Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
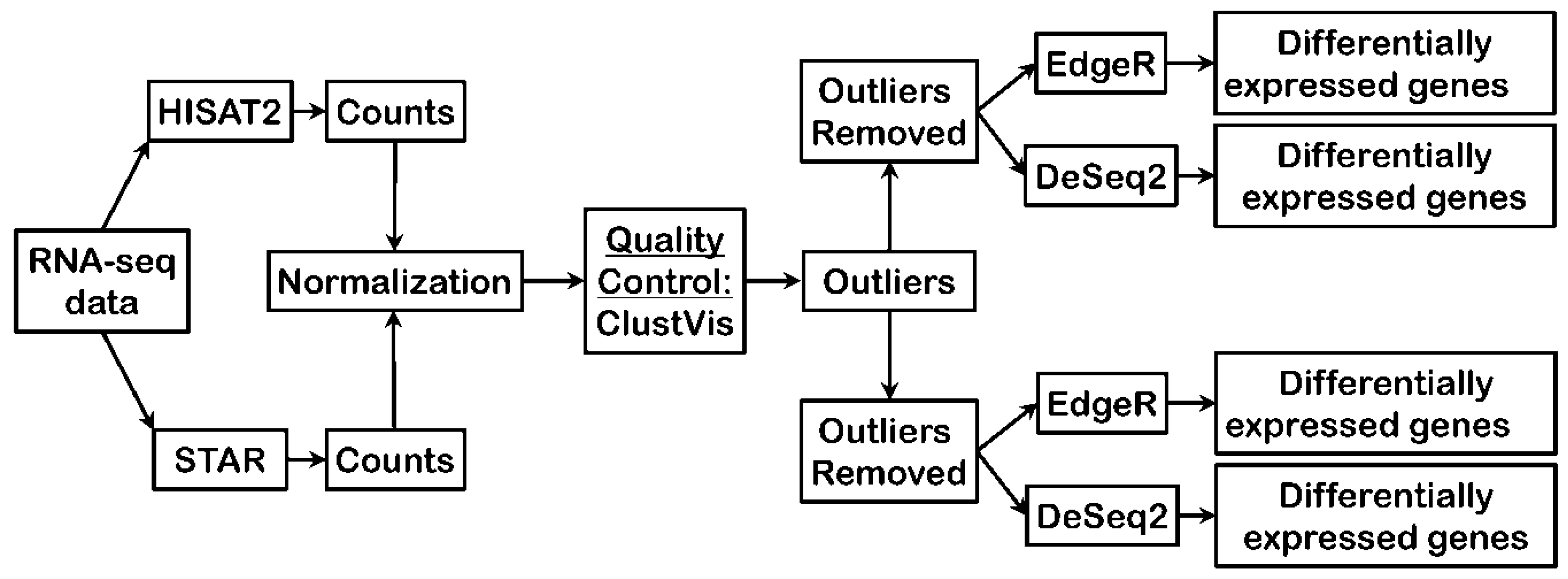
2. Materials and Methods
2.1. Breast Cancer Samples
2.2. RNA-seq Reads Alignment
2.2.1. STAR
–seedSearchStartLmax 50 –seedSearchStartLmaxOverLread 1.0 –seedSearchLmax 0 –seedMultimapNmax 10000 –seedPerReadMax 1000 –seedPerWindowMax 50 –seedNonoLociPerWindow 10 –alignIntronMin 21 –alignIntronMax 0 –alignMatesGapMax 0 –alignSJoverhangMin 5 –alignSJDBoverhangmin 3 –alignSpliceMateMapLmin 0–alignSplicedMateMapeLminOverLmate 0.66 –alignWindowsPerReadNmax 10000 –alignTranscriptsPerWindowNmax 100 –alignTranscriptsPerReadNmax 10000 –alignEndsType Local
2.2.2. HISAT2
–mp MX=6, MN=2 –sp MX=2,MN=1 –np 1 –rdg 5,3 –rfg 5,3 –score-min L,0,-0.2 –pen-cansplice 0 –pen-noncansplice 12 –pen-canintronlen G,-8,1 –pen-noncanintronlen G,-8,1 –min-intronlen 20–max-intronlen 500000
2.3. Gene Expression Counts
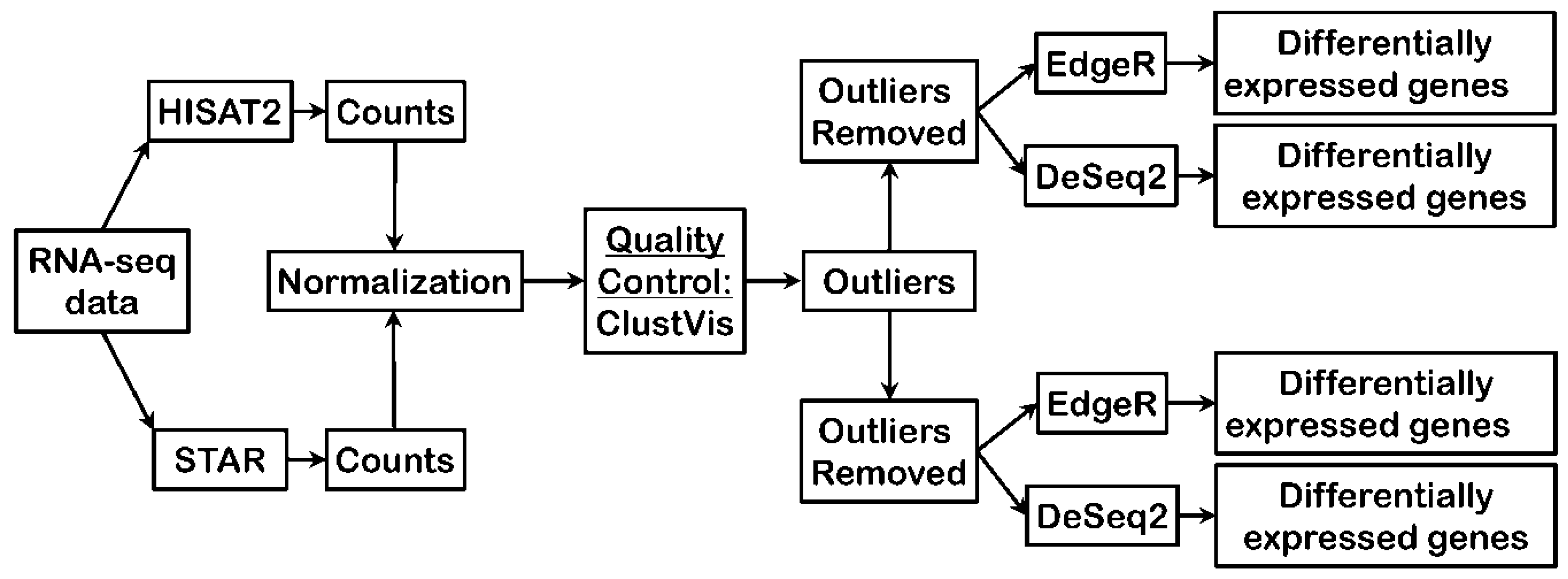
2.4. Data Normalization and Quality Control
