
Asthma-Associated COPD Etiotype: Clinical Features and Inflammatory Patterns in Biological Samples

 ,
,
Abstract
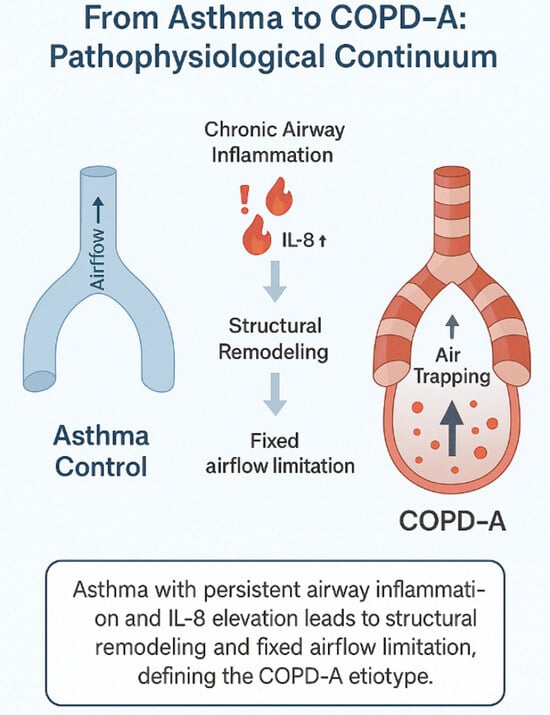
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Proceedings
2.3. Biochemical Analyses
2.4. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACO | Asthma–COPD Overlap |
| ACQ | Asthma Control Questionnaire |
| ACT | Asthma Control Test |
| aOR | Adjusted Odds Ratio |
| AQLQ | Asthma Quality Of Life Questionnaire |
| AR | Airway Remodeling |
| BD | Bronchodilator |
| BMI | Body Mass Index |
| CI | Confidence Interval |
| COPD | Chronic Obstructive Pulmonary Disease |
| FEV1 | Forced Expiratory Value In The First Second. |
| FVC | Forced Vital Capacity |
| GINA | Global Initiative For Asthma |
| GOLD | Chronic Obstructive Pulmonary Disease |
| IL | Interleukin |
| IS | Induced Sputum |
| KOLD | Korean Obstructive Lung Disease |
| NL | Nasal Lavage |
| SD | Standard Deviation |
| TNF | Tumor Necrosis Factor |
References
- Global Strategy for Asthma Management and Prevention (2024 Update): Global Initiative for Asthma (GINA). Available online: www.ginasthma.org (accessed on 24 March 2025).
- Global Strategy for Prevention, Diagnosis and Management of COPD: 2025 Report: Global Initiative for Chronic Obstructive Lung Disease (GOLD). Available online: https://goldcopd.org/ (accessed on 24 March 2025).
- Leung, C.; Sin, D.D. Asthma-COPD Overlap: What Are the Important Questions? Chest 2022, 161, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.; Fabbri, L.; Criner, G.; Martinez, F.J.; Mannino, D.; Vogelmeier, C.; Montes de Oca, M.; Papi, A.; Sin, D.D.; Han, M.K.; et al. Definition and Nomenclature of Chronic Obstructive Pulmonary Disease: Time for Its Revision. Am. J. Respir. Crit. Care Med. 2022, 206, 1317–1325. [Google Scholar] [CrossRef]
- Bhatt, S.P.; Agusti, A.; Bafadhel, M.; Christenson, S.A.; Bon, J.; Donaldson, G.C.; Sin, D.D.; Wedzicha, J.A.; Martinez, F.J. Phenotypes, Etiotypes, and Endotypes of Exacerbations of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2023, 208, 1026–1041. [Google Scholar] [CrossRef] [PubMed]
- de Marco, R.; Accordini, S.; Marcon, A.; Cerveri, I.; Antó, J.M.; Gislason, T.; Heinrich, J.; Janson, C.; Jarvis, D.; Kuenzli, N.; et al. Risk factors for chronic obstructive pulmonary disease in a European cohort of young adults. Am. J. Respir. Crit. Care Med. 2011, 183, 891–897. [Google Scholar] [CrossRef]
- Lange, P.; Parner, J.; Vestbo, J.; Schnohr, P.; Jensen, G. A 15-year follow-up study of ventilatory function in adults with asthma. N. Engl. J. Med. 1998, 339, 1194–1200. [Google Scholar] [CrossRef]
- Silva, G.E.; Sherrill, D.L.; Guerra, S.; Barbee, R.A. Asthma as a risk factor for COPD in a longitudinal study. Chest 2004, 126, 59–65. [Google Scholar] [CrossRef]
