
Preclinical Animal Models to Investigate the Role of Nav1.7 Ion Channels in Pain



Abstract
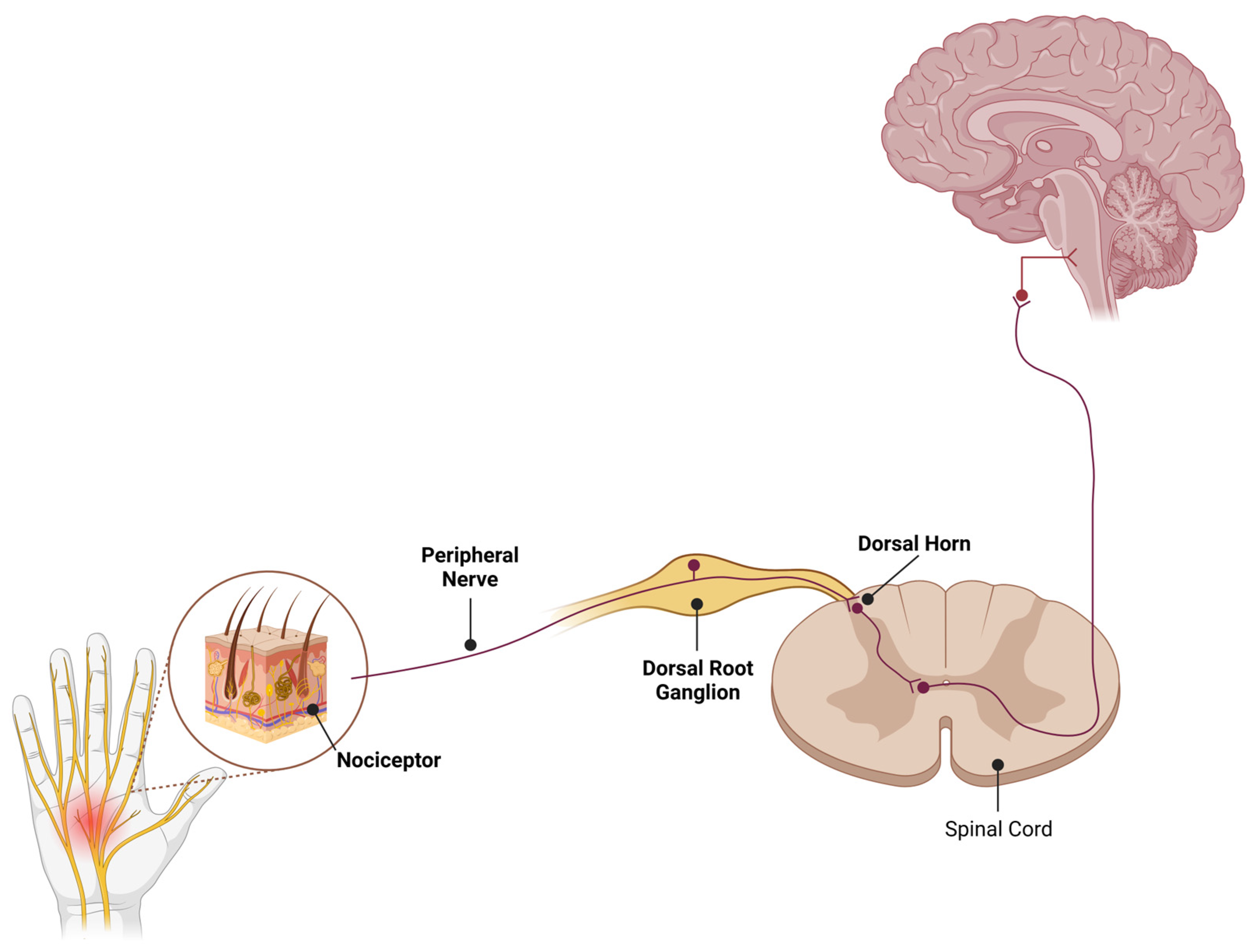
1. Introduction
2. Targeting Nav1.7 for Pain Applications
3. Preclinical Discovery of Analgesic Compounds
3.1. Animal Models of Pain
3.1.1. Complete Freund’s Adjuvant (CFA) and Carrageenan-Induced Pain Models
3.1.2. Formalin Test
3.1.3. Chronic Constriction Injury (CCI) Model
3.1.4. Spared Nerve Injury (SNI) Model
3.1.5. Spinal Nerve Ligation (SNL) Model
3.1.6. Spinal Cord Injury (SCI) Models
3.1.7. Chemotherapy-Induced Peripheral Neuropathy (CIPN) Models
3.1.8. Plantar Incision Model of Postoperative Pain
3.1.9. Osteoarthritis (OA) Models
3.2. Mechanical and Thermal Evoked Tests
4. Nav1.7-Mediated Reversal of Hyperalgesia in CFA and Carrageenan-Induced Pain Models
5. Role of Nav1.7 in Mediating Pain Behaviors in the Formalin Test
6. Efficacy Studies and Role of Nav1.7 in Neuropathic Pain Models
6.1. CCI Model
6.2. SNI Model
6.3. SNL Model
6.4. SCI Models
6.5. CIPN Models
7. Nav1.7 in the Plantar Incision Model of Post Operative Pain
8. Role of Nav1.7 in OA Models
9. Challenges and Future Direction
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CCI | Chronic constriction injury |
| Cdk5 | Cyclin-dependent kinase 5 |
| CFA | Complete Freund’s adjuvant |
| CIP | Congenital insensitivity to pain |
| CIPN | Chemotherapy-induced peripheral neuropathy |
| CNS | Central nervous system |
| CRMP2 | Collapsin response mediator protein 2 |
| DRG | Dorsal root ganglia |
| EM | Erythromelalgia |
| ERK1/2 | Extracellular signal-related kinase 1/2 |
| MIA | Monoiodoacetate arthritis |
| MAPK | Mitogen-activated protein kinase |
| NAD | Nicotinamide adenine dinucleotide |
| Nav1.7 | Voltage-gated sodium channel subtype 1.7 |
| Navs | Voltage-gated sodium channels |
| OA | Osteoarthritis |
| OD1 | First α toxin from Odonthobuthus doriae scorpion |
| PEPD | Paroxysmal extreme pain disorder |
| PNS | Peripheral nervous system |
| SCI | Spinal cord injury |
| SGK1 | Serum glucocorticoid-regulated kinase 1 |
| SNI | Spared nerve injury |
| SNL | Spinal nerve ligation |
| SUMO | Small ubiquitin-like modifier |
| TrkA | Tropomyosin receptor kinase A |
| TRP | Transient receptor potential |
| VSD | Voltage-sensing domain |
References
- Rice, A.S.C.; Smith, B.H.; Blyth, F.M. Pain and the global burden of disease. Pain 2016, 157, 791–796. [Google Scholar] [CrossRef]
- Marshall, B.; Bland, M.K.; Hulla, R.; Gatchel, R.J. Considerations in addressing the opioid epidemic and chronic pain within the USA. Pain Manag. 2019, 9, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Banderali, U.; Moreno, M.; Martina, M. The elusive Nav1.7: From pain to cancer. Curr. Top. Membr. 2023, 92, 47–69. [Google Scholar] [CrossRef]
