520 | 199 | 1.4 | <0.001 | 0.001 |
| CTAGE15 | chr7:143268761-143269080 | 319 | 1.38 | <0.001 | 0.001 |
| VIPR1 | chr3:42531141-42531360 | 219 | 1.37 | <0.001 | 0.026 |
| TMEM204 | chr16:1584901-1585100 | 199 | 1.31 | <0.001 | 0.002 |
| RNF103-CHMP3 | chr2:86948981-86949200 | 219 | 1.31 | <0.001 | 0.010 |
| PARVB | chr22:44394581-44394780 | 199 | 1.26 | <0.001 | 0.004 |
| POTED | chr21:14980641-14980880 | 239 | 1.23 | <0.001 | 0.001 |
| STAG2 | chrX:123095761-123095980 | 219 | 1.21 | <0.001 | 0.008 |
| KCNQ3 | chr8:133459461-133459680 | 219 | 1.19 | <0.001 | 0.026 |
| TBCE | chr1:235532261-235532520 | 259 | 1.16 | <0.001 | 0.001 |
| GAREML | chr2:26393901-26394160 | 259 | 1.14 | <0.001 | 0.003 |
| SEPT12 | chr16:4838741-4839000 | 259 | 1.13 | <0.001 | 0.003 |
| OR6C3 | chr12:55727101-55727440 | 339 | 1.11 | <0.001 | 0.011 |
| PPP1R32 | chr11:61247661-61247920 | 259 | 1.1 | <0.001 | 0.003 |
| ZNF169 | chr9:97023241-97023440 | 199 | 1.07 | <0.001 | 0.024 |
| TAS1R1 | chr1:6616841-6617200 | 359 | 1.07 | <0.001 | 0.001 |
| Hypomethylated DMRs | |||||
| TPD52L2 | chr20:62497561-62497920 | 359 | −1 | <0.001 | 0.003 |
| GAGE7 | chrX:49217161-49217580 | 419 | −1.15 | <0.001 | 0.011 |
| NUDT10 | chrX:51075781-51076040 | 259 | −1.37 | <0.001 | 0.001 |
| OPN1MW2 | chrX:153446941-153447140 | 199 | −1.39 | <0.001 | 0.008 |
| OPN1MW | chrX:153446941-153447140 | 199 | −1.39 | <0.001 | 0.008 |
| BRDT | chr1:92415321-92415520 | 199 | −1.39 | <0.001 | 0.023 |
| ELAC2 | chr17:12919641-12919840 | 199 | −1.45 | <0.001 | 0.012 |
| SLC25A35 | chr17:8196461-8196680 | 219 | −1.52 | <0.001 | 0.004 |
| C18orf8 | chr18:21081741-21081940 | 199 | −1.55 | <0.001 | 0.002 |
| SLC28A1 | chr15:85429461-85429660 | 199 | −1.67 | <0.001 | 0.008 |
| FBXW8 | chr12:117350081-117350320 | 239 | −1.79 | <0.001 | 0.008 |
| Hypermethylated | |||||
|---|---|---|---|---|---|
| Gene Name | Genomic Coordinates | DMR Length | log2FC | p-Value | q-Value |
| SLC26A9 | chr1:205895421-205895620 | 199 | 1.91 | <0.001 | 0.001 |
| FAM223A | chrX:153859601-153859800 | 199 | 1.82 | <0.001 | 0.001 |
| SDK2 | chr17:71432461-71432680 | 219 | 1.69 | <0.001 | 0.001 |
| XAGE1B | chrX:52260741-52261020 | 279 | 1.66 | <0.001 | 0.001 |
| SCRIB | chr8:144877441-144877660 | 219 | 1.62 | <0.001 | 0.001 |
| KIAA1467 | chr12:13198981-13199200 | 219 | 1.62 | <0.001 | 0.001 |
| AK092098 | chr11:63591421-63591720 | 299 | 1.58 | <0.001 | 0.001 |
| ATP5J | chr21:27105221-27105420 | 199 | 1.56 | <0.001 | 0.004 |
| AF420437 | chr1:146216561-146217120 | 559 | 1.49 | <0.001 | 0.002 |
| ZNF346 | chr5:176448161-176448360 | 199 | 1.47 | <0.001 | 0.004 |
| AX747590 | chr8:12435501-12435760 | 259 | 1.46 | <0.001 | 0.001 |
| AK128525 | chr2:89160101-89160340 | 239 | 1.45 | <0.001 | 0.001 |
| XLOC_007349 | chr9:38128521-38128740 | 219 | 1.4 | <0.001 | 0.001 |
| FKBP8 | chr19:18655321-18655520 | 199 | 1.4 | <0.001 | 0.001 |
| LOC101927468 | chr1:147717321-147717520 | 199 | 1.38 | <0.001 | 0.001 |
| CTAGE15 | chr7:143268761-143269080 | 319 | 1.38 | <0.001 | 0.001 |
| AF258560 | chr16:24930681-24930880 | 199 | 1.38 | <0.001 | 0.005 |
| LOXL2 | chr8:23190561-23190780 | 219 | 1.35 | <0.001 | 0.003 |
| AC016644.1 | chr5:56238121-56238320 | 199 | 1.35 | <0.001 | 0.019 |
| RP11-14N7.2 | chr1:148934661-148934860 | 199 | 1.34 | <0.001 | 0.006 |
| AP001476.4 | chr21:47470561-47470760 | 199 | 1.34 | <0.001 | 0.020 |
| RP3-399L15.2 | chr6:114858501-114858700 | 199 | 1.31 | <0.001 | 0.011 |
| AK310441 | chr1:148876821-148877060 | 239 | 1.28 | <0.001 | 0.001 |
| RP11-423O2.7 | chr1:142958401-142958660 | 259 | 1.25 | <0.001 | 0.017 |