2.4.1. Data Normalization
2.4.2. Quality Control
2.5. Differential Gene Expression Analysis
2.5.1. DESeq2
2.5.2. edgeR
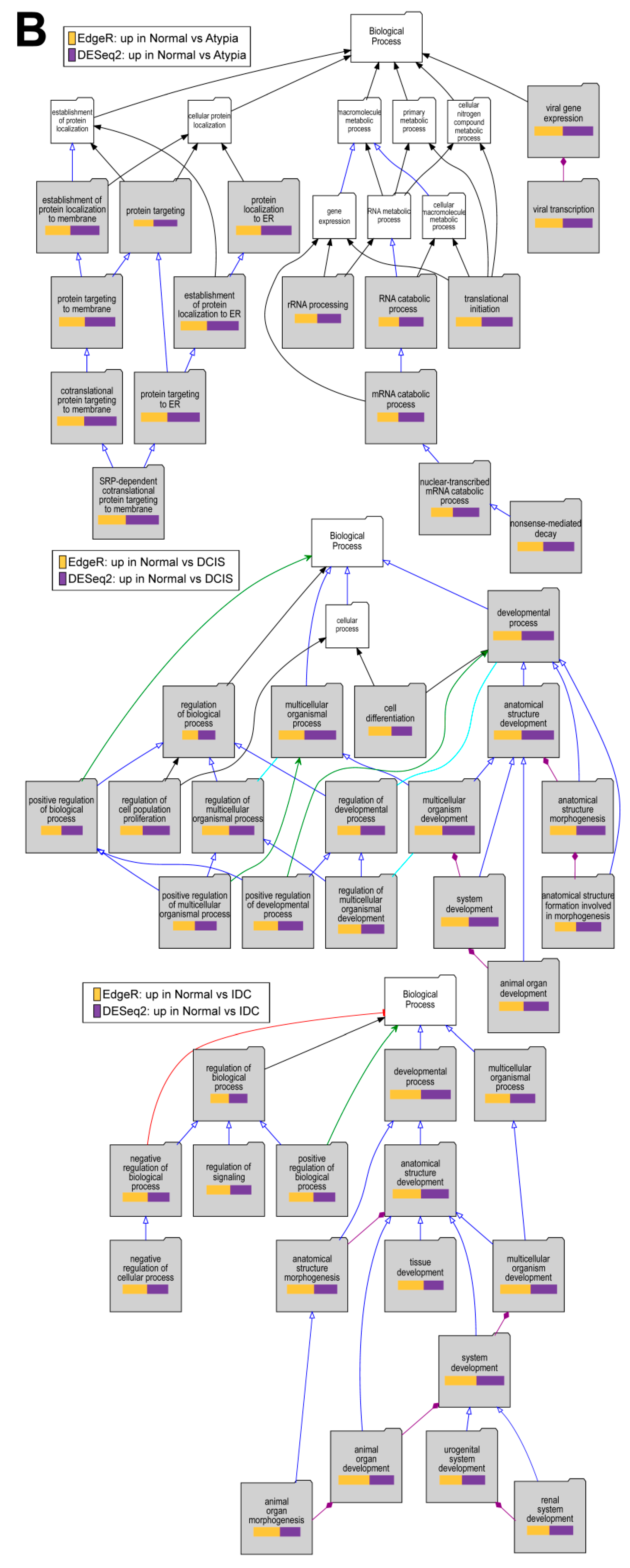
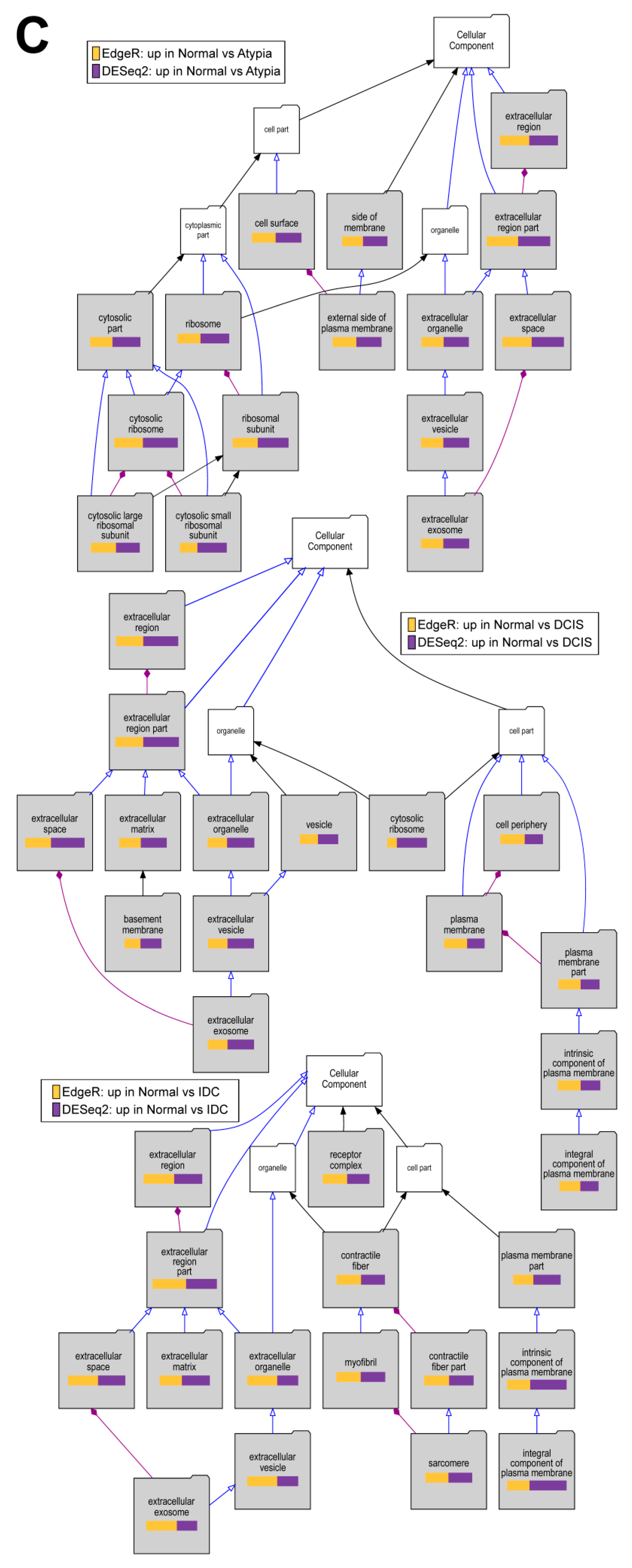
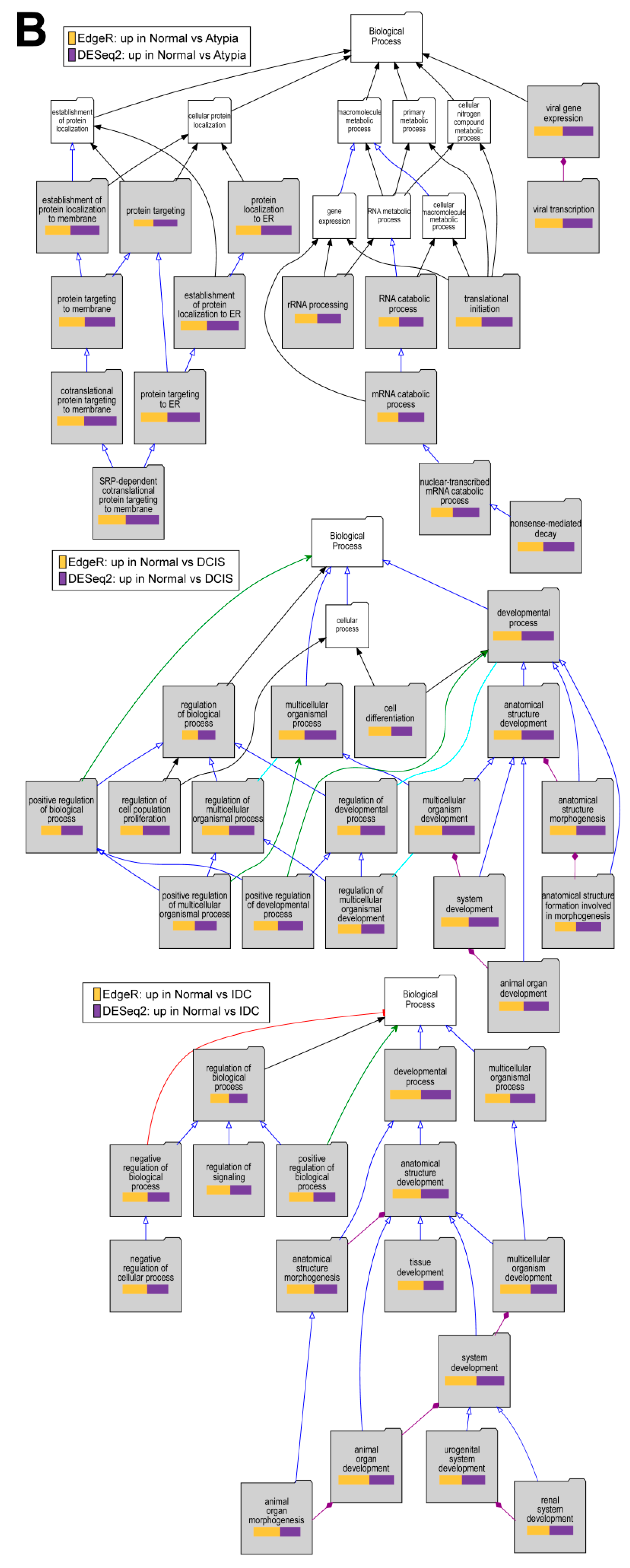
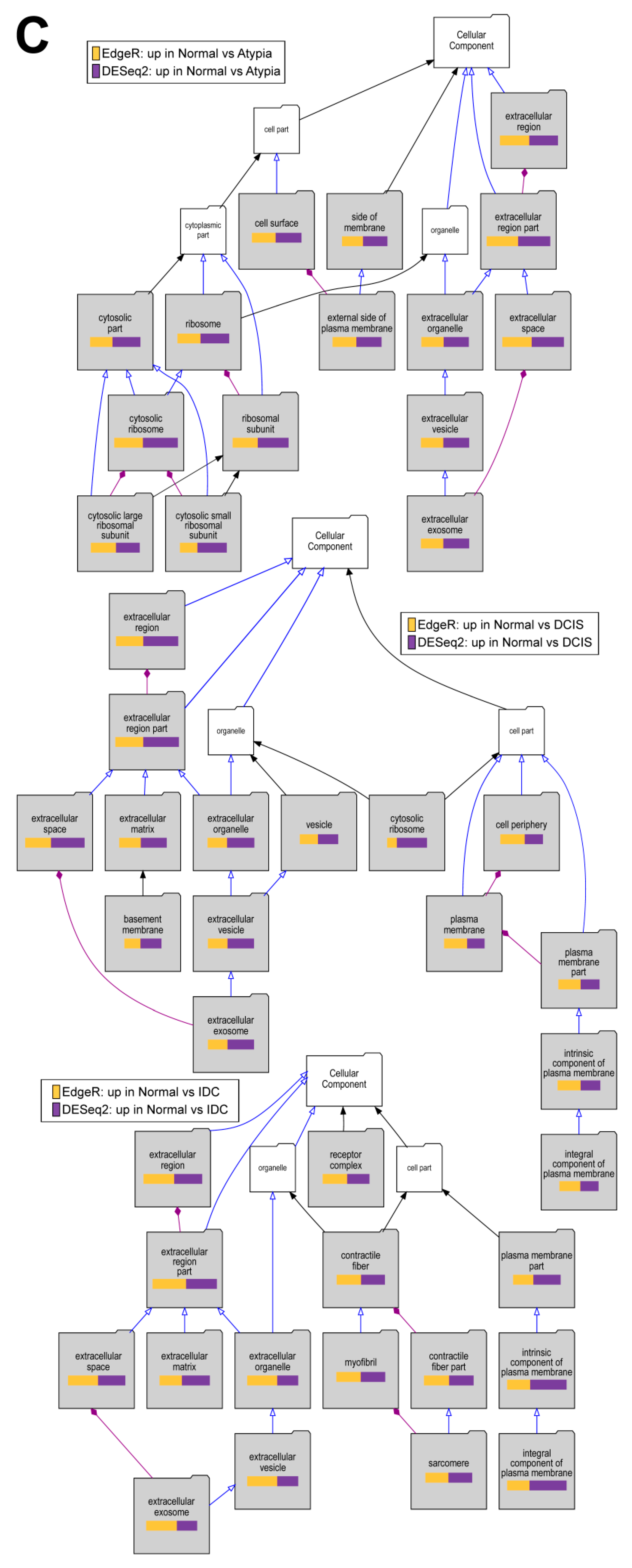
2.6. Gene Enrichment Analysis Using Visual Annotation Display (VLAD)
3. Results
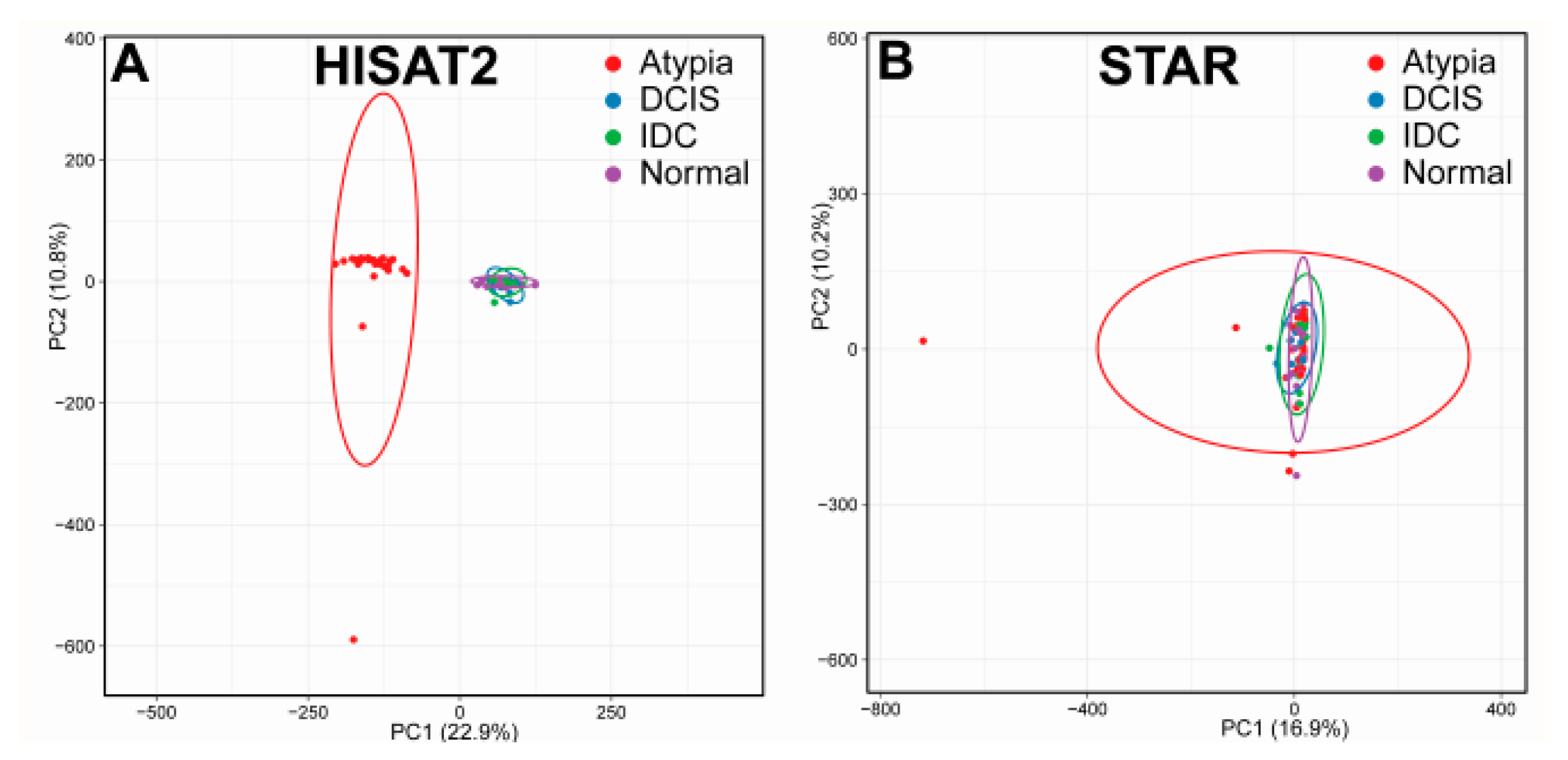
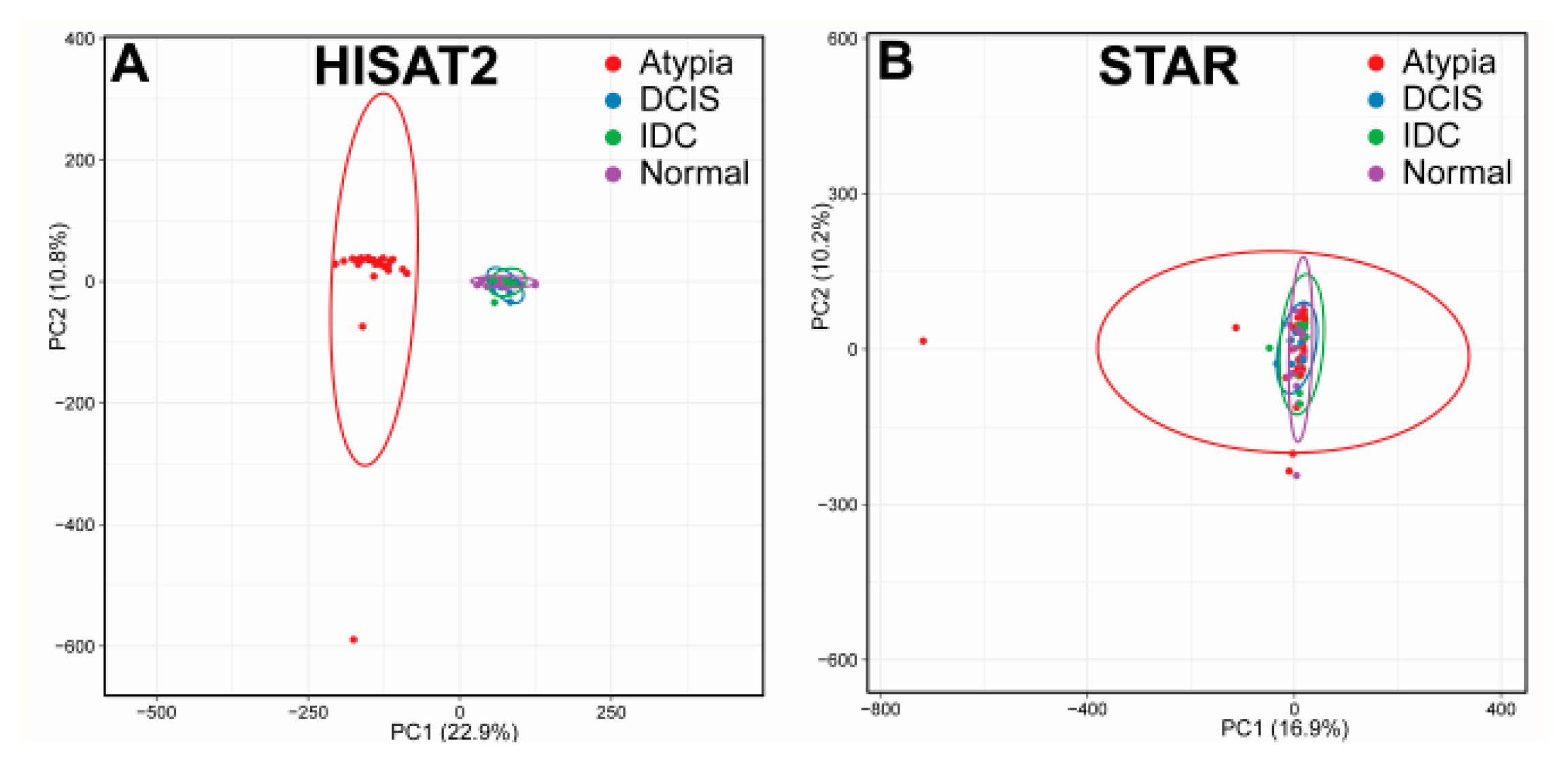
3.1. Bioinformatics Pipeline and Quality Control
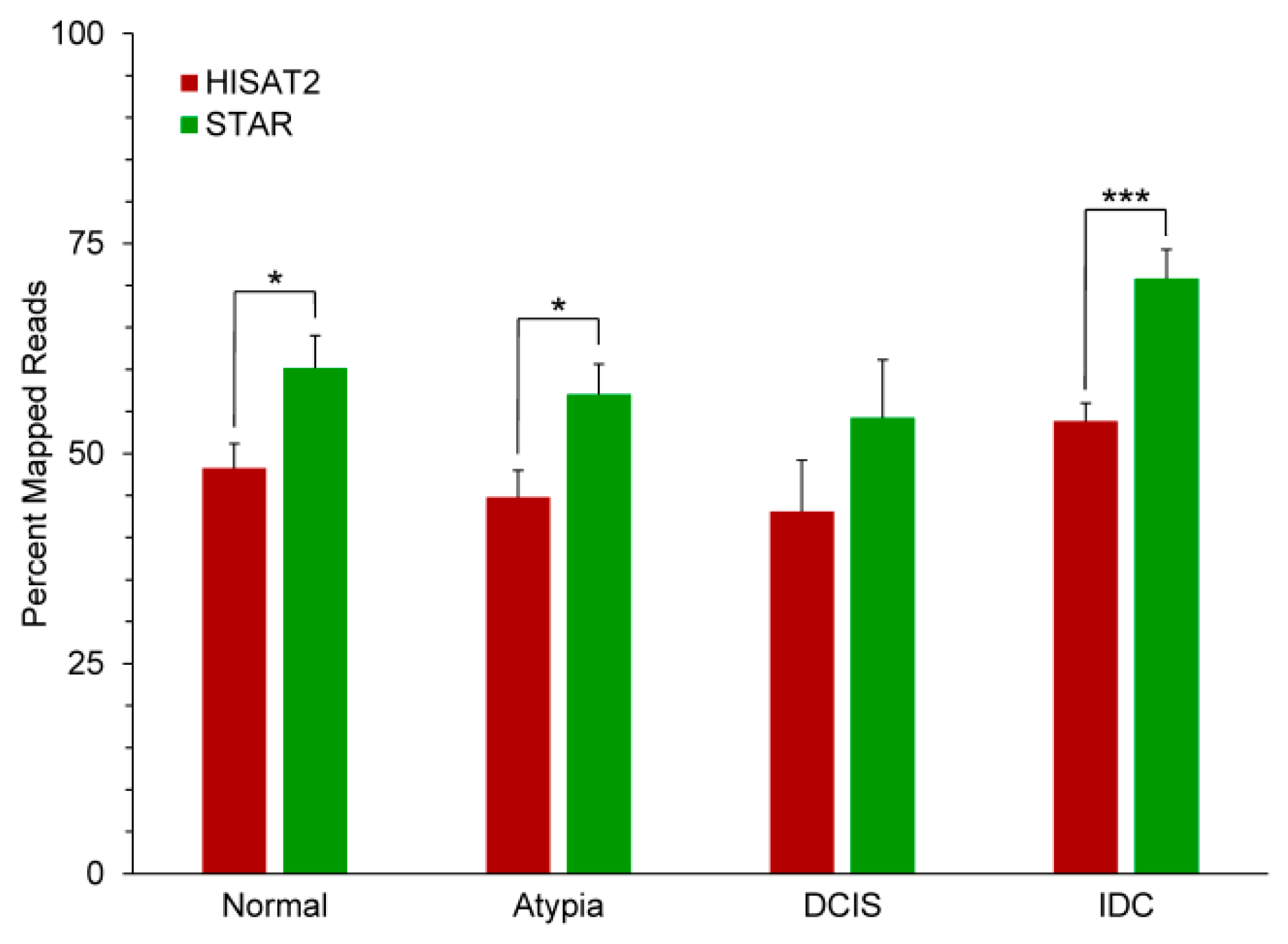
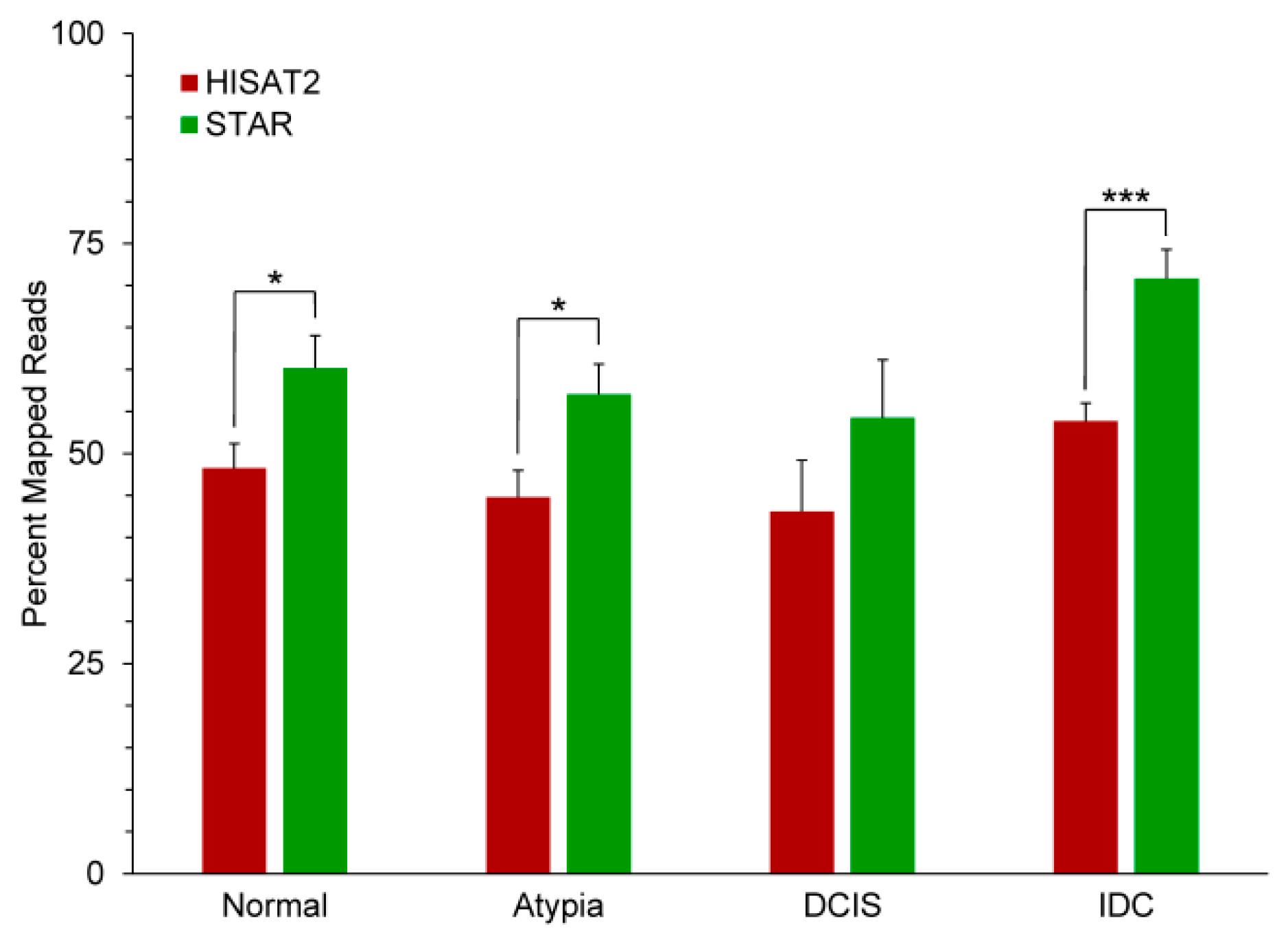
3.2. Outputs of Aligners
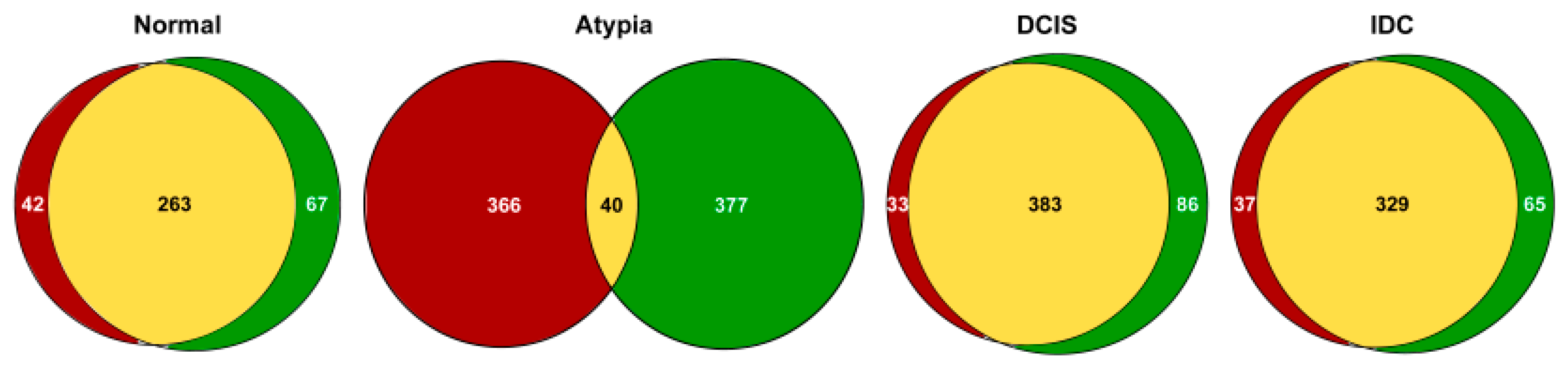
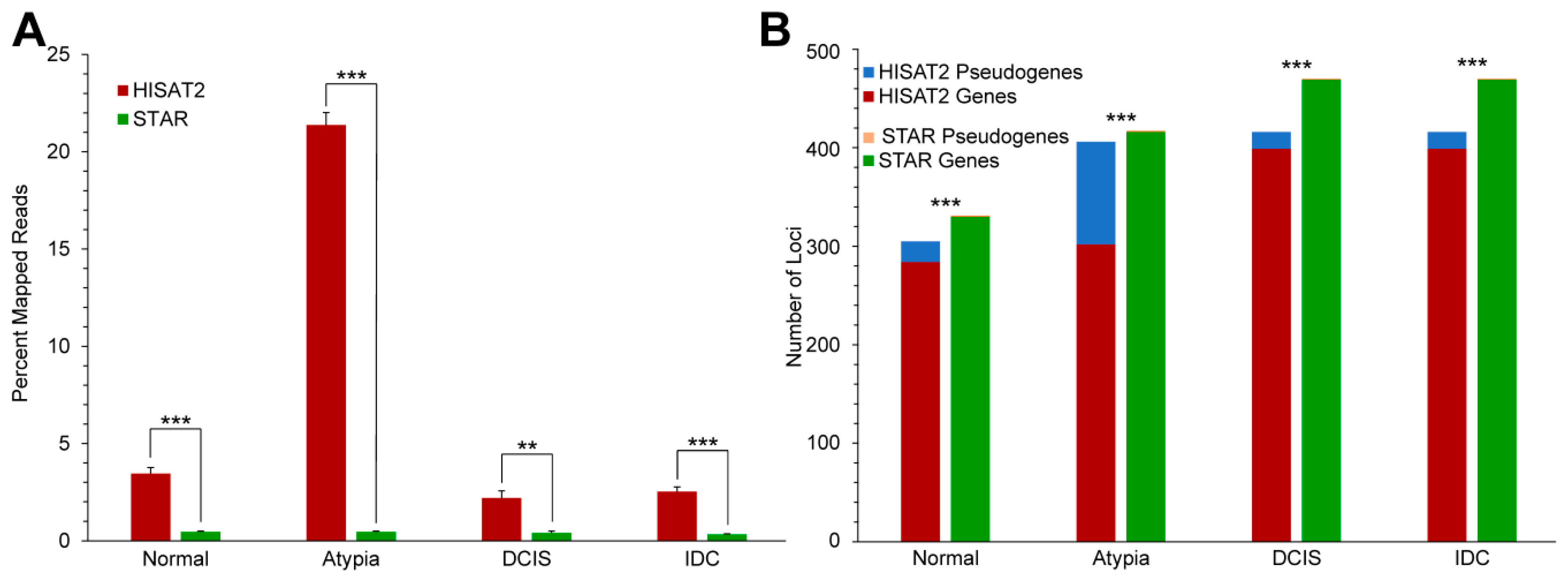
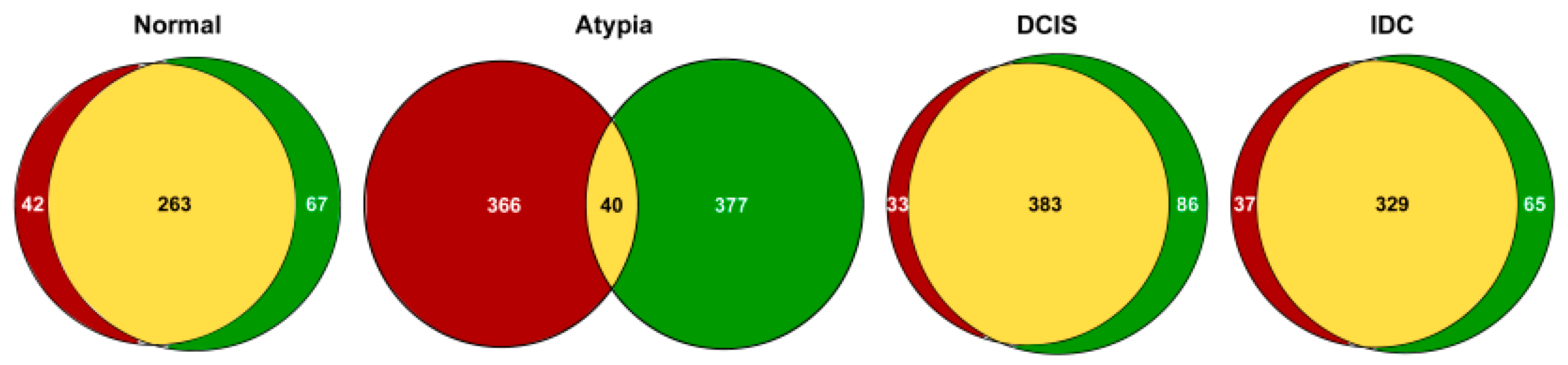
3.3. Gene Expression Profiling
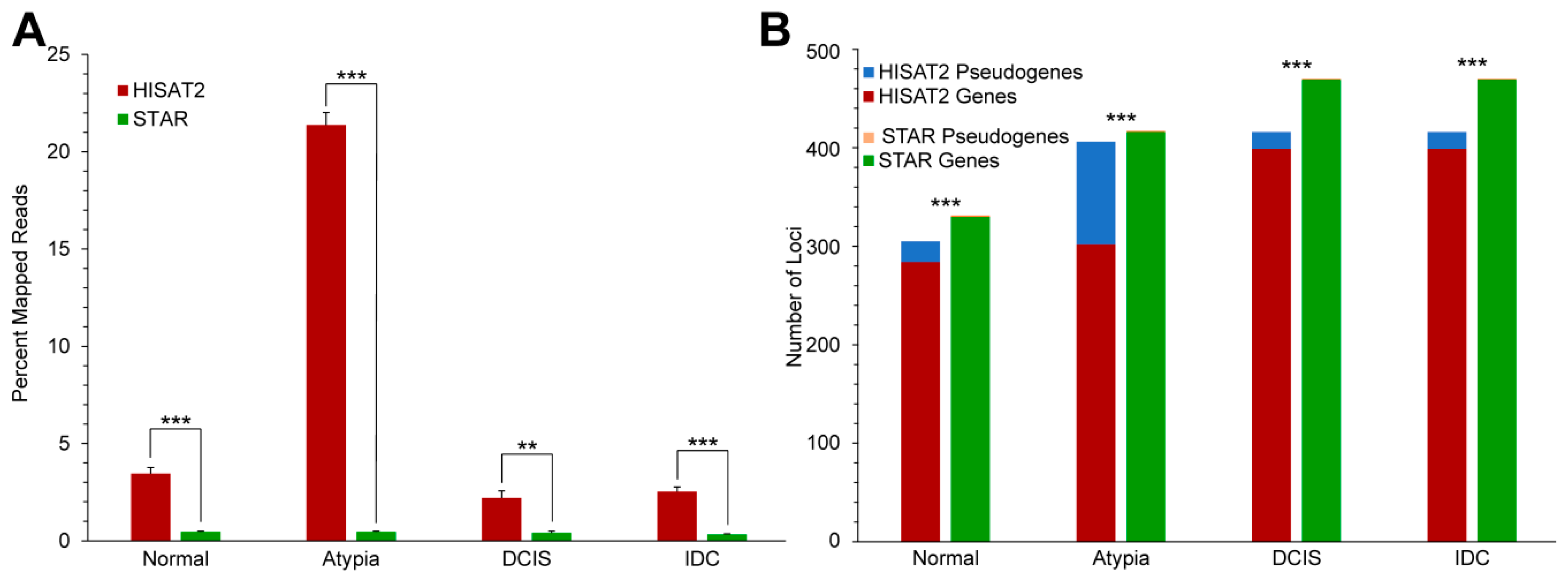
3.3.1. Highly Expressed Genes
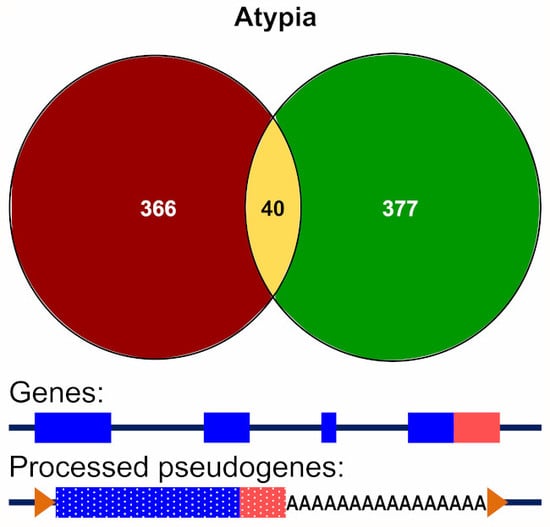
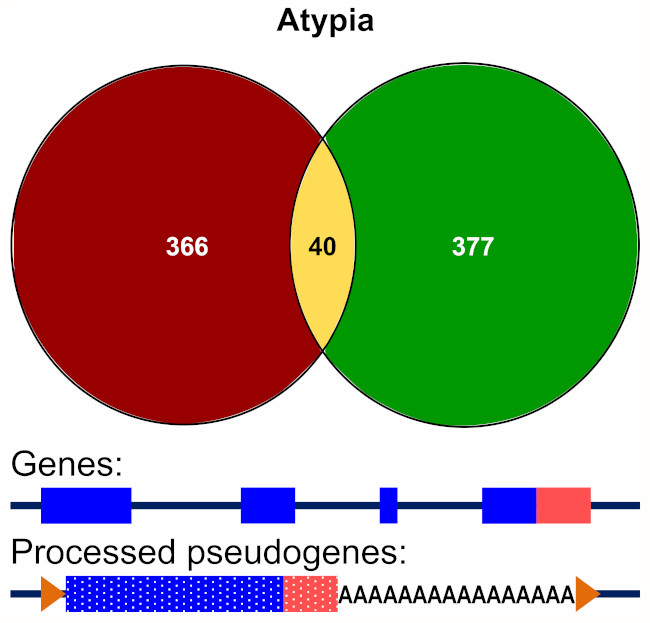
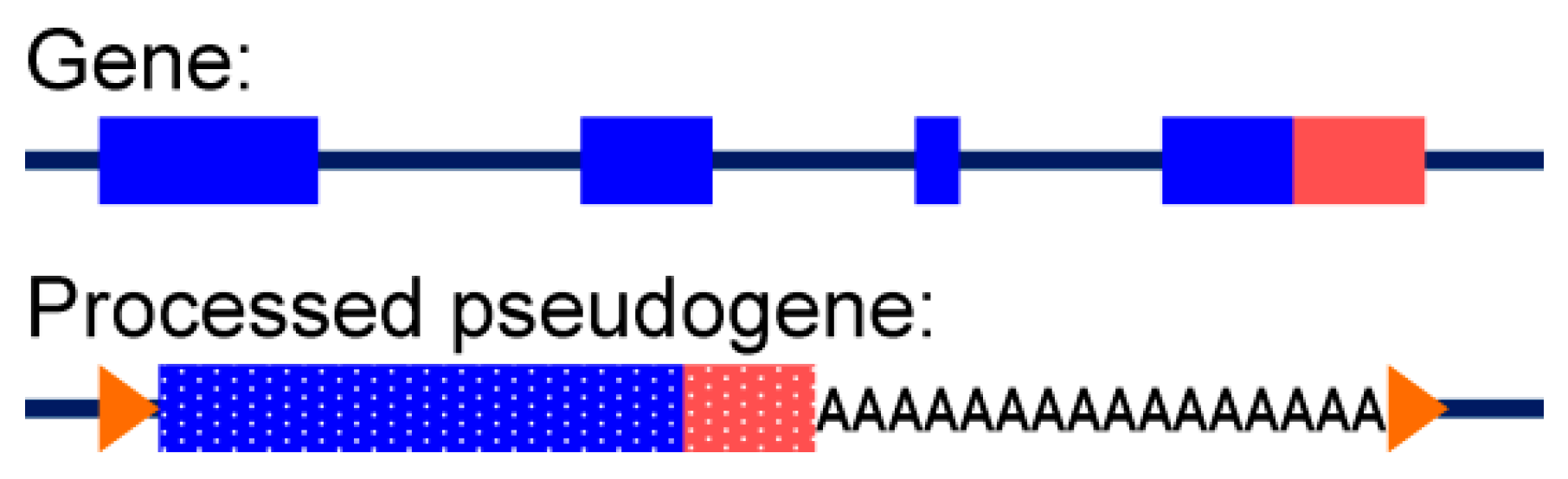