- Humphreys, K.; Shover, C.L.; Andrews, C.M.; Bohnert, A.S.B.; Brandeau, M.L.; Caulkins, J.P.; Chen, J.H.; Cuéllar, M.F.; Hurd, Y.L.; Juurlink, D.N.; et al. Responding to the opioid crisis in North America and beyond: Recommendations of the Stanford-Lancet Commission. Lancet 2022, 399, 555–604. [Google Scholar] [CrossRef]
- Woller, S.A.; Eddinger, K.A.; Corr, M.; Yaksh, T.L. An overview of pathways encoding nociception. Clin. Exp. Rheumatol. 2017, 35, 40–46. [Google Scholar] [PubMed]
- Koivisto, A.P.; Voets, T.; Iadarola, M.J.; Szallasi, A. Targeting TRP channels for pain relief: A review of current evidence from bench to bedside. Curr. Opin. Pharmacol. 2024, 75, 102447. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.; Garraway, S.M. A review of dorsal root ganglia and primary sensory neuron plasticity mediating inflammatory and chronic neuropathic pain. Neurobiol. Pain 2024, 15, 100151. [Google Scholar] [CrossRef]
- Kanellopoulos, A.H.; Matsuyama, A. Voltage-gated sodium channels and pain-related disorders. Clin. Sci. 2016, 130, 2257–2265. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The Na(V)1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2013, 1, 49–62. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Gollob, M.H.; Darbar, D. Voltage-gated sodium channels: Biophysics, pharmacology, and related channelopathies. Front. Pharmacol. 2012, 3, 124. [Google Scholar] [CrossRef]
- Awad, S.S.; Lightowlers, R.N.; Young, C.; Chrzanowska-Lightowlers, Z.M.; Lomo, T.; Slater, C.R. Sodium channel mRNAs at the neuromuscular junction: Distinct patterns of accumulation and effects of muscle activity. J. Neurosci. 2001, 21, 8456–8463. [Google Scholar] [CrossRef]
- Ogata, N.; Ohishi, Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 2002, 88, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.G.; Westenbroek, R.E.; Maass, A.H.; Lange, V.; Renner, A.; Wischmeyer, E.; Bonz, A.; Muck, J.; Ertl, G.; Catterall, W.A.; et al. Distribution and function of sodium channel subtypes in human atrial myocardium. J. Mol. Cell Cardiol. 2013, 61, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.; D’Hooge, R.; Meert, T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 2010, 208, 149–157. [Google Scholar] [CrossRef] [PubMed]
- McDermott, L.A.; Weir, G.A.; Themistocleous, A.C.; Segerdahl, A.R.; Blesneac, I.; Baskozos, G.; Clark, A.J.; Millar, V.; Peck, L.J.; Ebner, D.; et al. Defining the Functional Role of NaV1.7 in Human Nociception. Neuron 2019, 101, 905–919. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef]
- Hong, S.; Morrow, T.J.; Paulson, P.E.; Isom, L.L.; Wiley, J.W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 2004, 279, 29341–29350. [Google Scholar] [CrossRef]
- Mukai, M.; Sakuma, Y.; Suzuki, M.; Orita, S.; Yamauchi, K.; Inoue, G.; Aoki, Y.; Ishikawa, T.; Miyagi, M.; Kamoda, H.; et al. Evaluation of behavior and expression of NaV1.7 in dorsal root ganglia after sciatic nerve compression and application of nucleus pulposus in rats. Eur. Spine J. 2014, 23, 463–468. [Google Scholar] [CrossRef]