- Tiotiu, A.; Steiropoulos, P.; Novakova, S.; Nedeva, D.; Novakova, P.; Chong-Neto, H.; Fogelbach, G.G.; Kowal, K. Airway Remodeling in Asthma: Mechanisms, Diagnosis, Treatment, and Future Directions. Arch. Bronconeumol. 2025, 61, 31–40. [Google Scholar] [CrossRef]
- Ojiaku, C.A.; Yoo, E.J.; Panettieri, R.A., Jr. Transforming Growth Factor β1 Function in Airway Remodeling and Hyperresponsiveness. The Missing Link? Am. J. Respir. Cell Mol. Biol. 2017, 56, 432–442. [Google Scholar] [CrossRef]
- Tiotiu, A. Biomarkers in asthma: State of the art. Asthma Res. Pract. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway Remodeling in Asthma. Front. Med. 2020, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Tatler, A.L. Pathobiology of Airway Remodeling in Asthma: The Emerging Role of Integrins. J. Asthma Allergy 2022, 15, 595–610. [Google Scholar] [CrossRef]
- Lezmi, G.; Gosset, P.; Deschildre, A.; Abou-Taam, R.; Mahut, B.; Beydon, N.; de Blic, J. Airway Remodeling in Preschool Children with Severe Recurrent Wheeze. Am. J. Respir. Crit. Care Med. 2015, 192, 164–171. [Google Scholar] [CrossRef] [PubMed]
- de Farias, C.F.; Amorim, M.M.; Dracoulakis, M.; Caetano, L.B.; Santoro, I.L.; Fernandes, A.L. Nasal lavage, blood or sputum: Which is best for phenotyping asthma? Respirology 2017, 22, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Amorim, M.M.; Araruna, A.; Caetano, L.B.; Cruz, A.C.; Santoro, L.L.; Fernandes, A.L. Nasal eosinophilia: An indicator of eosinophilic inflammation in asthma. Clin. Exp. Allergy 2010, 40, 867–874, Erratum in Clin. Exp. Allergy 2010, 40, 1711. [Google Scholar] [CrossRef] [PubMed]
- Corrêa da Silva, L.M.; Corrêa da Silva, L.C. Validação do questionário de qualidade de vida em asma (Juniper) para o português brasileiro. Rev. AMRIGS 2007, 51, 31–37. [Google Scholar]
- Leite, M.; Ponte, E.V.; Petroni, J.; D’Oliveira Júnior, A.; Pizzichini, E.; Cruz, A.A. Evaluation of the asthma control questionnaire validated for use in Brazil. J. Bras. Pneumol. 2008, 34, 756–763. [Google Scholar] [CrossRef]
- Roxo, J.P.; Ponte, E.V.; Ramos, D.C.; Pimentel, L.; D’Oliveira Júnior, A.; Cruz, A.A. Validação do Teste de Controle da Asma em português para uso no Brasil: Validation for use in Brazil [Portuguese-language version of the Asthma Control Test]. J. Bras. Pneumol. 2010, 36, 159–166. (In Portuguese) [Google Scholar] [CrossRef]
- Albuquerque, A.L.P.; Berton, D.C.; Campos, E.V.M.F.Á.S.; Queiroga-Júnior, F.J.P.; Santana, A.N.C.; Wong, B.M.S.; Batista, D.R.; Melo, F.X.; Didier-Neto, F.M.F.; Barros, J.A.; et al. New spirometry recommendations from the Brazilian Thoracic Association—2024 update. J. Bras. Pneumol. 2025, 50, e20240169. [Google Scholar] [CrossRef]
- Hargreave, F.E.; Pizzichini, E.; Pizzichini, M. Induced sputum examination. J. Allergy Clin. Immunol. 1998, 101 Pt 1, 569–570. [Google Scholar] [CrossRef]