3.3.2. Alignment to Pseudogenes
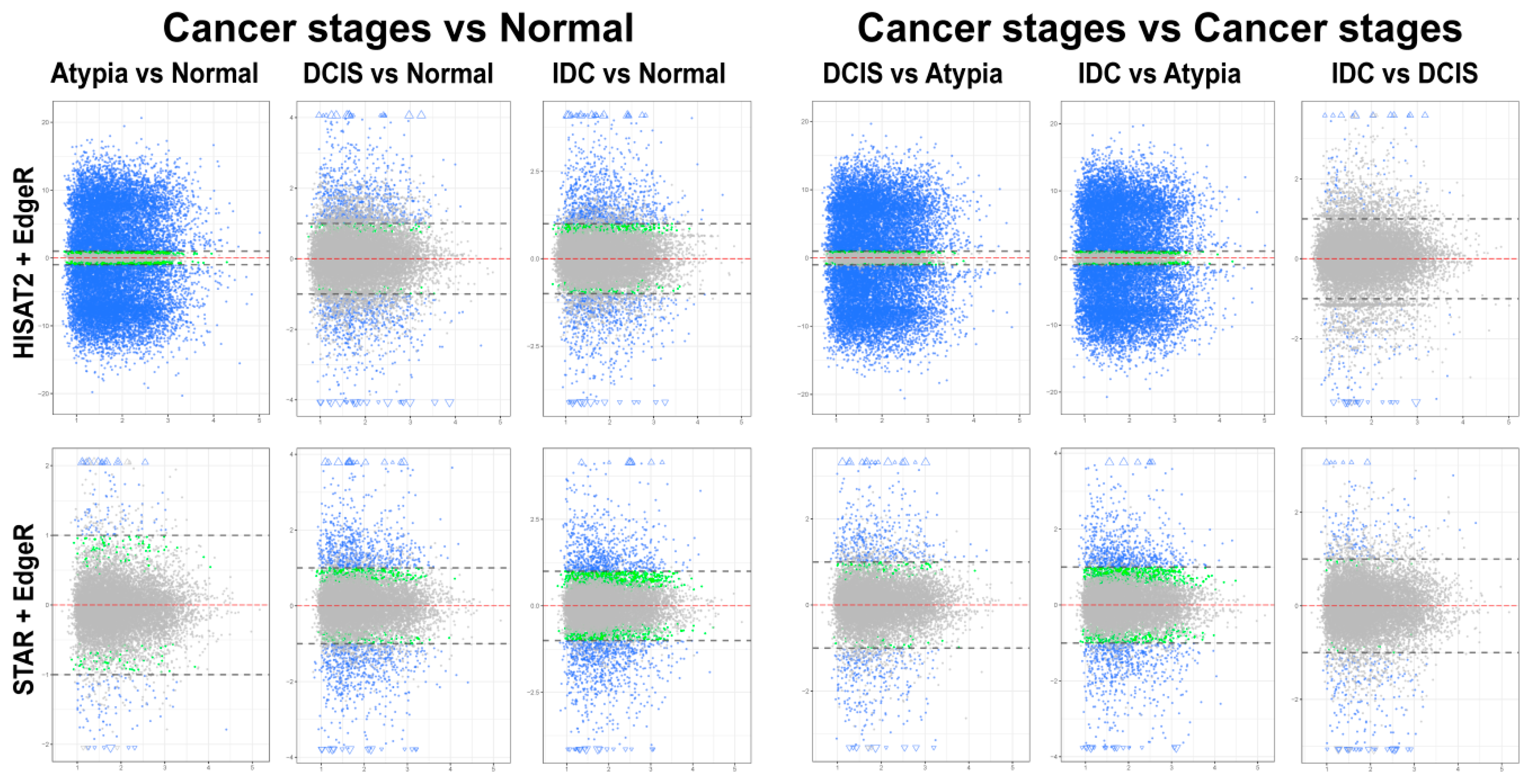
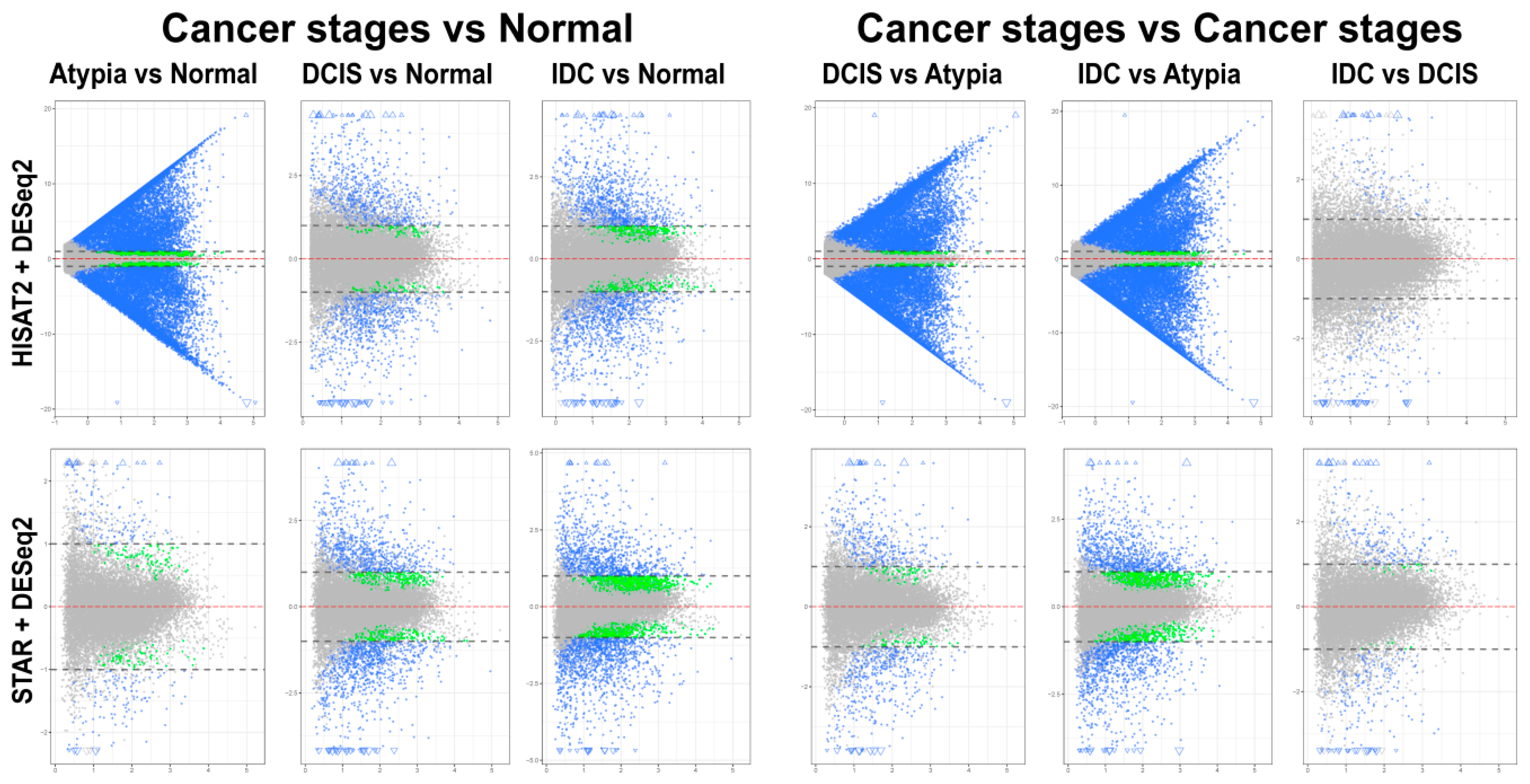
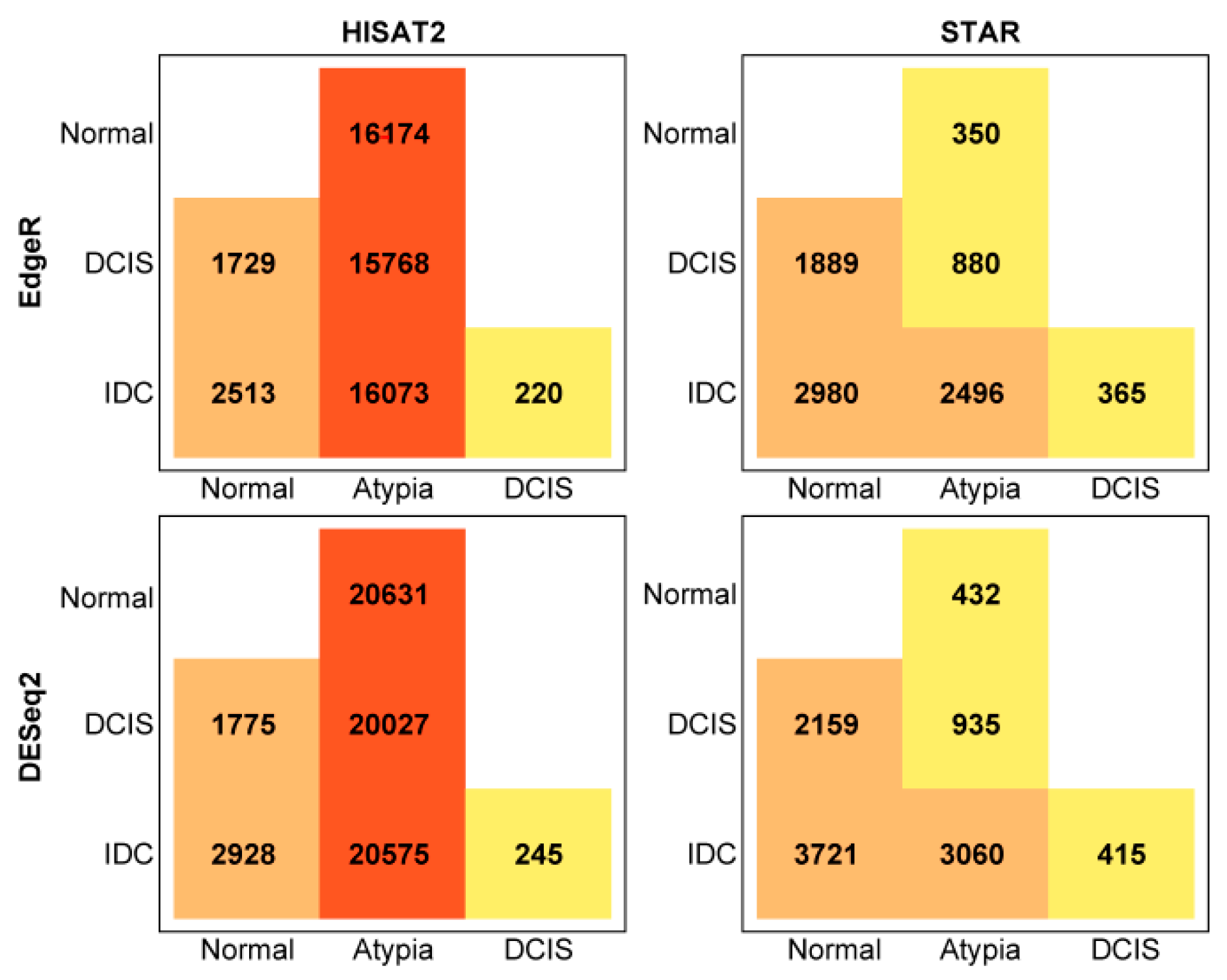
3.4. Differential Gene Expression Analysis
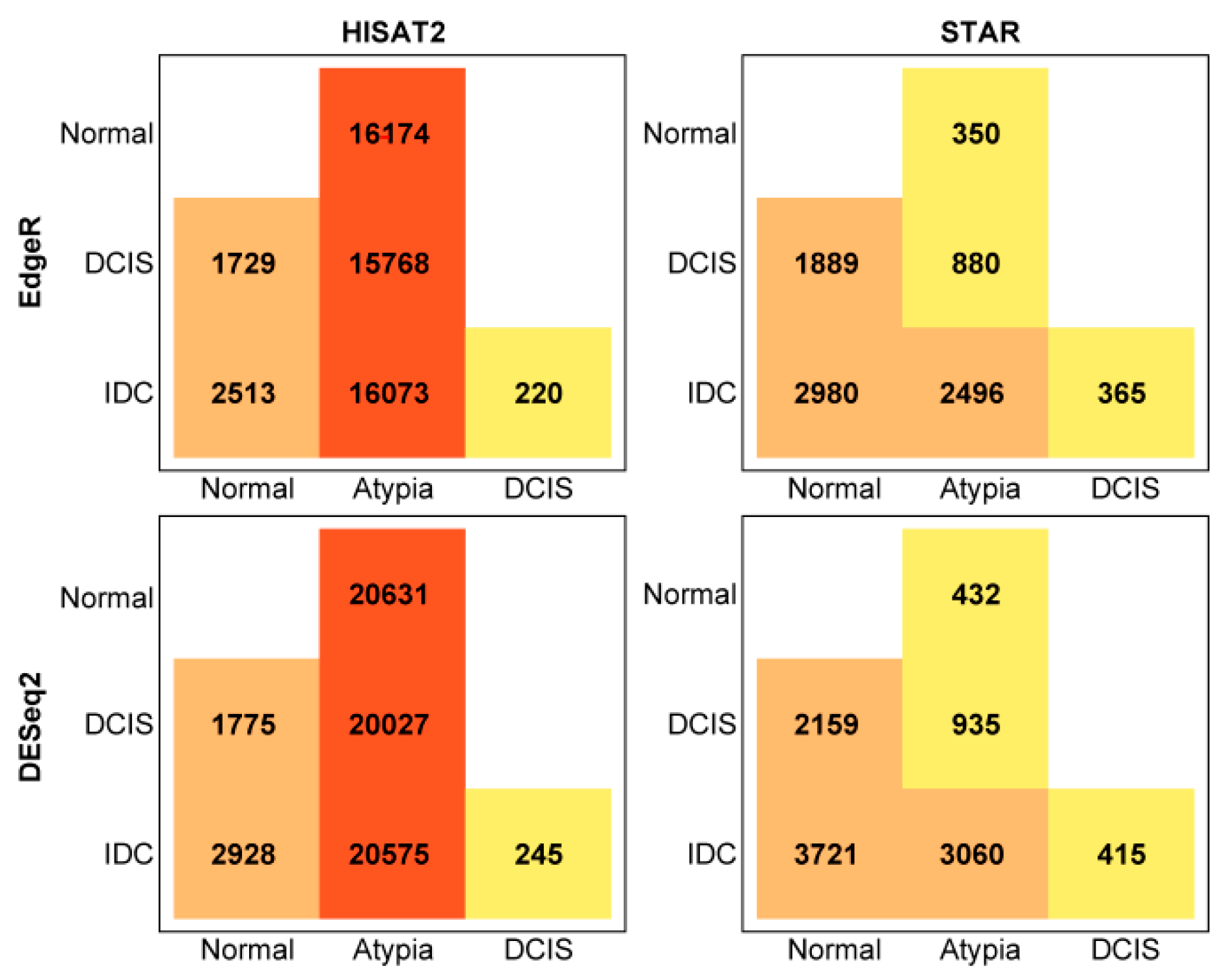
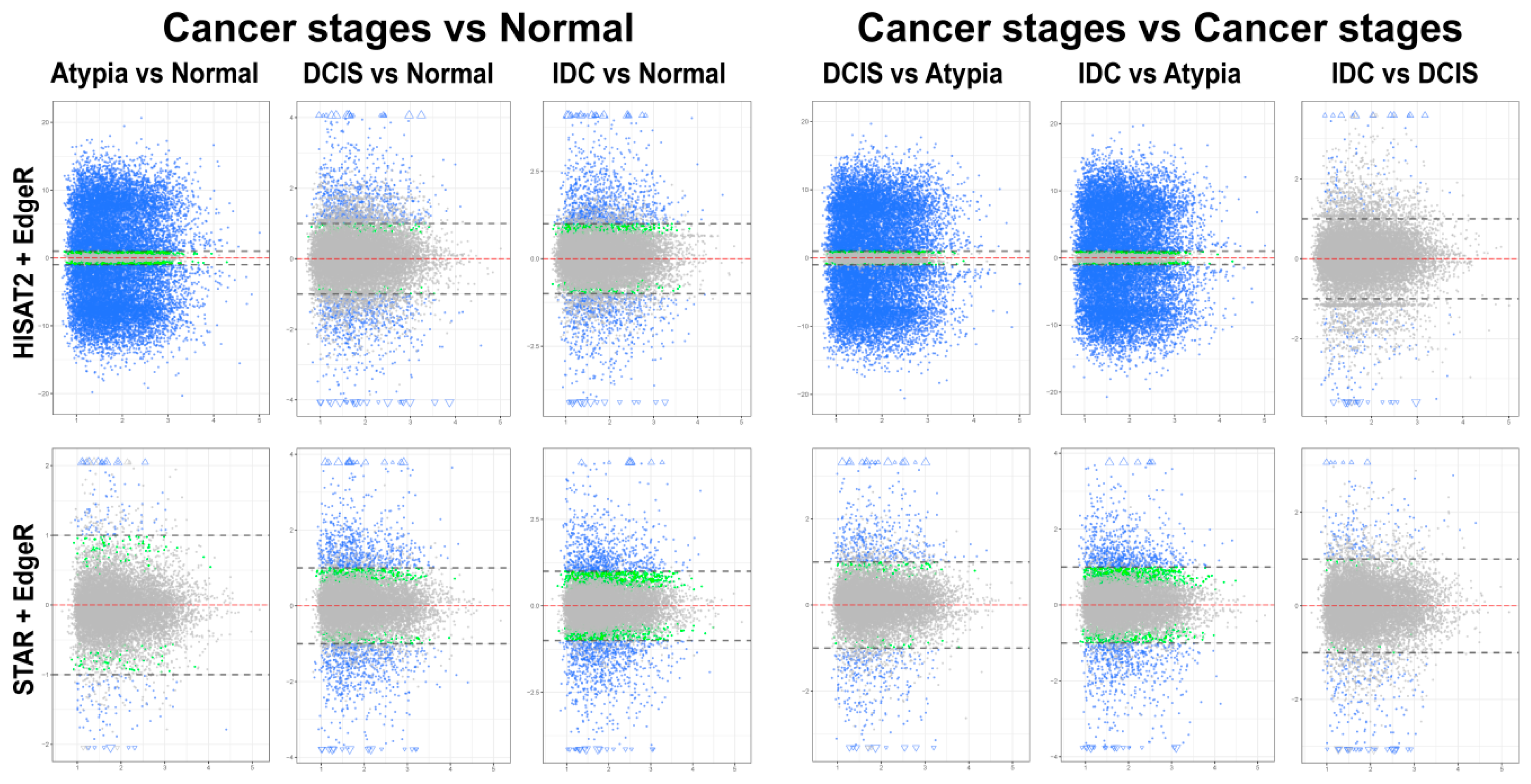
3.4.1. edgeR Following HISAT2 or STAR
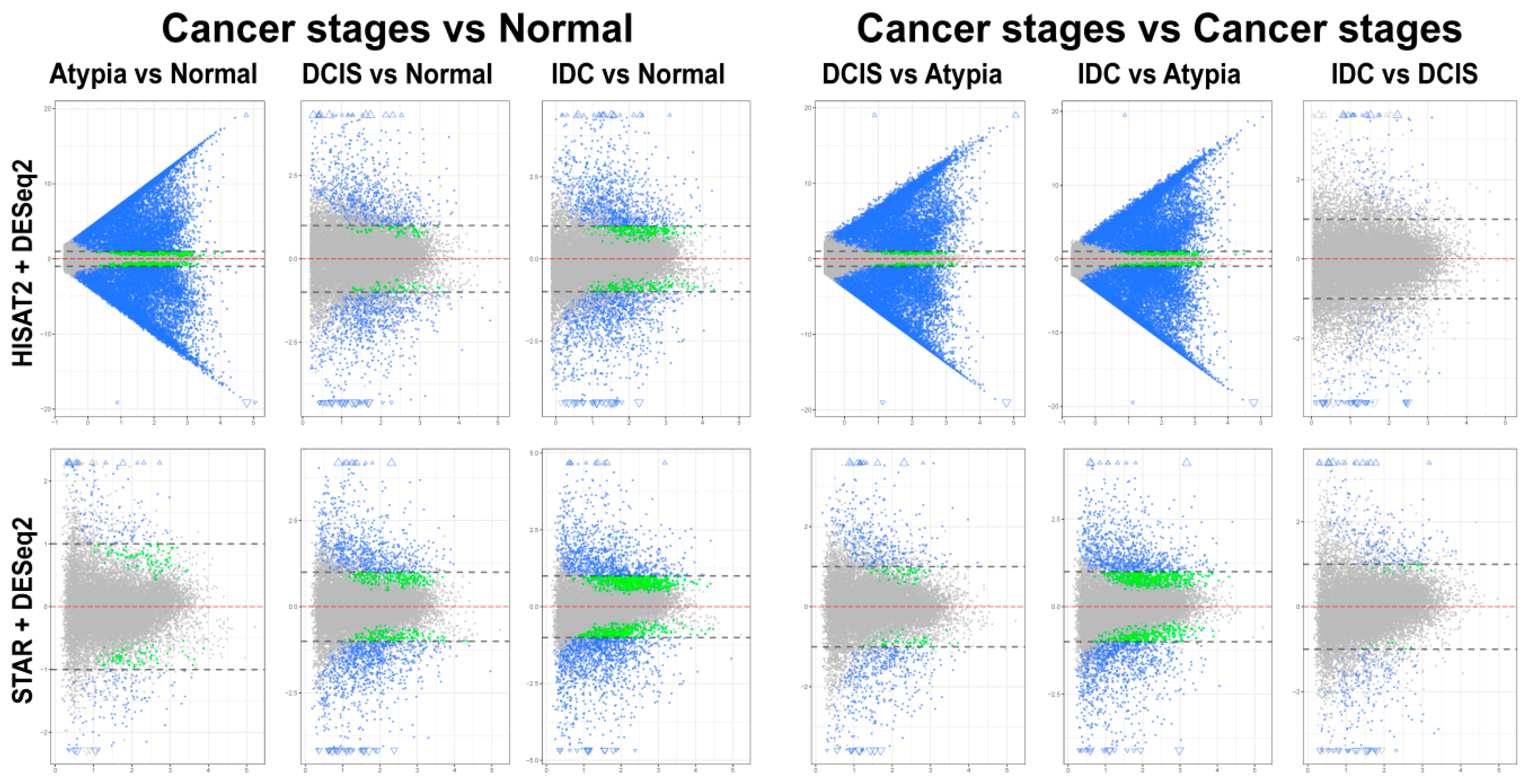
3.4.2. DESeq2 Following HISAT2 and STAR
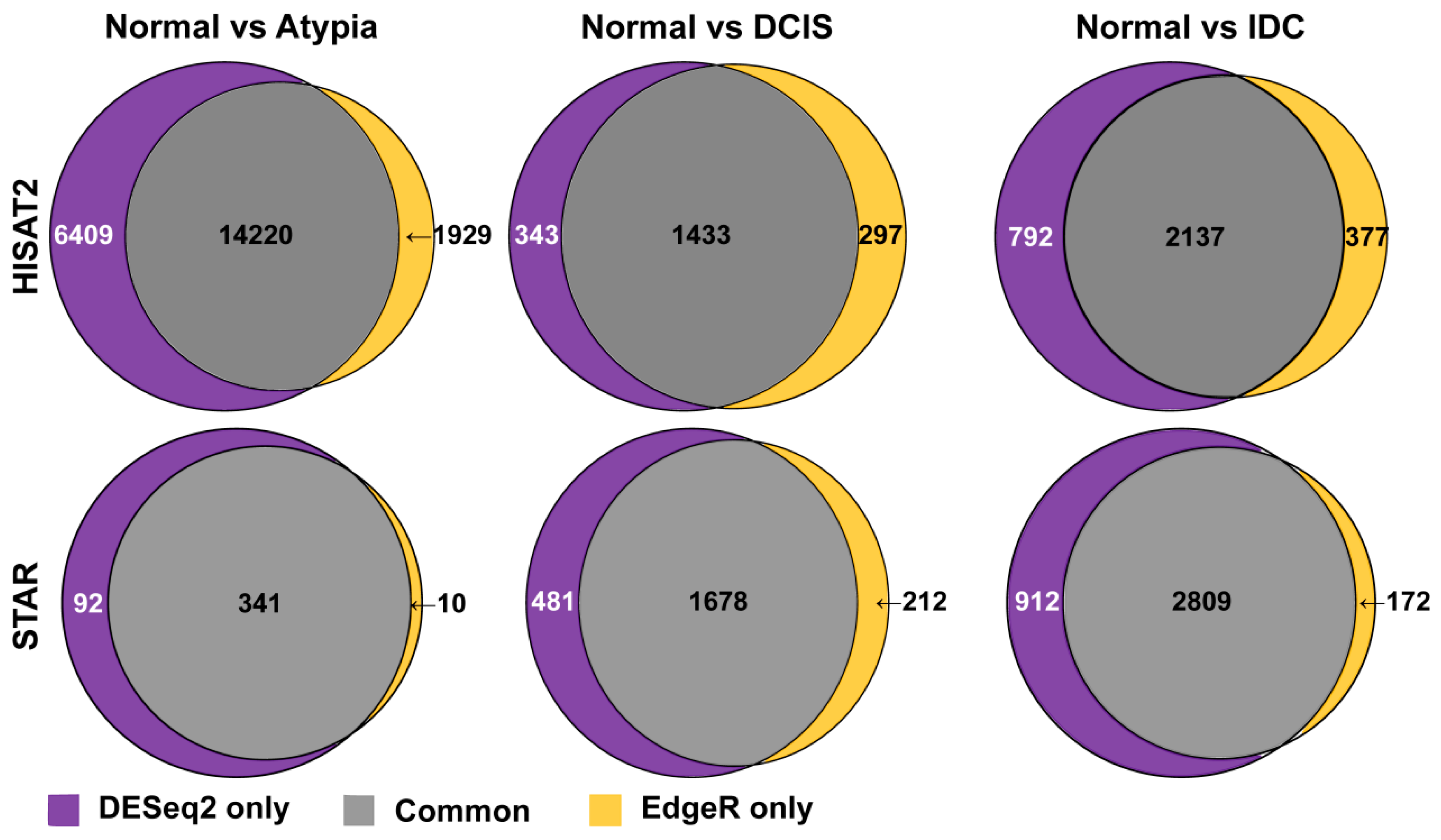
3.4.3. Comparison of DESeq2 and edgeR Results
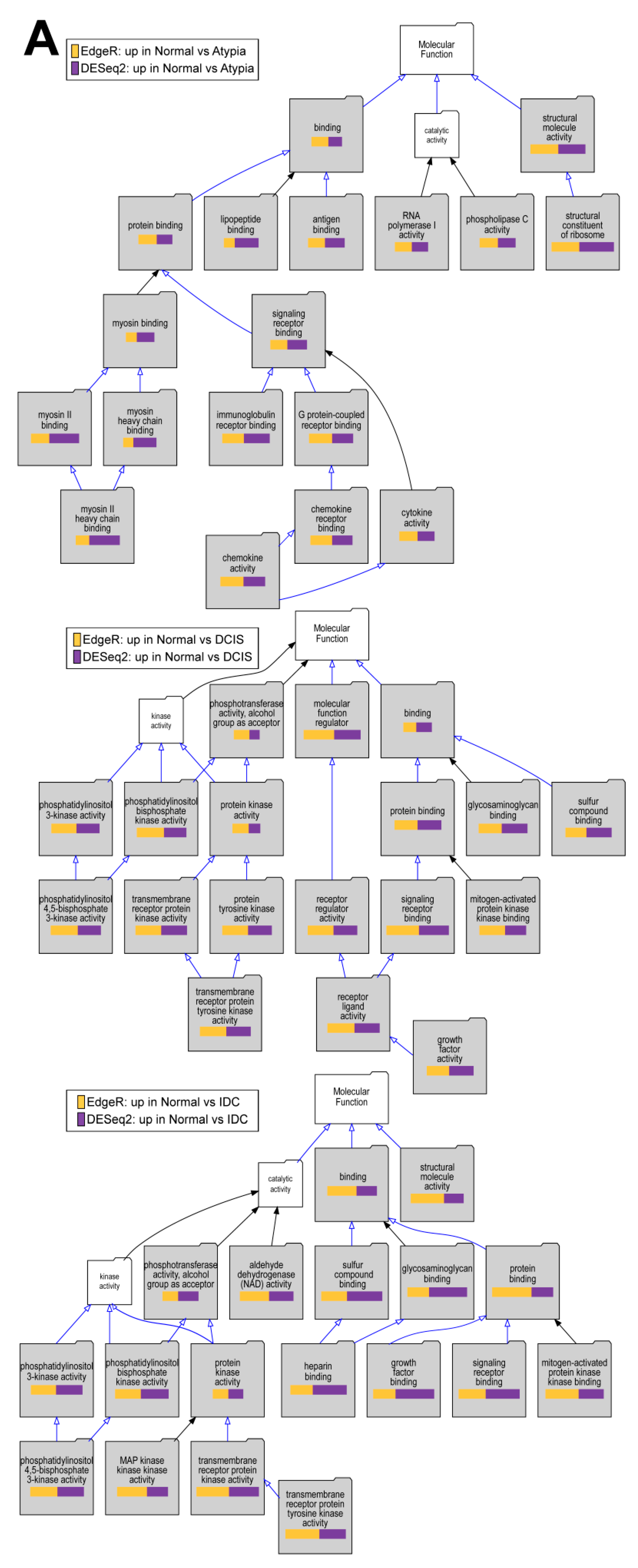
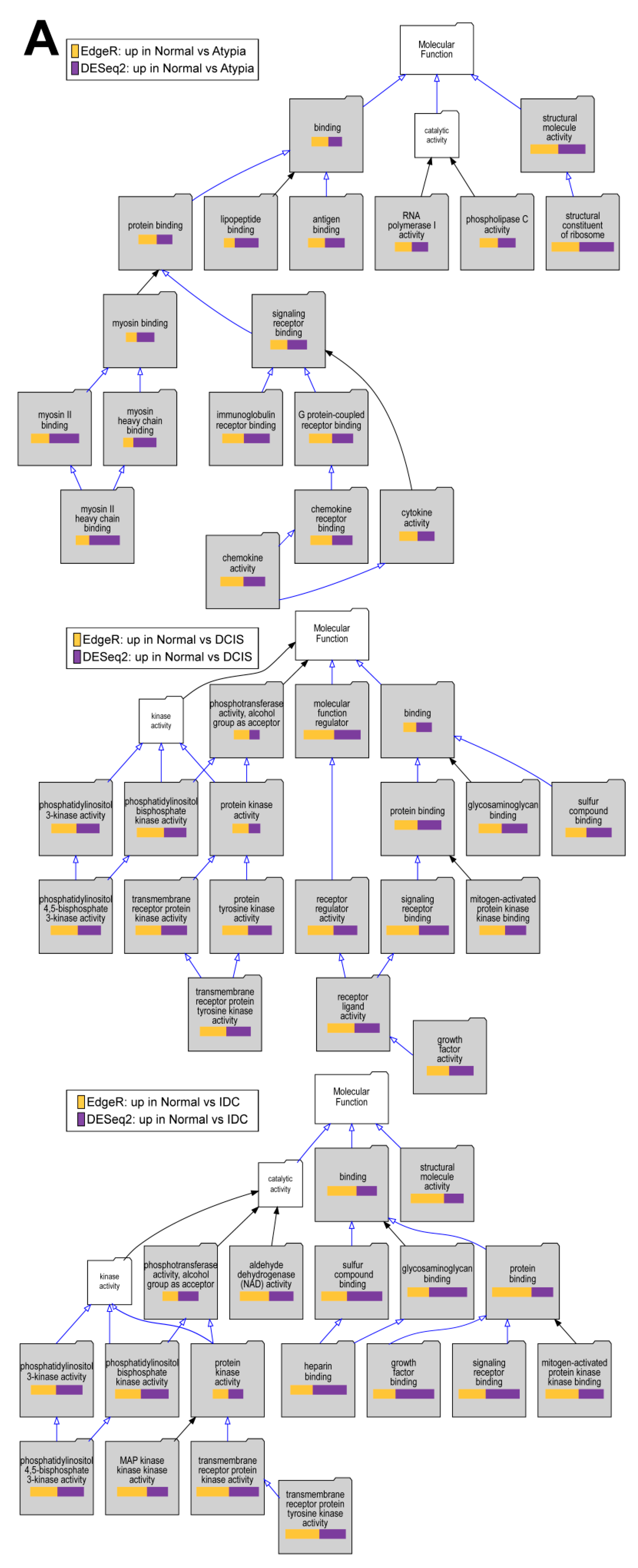
3.4.4. Downstream Analysis of Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hawkins, R.D.; Hon, G.C.; Ren, B. Next-generation genomics: An integrative approach. Nat. Rev. Genet. 2010, 11, 476. [Google Scholar] [CrossRef] [PubMed]
- Senkus, E.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rutgers, E.; Zackrisson, S.; Cardoso, F. Primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v8–v30. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Members, P.; André, F. Tailoring therapies—Improving the management of early breast cancer: St Gallen international expert consensus on the primary therapy of early breast cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef]
- NCBI. SRA Database Growth. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sragrowth/ (accessed on 22 February 2019).
- Ben-Ezra, J.; Johnson, D.A.; Rossi, J.; Cook, N.; Wu, A. Effect of fixation on the amplification of nucleic acids from paraffin-embedded material by the polymerase chain reaction. J. Histochem. Cytochem. 1991, 39, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Ohnishi, T.; Kawamoto, S.; Monden, M.; Okubo, K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999, 27, 4436–4443. [Google Scholar] [CrossRef]
- Srinivasan, M.; Sedmak, D.; Jewell, S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am. J. Pathol. 2002, 161, 1961–1971. [Google Scholar] [CrossRef]
- Buckingham, L. Molecular Diagnostics: Fundamentals, Methods and Clinical Applications; FA Davis: Philadelphia, PA, USA, 2011. [Google Scholar]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Google Scholar. Available online: https://scholar.google.com/ (accessed on 22 February 2019).
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Galaxy Team. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.L.; Li, J.; Guo, X.; Sweeney, R.T.; Varma, S.; Zhu, S.X.; Li, R.; Tibshirani, R.; West, R.B. A shared transcriptional program in early breast neoplasias despite genetic and clinical distinctions. Genome Biol. 2014, 15, R71. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Simpson, J.T.; Durbin, R. Efficient construction of an assembly string graph using the FM-index. Bioinformatics 2010, 26, i367–i373. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- McDermaid, A.; Monier, B.; Zhao, J.; Ma, Q. Vidger: An R package for integrative interpretation of differential gene expression results of RNA-seq data. bioRxiv 2018. [Google Scholar]