- Laedermann, C.J.; Cachemaille, M.; Kirschmann, G.; Pertin, M.; Gosselin, R.D.; Chang, I.; Albesa, M.; Towne, C.; Schneider, B.L.; Kellenberger, S.; et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Investig. 2013, 123, 3002–3013. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; Chapman, M.L.; Marron, B.E.; Prime, R.; Storer, R.I.; Swain, N.A. Recent progress in sodium channel modulators for pain. Bioorg. Med. Chem. Lett. 2014, 24, 3690–3699. [Google Scholar] [CrossRef]
- Deuis, J.R.; Wingerd, J.S.; Winter, Z.; Durek, T.; Dekan, Z.; Sousa, S.R.; Zimmermann, K.; Hoffmann, T.; Weidner, C.; Nassar, M.A.; et al. Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins 2016, 8, 78. [Google Scholar] [CrossRef]
- Mueller, A.; Starobova, H.; Morgan, M.; Dekan, Z.; Cheneval, O.; Schroeder, C.I.; Alewood, P.F.; Deuis, J.R.; Vetter, I. Antiallodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute postsurgical pain: Evidence for analgesic synergy with opioids and baclofen. Pain 2019, 160, 1766–1780. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, J.M.; Palmer, J.; Morisset, V.; Giblin, G.M.; Obermann, M.; Ettlin, D.A.; Cruccu, G.; Bendtsen, L.; Estacion, M.; Derjean, D.; et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: A double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017, 16, 291–300. [Google Scholar] [CrossRef]
- McDonnell, A.; Collins, S.; Ali, Z.; Iavarone, L.; Surujbally, R.; Kirby, S.; Butt, R.P. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 2018, 159, 1465–1476. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Yarov-Yarovoy, V. Towards Structure-Guided Development of Pain Therapeutics Targeting Voltage-Gated Sodium Channels. Front. Pharmacol. 2022, 13, 842032. [Google Scholar] [CrossRef]
- Eagles, D.A.; Chow, C.Y.; King, G.F. Fifteen years of NaV 1.7 channels as an analgesic target: Why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy? Br. J. Pharmacol. 2022, 179, 3592–3611. [Google Scholar] [CrossRef]
- Neff, R.A.; Wickenden, A.D. Selective Targeting of Nav1.7 with Engineered Spider Venom-Based Peptides. Channels 2021, 15, 179–193. [Google Scholar] [CrossRef]
- Ouyang, X.; Su, M.; Xue, D.; Dong, L.; Niu, H.; Li, W.; Liu, Y.; Wang, K.; Shao, L. Design, synthesis, and biological evaluation of acyl sulfonamide derivatives with spiro cycles as NaV1.7 inhibitors for antinociception. Bioorg. Med. Chem. 2023, 86, 117290. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, X.; Li, L.; Huang, Y.; Cui, C.; Hu, Q.; Xu, H.; Yin, B.; Chen, X.; Zhao, D.; et al. Recent advances in small molecule Nav 1.7 inhibitors for cancer pain management. Bioorg. Chem. 2024, 150, 107605. [Google Scholar] [CrossRef] [PubMed]