- Pizzichini, E.; Pizzichini, M.M.; Kidney, J.C.; Efthimiadis, A.; Hussack, P.; Popov, T.; Cox, G.; Dolovich, J.; O’Byrne, P.; Hargreave, F.E. Induced sputum, bronchoalveolar lavage and blood from mild asthmatics: Inflammatory cells, lymphocyte subsets and soluble markers compared. Eur. Respir. J. 1998, 11, 828–834. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.; Kim, Y.J.; Lee, S.W.; Lee, J.S.; Oh, Y.M. COPD Risk Factor Profiles in General Population and Referred Patients: Potential Etiotypes. Int. J. Chronic Obstr. Pulm. Dis. 2023, 18, 2509–2520. [Google Scholar] [CrossRef]
- Hosseini, M.; Almasi-Hashiani, A.; Sepidarkish, M.; Maroufizadeh, S. Global prevalence of asthma-COPD overlap (ACO) in the general population: A systematic review and meta-analysis. Respir. Res. 2019, 20, 229. [Google Scholar] [CrossRef]
- Marcon, A.; Locatelli, F.; Dharmage, S.C.; Svanes, C.; Heinrich, J.; Leynaert, B.; Burney, P.; Corsico, A.; Caliskan, G.; Calciano, L.; et al. The coexistence of asthma and COPD: Risk factors, clinical history and lung function trajectories. Eur. Respir. J. 2021, 58, 2004656. [Google Scholar] [CrossRef]
- Cho, S.J.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef]
- Vigna, M.; Aiello, M.; Bertorelli, G.; Crisafulli, E.; Chetta, A. Flow and volume response to bronchodilator in patients with COPD. Acta Biomed. 2018, 89, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Trayer, J.; Isaza-Correa, J.; Kelly, L.; Kelleher, M.; Hourihane, J.; Byrne, A.; Molloy, E. The role of neutrophils in allergic disease. Clin. Exp. Immunol. 2025, 219, uxae126. [Google Scholar] [CrossRef] [PubMed]
- Mulvanny, A.; Pattwell, C.; Beech, A.; Southworth, T.; Singh, D. Validation of Sputum Biomarker Immunoassays and Cytokine Expression Profiles in COPD. Biomedicines 2022, 10, 1949. [Google Scholar] [CrossRef] [PubMed]
- Mulvanny, A.; Beech, A.; Li, J.; Lea, S.; Singh, D. Sputum Cytokine Repeatability in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2025, 20, 3779–3790. [Google Scholar] [CrossRef]

{kind=link}
| Variables | Non-COPD-A (n = 52) | COPD-A (n = 42) | p-Value |
|---|---|---|---|
| Sex (n, %) | F: 39 (75) M: 13 (25) | F: 28 (66.7) M: 14 (33.3) | 0.49 |
| Age (years) | 48.7 ± 16.0 | 60.5 ± 12.4 | <0.001 |
| Ever-smokers (n, %) | 4 (7.7) | 5 (11.9) | 0.51 |
| BMI (kg/m2) | 27.7 ± 5.1 | 26.5 ± 3.8 | 0.19 |
| Asthma onset age (years) | 15.5 (1–62) | 19.5 (1–56) | 0.23 |
| Asthma lifetime history (years) | 30.1 (5.0–68.4) | 39.8 (17.4–71.3) | <0.001 |
| Allergic rhinitis (n, %) | 31 (59.6) | 14 (34.1) | 0.021 |
| GINA treatment step (n, %) | Step 3/4: 35 (67.3) Step 5: 17 (32.7) | Step 3/4: 21 (50.0) Step 5: 21 (50.0) | 0.097 |
| Daily inhaled budesonide (μg) | 800 (200–1600) | 1200 (400–1200) | 0.014 |