- McCullagh, P.; Nelder, J.A. Generalized Linear Models; CRC press: Boca Raton, FL, USA, 1989; Volume 37. [Google Scholar]
- Richardson, J.E.; Bult, C.J. Visual annotation display (VLAD): A tool for finding functional themes in lists of genes. Mamm. Genome 2015, 26, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Goldsmith, C.-A.W.; Eppig, J.T. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2004, 6, R7. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Ashburner, M.; Rosse, C.; Bard, J.; Bug, W.; Ceusters, W.; Goldberg, L.J.; Eilbeck, K.; Ireland, A.; Mungall, C.J.; et al. The OBO foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007, 25, 1251. [Google Scholar] [CrossRef]
- PDQ Cancer Genetics Editorial Board. Genetics of breast and gynecologic cancers (PDQ®). In PDQ Cancer Information Summaries [Internet]; National Cancer Institute (US): Bethesda, MD, USA, 2018. [Google Scholar]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with Tophat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Baruzzo, G.; Hayer, K.E.; Kim, E.J.; Di Camillo, B.; FitzGerald, G.A.; Grant, G.R. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 2016, 14, 135. [Google Scholar] [CrossRef]
- Engström, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; The, R.C.; Alioto, T.; Behr, J.; Bertone, P.; Bohnert, R.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Pachter, L. Models for Transcript Quantification from RNA-seq. Available online: https://arxiv.org/abs/1104.3889 (accessed on 26 February 2019).
- Chhangawala, S.; Rudy, G.; Mason, C.E.; Rosenfeld, J.A. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. 2015, 16, 131. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.; Smyth, G.K. Differential expression analysis of complex RNA-seq experiments using edgeR. In Statistical Analysis of Next Generation Sequencing Data; Springer: New York, NY, USA, 2014; pp. 51–74. [Google Scholar]
- Goh, W.W.B.; Wang, W.; Wong, L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raplee, I.D.; Evsikov, A.V.; Marín de Evsikova, C. Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research. J. Pers. Med. 2019, 9, 18. https://doi.org/10.3390/jpm9020018
Raplee ID, Evsikov AV, Marín de Evsikova C. Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research. Journal of Personalized Medicine. 2019; 9(2):18. https://doi.org/10.3390/jpm9020018
Chicago/Turabian StyleRaplee, Isaac D., Alexei V. Evsikov, and Caralina Marín de Evsikova. 2019. "Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research" Journal of Personalized Medicine 9, no. 2: 18. https://doi.org/10.3390/jpm9020018
APA StyleRaplee, I. D., Evsikov, A. V., & Marín de Evsikova, C. (2019). Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research. Journal of Personalized Medicine, 9(2), 18. https://doi.org/10.3390/jpm9020018