- Dormer, A.; Narayanan, M.; Schentag, J.; Achinko, D.; Norman, E.; Kerrigan, J.; Jay, G.; Heydorn, W. A Review of the Therapeutic Targeting of SCN9A and Nav1.7 for Pain Relief in Current Human Clinical Trials. J. Pain Res. 2023, 16, 1487–1498. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef]
- Minett, M.S.; Falk, S.; Santana-Varela, S.; Bogdanov, Y.D.; Nassar, M.A.; Heegaard, A.M.; Wood, J.N. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014, 6, 301–312. [Google Scholar] [CrossRef]
- Wheeler, D.W.; Lee, M.C.; Harrison, E.K.; Menon, D.K.; Woods, C.G. Case Report: Neuropathic pain in a patient with congenital insensitivity to pain. F1000Research 2014, 3, 135. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef] [PubMed]
- McCarson, K.E.; Fehrenbacher, J.C. Models of Inflammation: Carrageenan- or Complete Freund’s Adjuvant (CFA)-Induced Edema and Hypersensitivity in the Rat. Curr. Protoc. 2021, 1, e202. [Google Scholar] [CrossRef]
- Eijkelkamp, N.; Heijnen, C.J.; Carbajal, A.G.; Willemen, H.L.; Wang, H.; Minett, M.S.; Wood, J.N.; Schedlowski, M.; Dantzer, R.; Kelley, K.W.; et al. G protein-coupled receptor kinase 6 acts as a critical regulator of cytokine-induced hyperalgesia by promoting phosphatidylinositol 3-kinase and inhibiting p38 signaling. Mol. Med. 2012, 18, 556–564. [Google Scholar] [CrossRef]
- Liang, L.; Fan, L.; Tao, B.; Yaster, M.; Tao, Y.X. Protein kinase B/Akt is required for complete Freund’s adjuvant-induced upregulation of Nav1.7 and Nav1.8 in primary sensory neurons. J. Pain 2013, 14, 638–647. [Google Scholar] [CrossRef]
- Marchand, F.; Perretti, M.; McMahon, S.B. Role of the immune system in chronic pain. Nat. Rev. Neurosci. 2005, 6, 521–532. [Google Scholar] [CrossRef]
- Dunham, J.P.; Kelly, S.; Donaldson, L.F. Inflammation reduces mechanical thresholds in a population of transient receptor potential channel A1-expressing nociceptors in the rat. Eur. J. Neurosci. 2008, 27, 3151–3160. [Google Scholar] [CrossRef] [PubMed]
- Tjølsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.M.; Cater, H.L.; Thakur, M.; Wells, S.; McMahon, S.B. A refinement to the formalin test in mice. F1000Research 2019, 8, 891. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef]
- Hoffmann, T.; Klemm, F.; IKichko, T.; Sauer, S.K.; Kistner, K.; Riedl, B.; Raboisson, P.; Luo, L.; Babes, A.; Kocher, L.; et al. The formalin test does not probe inflammatory pain but excitotoxicity in rodent skin. Physiol. Rep. 2022, 10, e15194. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Austin, P.J.; Wu, A.; Moalem-Taylor, G. Chronic constriction of the sciatic nerve and pain hypersensitivity testing in rats. J. Vis. Exp. 2012, 61, 3393. [Google Scholar] [CrossRef]