| ACT | 23 (15–25) | 23 (9–25) | 0.64 |
| ACQ | 0.64 (0–2.71) | 1.00 (0.43–3.71) | 0.003 |
| AQLQ | 6.00 (3.45–7.00) | 6.23 (2.88–7.00) | 0.87 |
| Variables | Non-COPD-A (n = 52) | COPD-A (n = 42) | p-Value |
|---|---|---|---|
| FVC (L) | 3.13 ± 0.81 | 2.72 ± 0.88 | 0.022 |
| FVC (% predicted) | 91.5 ± 11.7 | 82.6 ± 15.6 | 0.002 |
| FEV1 (L) | 2.33 ± 0.64 | 1.62 ± 0.57 | <0.001 |
| FEV1 (% predicted) | 83.2 ± 11.0 | 62.2 ± 12.7 | <0.001 |
| FEV1/FVC (%) | 74.6 ± 5.4 | 60.0 ± 7.8 | <0.001 |
| Post-BD FVC (L) | 3.18 ± 0.79 | 2.91 ± 0.89 | 0.12 |
| Post-BD FVC (% predicted) | 93.3 ± 12.0 | 88.7 ± 15.2 | 0.10 |
| Post-BD FEV1 (L) | 2.44 ± 0.64 | 1.78 ± 0.59 | <0.001 |
| Post-BD FEV1 (% predicted) | 87.1 ± 11.2 | 68.1 ± 12.8 | <0.001 |
| Post-BD FEV1/FVC (%) | 76.6 ± 4.8 | 61.2 ± 8.0 | <0.001 |
| Delta (Post-Pre) FVC (mL) | +50.0 (−340 to +360) | +160.0 (−250 to +1400) | 0.003 |
| Delta (Post-Pre) FVC (% predicted) | +1.0 (−9.0 to +11.0) | +5.0 (−6.0 to +33.0) | 0.002 |
| Delta (Post-Pre) FEV1 (mL) | +90.0 (−230 to +480) | +125 (−70 to +1000) | 0.29 |
| Delta (Post-Pre) FEV1 (% predicted) | +3.0 (−7.0 to +19.0) | +6.0 (−3.0 to +30.0) | 0.21 |
| Positive Response to BD (n,%) | 13 (25.0%) | 23 (54.8%) | 0.005 |
| Variables | Non-COPD-A (n = 50) | COPD-A (n = 39) | p-Value |
|---|---|---|---|
| Neutrophils (%) | 45 (0–98) | 30 (0–100) | 0.31 |
| Eosinophils (%) | 2.5 (0.0–37.0) | 1 (0–23) | 0.38 |
| Lymphomononuclear cells (%) | 1 (0–17) | 0 (0–12) | 0.45 |
| IL-5 (pg/mL) | 0.37 (0.03–6.50) | 0.32 (0–6.25) | 0.18 |
| IL-8 (pg/mL) | 3.92 (0–100.21) | 2.63 (0.11–33.45) | 0.09 |
| IL-13 (pg/mL) | 8.82 (0–203.25) | 8.06 (0–94.19) | 0.30 |
| IL-17A (pg/mL) | 0.67 (0–37.11) | 0.50 (0–8.32) | 0.48 |
| IL-17F (pg/mL) | 0.008 (0–0.11) | 0.009 (0–0.042) | 0.99 |
| IL-25 (pg/mL) | 0.002 (0–0.042) | 0 (0–0.011) | 0.31 |
| IL-33 (pg/mL) | 1.59 (0–50.63) | 0.60 (0–14.40) | 0.14 |
| TNF (pg/mL) | 0.74 (0–8.42) | 0.43 (0–3.65) | 0.22 |
| Variables | Non-COPD-A (n = 47) | COPD-A (n = 36) | p-Value |
|---|---|---|---|
| Neutrophils (%) | 76 (0–98) | 77.5 (0–99) | 0.26 |
| Eosinophils (%) | 2 (0–40) | 1.5 (0–36) | 0.97 |
| Lymphomononuclear cells (%) | 0 (0–3) | 0 (0–3) | 0.78 |
| IL-5 pg/mL (pg/mL) * | 1.48 (0.13–6.57) | 2.11 (0.06–33.75) | 0.16 |
| IL-8 pg/mL (pg/mL) ** | 2.50 (0.29–25.40) | 7.66 (0.16–826.50) | 0.024 |
| IL-13 (pg/mL) + | 19.89 (0–136.38) | 20.96 (0–166.35) | 0.97 |
| IL-17A (pg/mL) ++ | 0.76 (0–5.33) | 1.30 (0–24.77) | 0.25 |
| IL-17F (pg/mL) ++ | 0.010 (0–0.045) | 0.012 (0–0.064) | 0.18 |
| IL-25 (pg/mL) ¶ | 0.002 (0–0.009) | 0.003 (0–0.019) | 0.28 |
| IL-33 (pg/mL) ¶¶ | 2.60 (0.13–9.25) | 2.27 (0–37.28) | 0.71 |
| TNF (pg/mL) * | 1.14 (0.07–5.44) | 1.37 (0.07–115.21) | 0.25 |
| Variables | Non-COPD-A (n = 50) | COPD-A (n = 40) | p-Value |