| LOC101928402 | chrX:123095761-123095980 | 219 | 1.21 | <0.001 | 0.008 |
| LINC00521 | chr14:94461821-94462080 | 259 | 1.18 | <0.001 | 0.006 |
| TBCE | chr1:235532261-235532520 | 259 | 1.16 | <0.001 | 0.001 |
| SMIM22 | chr16:4838741-4839000 | 259 | 1.13 | <0.001 | 0.003 |
| XLOC_l2_000395 | chr1:142839541-142839880 | 339 | 1.11 | <0.001 | 0.026 |
| SEMA4C | chr2:97531721-97531960 | 239 | 1.11 | <0.001 | 0.012 |
| LOC101929378 | chr2:157111641-157111940 | 299 | 1.08 | <0.001 | 0.010 |
| ZNF169 | chr9:97023241-97023440 | 199 | 1.07 | <0.001 | 0.024 |
| GNPTG | chr16:1409001-1409360 | 359 | 1.07 | <0.001 | 0.004 |
| TIMELESS | chr12:56816721-56817100 | 379 | 1.05 | <0.001 | 0.002 |
| RP13-638C3.3 | chr17:80544641-80544940 | 299 | 1.02 | <0.001 | 0.009 |
| XLOC_009584 | chr11:123084121-123084460 | 339 | 1 | <0.001 | 0.006 |
| Hypomethylated | |||||
| SSH1 | chr12:109199901-109200160 | 259 | −1.12 | <0.001 | 0.004 |
| XLOC_005639 | chr6:21980601-21980860 | 259 | −1.13 | <0.001 | 0.025 |
| RP11-458D21.1 | chr1:145380441-145380780 | 339 | −1.19 | <0.001 | 0.011 |
| LOC100506603 | chr14:77252181-77252640 | 459 | −1.19 | <0.001 | 0.002 |
| AK125727 | chr14:77252181-77252640 | 459 | −1.19 | <0.001 | 0.002 |
| AP001476.3 | chr21:47477561-47477760 | 199 | −1.24 | <0.001 | 0.025 |
| BC034416 | chr3:180586661-180586880 | 219 | −1.31 | <0.001 | 0.007 |
| RN7SL367P | chr16:1946361-1946700 | 339 | −1.35 | <0.001 | 0.004 |
| RP11-586K12.4 | chr16:32752701-32752900 | 199 | −1.37 | <0.001 | 0.005 |
| EIF3B | chr7:2412041-2412260 | 219 | −1.37 | <0.001 | 0.004 |
| ANKIB1 | chr7:91999241-91999440 | 199 | −1.37 | <0.001 | 0.013 |
| XLOC_010373 | chr13:45618701-45618900 | 199 | −1.38 | <0.001 | 0.013 |
| RP11-510M2.5 | chr16:71577621-71577820 | 199 | −1.38 | <0.001 | 0.004 |
| OPN1MW | chrX:153446941-153447140 | 199 | −1.39 | <0.001 | 0.008 |
| ELAC2 | chr17:12919641-12919840 | 199 | −1.45 | <0.001 | 0.012 |
| SLC25A35 | chr17:8196461-8196680 | 219 | −1.52 | <0.001 | 0.004 |
| AK095057 | chr5:179268841-179269080 | 239 | −1.53 | <0.001 | 0.024 |
| C18orf8 | chr18:21081741-21081940 | 199 | −1.55 | <0.001 | 0.002 |
| RP11-168K11.3 | chr9:116382121-116382460 | 339 | −1.62 | <0.001 | 0.005 |
| LL22NC03-N27C7.1 | chr22:24081461-24081680 | 219 | −1.63 | <0.001 | 0.009 |
| CRAMP1L | chr16:1716841-1717040 | 199 | −1.76 | <0.001 | 0.014 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solomon, W.L.; Hector, S.B.E.; Raghubeer, S.; Erasmus, R.T.; Kengne, A.P.; Matsha, T.E. Genome-Wide DNA Methylation and LncRNA-Associated DNA Methylation in Metformin-Treated and -Untreated Diabetes. Epigenomes 2020, 4, 19. https://doi.org/10.3390/epigenomes4030019
Solomon WL, Hector SBE, Raghubeer S, Erasmus RT, Kengne AP, Matsha TE. Genome-Wide DNA Methylation and LncRNA-Associated DNA Methylation in Metformin-Treated and -Untreated Diabetes. Epigenomes. 2020; 4(3):19. https://doi.org/10.3390/epigenomes4030019
Chicago/Turabian StyleSolomon, Wendy L., Stanton B. E. Hector, Shanel Raghubeer, Rajiv T. Erasmus, Andre P. Kengne, and Tandi E. Matsha. 2020. "Genome-Wide DNA Methylation and LncRNA-Associated DNA Methylation in Metformin-Treated and -Untreated Diabetes" Epigenomes 4, no. 3: 19. https://doi.org/10.3390/epigenomes4030019
APA StyleSolomon, W. L., Hector, S. B. E., Raghubeer, S., Erasmus, R. T., Kengne, A. P., & Matsha, T. E. (2020). Genome-Wide DNA Methylation and LncRNA-Associated DNA Methylation in Metformin-Treated and -Untreated Diabetes. Epigenomes, 4(3), 19. https://doi.org/10.3390/epigenomes4030019