- Medeiros, P.; Dos Santos, I.R.; Júnior, I.M.; Palazzo, E.; da Silva, J.A.; Machado, H.R.; Ferreira, S.H.; Maione, S.; Coimbra, N.C.; de Freitas, R.L. An Adapted Chronic Constriction Injury of the Sciatic Nerve Produces Sensory, Affective, and Cognitive Impairments: A Peripheral Mononeuropathy Model for the Study of Comorbid Neuropsychiatric Disorders Associated with Neuropathic Pain in Rats. Pain Med. 2021, 22, 338–351. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Cai, W.; Zhao, Q.; Shao, J.; Zhang, J.; Li, L.; Ren, X.; Su, S.; Bai, Q.; Li, M.; Chen, X.; et al. MicroRNA-182 Alleviates Neuropathic Pain by Regulating Nav1.7 Following Spared Nerve Injury in Rats. Sci. Rep. 2018, 8, 16750. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Xu, X.; Chen, Y.; Lin, C.; Lin, F.; Liu, R. Effects of High-Voltage Pulsed Radiofrequency on the Ultrastructure and Nav1.7 Level of the Dorsal Root Ganglion in Rats with Spared Nerve Injury. Neuromodulation 2022, 25, 980–988. [Google Scholar] [CrossRef]
- Bourquin, A.F.; Süveges, M.; Pertin, M.; Gilliard, N.; Sardy, S.; Davison, A.C.; Spahn, D.R.; Decosterd, I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 2006, 122, 14.e1–14.e14. [Google Scholar] [CrossRef]
- Swett, J.E.; Torigoe, Y.; Elie, V.R.; Bourassa, C.M.; Miller, P.G. Sensory neurons of the rat sciatic nerve. Exp. Neurol. 1991, 114, 82–103. [Google Scholar] [CrossRef]
- Rigaud, M.; Gemes, G.; Barabas, M.E.; Chernoff, D.I.; Abram, S.E.; Stucky, C.L.; Hogan, Q.H. Species and strain differences in rodent sciatic nerve anatomy: Implications for studies of neuropathic pain. Pain 2008, 136, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Laedermann, C.J.; Pertin, M.; Suter, M.R.; Decosterd, I. Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 2014, 10, 19. [Google Scholar] [CrossRef]
- Ho Kim, S.; Mo Chung, J. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Eschenfelder, S.; Blenk, K.H.; Jänig, W.; Häbler, H. Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain 2000, 84, 309–318. [Google Scholar] [CrossRef]
- Mabuchi, T.; Matsumura, S.; Okuda-Ashitaka, E.; Kitano, T.; Kojima, H.; Nagano, T.; Minami, T.; Ito, S. Attenuation of neuropathic pain by the nociceptin/orphanin FQ antagonist JTC-801 is mediated by inhibition of nitric oxide production. Eur. J. Neurosci. 2003, 7, 1384–1392. [Google Scholar] [CrossRef]
- Clifford, J.L.; Mares, A.; Hansen, J.; Averitt, D.L. Preemptive perineural bupivacaine attenuates the maintenance of mechanical and cold allodynia in a rat spinal nerve ligation model. BMC Anesthesiol. 2015, 15, 135. [Google Scholar] [CrossRef]
- Widerström-Noga, E. Neuropathic Pain and Spinal Cord Injury: Management, Phenotypes, and Biomarkers. Drugs 2023, 83, 1001–1025. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.J.; Xu, J.X.; Aldskogius, H.; Seiger, Å.; Wiesenfeld-Hallin, Z. Allodynia-like effects in rat after ischaemic spinal cord injury photochemically induced by laser irradiation. Pain 1991, 45, 175–185. [Google Scholar] [CrossRef]