|---|---|---|---|
| Leucocytes (cells/mm) | 6935 (4270–12,200) | 7380 (4360–17,800) | 0.25 |
| Neutrophils (cells/mm) | 4150.5 (1290–9638) | 4385 (2143–14,774) | 0.35 |
| Eosinophils (cells/mm) | 184 (0–1261) | 198.5 (0–836) | 0.88 |
| IL-5 (pg/mL) * | 4.83 (0–90.14) | 4.42 (0–34.19) | 0.18 |
| IL-8 (pg/mL) * | 9.48 (0–78.73) | 8.76 (1.41–23.66) | 0.97 |
| IL-13 (pg/mL) * | 770.61 (30.55–1495.47) | 639.96 (151.71–1548.95) | 0.30 |
| IL-17A (pg/mL) * | 1.65 (0–30.89) | 1.39 (0–27.02) | 0.33 |
| IL-17F (pg/mL) * | 0.02 (0–0.17) | 0.01 (0–0.09) | 0.11 |
| IL-25 (pg/mL) * | 0.01 (0–0.42) | 0.00 (0–0.26) | 0.31 |
| IL-33 (pg/mL) * | 0.00 (0–86.91) | 0.00 (0–61.32) | 0.30 |
| TNF (pg/mL) * | 14.17 (4.05–64.48) | 16.49 (7.03–48.33) | 0.27 |
| Covariates | Univariable Analysis | Multivariable Analysis | ||
|---|---|---|---|---|
| Odds Ratio (95% CI) | p-Value | Adjusted Odds Ratio (95% CI) | p-Value | |
| Age | 1.06 (1.02–1.09) | <0.001 | 1.08 (0.99–1.17) | 0.074 |
| Sex female | 0.67 (0.27–1.64) | 0.376 | 1.43 (0.17–11.98) | 0.739 |
| BMI | 0.94 (0.86–1.03) | 0.202 | 0.84 (0.65–1.09) | 0.194 |
| Asthma duration | 1.05 (1.02–1.08) | 0.001 | 1.04 (0.98–1.11) | 0.185 |
| ACQ score | 2.19 (1.12–4.30) | 0.022 | 2.50 (2.47–13.27) | 0.281 |
| Post-BD FEV1 (% predicted) | 0.86 (0.81–0.91) | <0.001 | 0.84 (0.74–0.95) | 0.006 |
| IS IL-8 ≥ 3.096 pg/mL | 3.81 (1.22–11.91) | 0.022 | 12.82 (1.41–116.47) | 0.023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Farias, C.F.; Baddini-Martinez, J.; Fernandes, A.L.G.; Amorim, M.M.; Dracoulakis, M.; Santos, M.A.; Caetano, L.B.; Filho, F.S.L. Asthma-Associated COPD Etiotype: Clinical Features and Inflammatory Patterns in Biological Samples. J. Pers. Med. 2025, 15, 615. https://doi.org/10.3390/jpm15120615
de Farias CF, Baddini-Martinez J, Fernandes ALG, Amorim MM, Dracoulakis M, Santos MA, Caetano LB, Filho FSL. Asthma-Associated COPD Etiotype: Clinical Features and Inflammatory Patterns in Biological Samples. Journal of Personalized Medicine. 2025; 15(12):615. https://doi.org/10.3390/jpm15120615
Chicago/Turabian Stylede Farias, Camyla Fernandez, José Baddini-Martinez, Ana Luisa Godoy Fernandes, Maria Marta Amorim, Michel Dracoulakis, Maria Amélia Santos, Lilian Ballini Caetano, and Fernando Sergio Leitão Filho. 2025. "Asthma-Associated COPD Etiotype: Clinical Features and Inflammatory Patterns in Biological Samples" Journal of Personalized Medicine 15, no. 12: 615. https://doi.org/10.3390/jpm15120615
APA Stylede Farias, C. F., Baddini-Martinez, J., Fernandes, A. L. G., Amorim, M. M., Dracoulakis, M., Santos, M. A., Caetano, L. B., & Filho, F. S. L. (2025). Asthma-Associated COPD Etiotype: Clinical Features and Inflammatory Patterns in Biological Samples. Journal of Personalized Medicine, 15(12), 615. https://doi.org/10.3390/jpm15120615