- Scheff, S.W.; Rabchevsky, A.G.; Fugaccia, I.; Main, J.A.; Lumpp, J.E., Jr. Experimental modeling of spinal cord injury: Characterization of a force-defined injury device. J. Neurotrauma 2003, 20, 179–193. [Google Scholar] [CrossRef]
- Yoon, Y.W.; Dong, H.; Arends, J.J.; Jacquin, M.F. Mechanical and cold allodynia in a rat spinal cord contusion model. Somatosens. Mot. Res. 2004, 21, 25–31. [Google Scholar] [CrossRef]
- Masri, R.; Keller, A. Chronic pain following spinal cord injury. Adv. Exp. Med. Biol. 2012, 760, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.L.; Minhas, N.K.; Jutzeler, C.R.; Erskine, E.L.; Liu, L.J.; Ramer, M.S. Neuropathic pain following traumatic spinal cord injury: Models, measurement, and mechanisms. J. Neurosci. Res. 2017, 95, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, F.Y.; Aleanizy, F.S.; El Tahir, E.; Alkahtani, H.M.; AlQuadeib, B.T. Paclitaxel. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier BV: Amsterdam, The Netherlands, 2019; Volume 44, pp. 205–238. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Staff, N.P.; Fehrenbacher, J.C.; Caillaud, M.; Damaj, M.I.; Segal, R.A.; Rieger, S. Pathogenesis of paclitaxel-induced peripheral neuropathy: A current review of in vitro and in vivo findings using rodent and human model systems. Exp. Neurol. 2020, 324, 113121. [Google Scholar] [CrossRef]
- Mahon, S.M.; Carr, E. Peripheral Neuropathy: Common Side Effect. Clin. J. Oncol. Nurs. 2021, 25. [Google Scholar] [CrossRef]
- Pennypacker, S.D.; Fonseca, M.M.; Morgan, J.W.; Dougherty, P.M.; Cubillos-Ruiz, J.R.; Strowd, R.E.; Romero-Sandoval, E.A. Methods and protocols for chemotherapy-induced peripheral neuropathy (CIPN) mouse models using paclitaxel. Methods Cell Biol. 2022, 168, 277–298. [Google Scholar] [CrossRef]
- Fregoso, G.; Wang, A.; Tseng, K.; Wang, J. Transition from Acute to Chronic Pain: Evaluating Risk for Chronic Postsurgical Pain. Pain. Physician 2019, 22, 479–488. [Google Scholar] [PubMed]
- Brennan, T.J.; Vandermeulen, E.P.; Gebhart, G.F. Characterization of a rat model of incisional pain. Pain 1996, 64, 493–502. [Google Scholar] [CrossRef]
- Pogatzki, E.M.; Raja, S.N. A mouse model of incisional pain. Anesthesiology 2003, 99, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Cowie, A.M.; Stucky, C.L. A Mouse Model of Postoperative Pain. Bio-Protocol 2019, 9, e3140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Ren, K.; Dubner, R. Osteoarthritis pain mechanisms: Basic studies in animal models. Osteoarthr. Cartil. 2013, 21, 1308–1315. [Google Scholar] [CrossRef]
- Zaki, S.; Blaker, C.L.; Little, C.B. OA foundations—Experimental models of osteoarthritis. Osteoarthr. Cartil. 2022, 30, 357–380. [Google Scholar] [CrossRef]
- Guzman, R.E.; Evans, M.G.; Bove, S.; Morenko, B.; Kilgore, K. Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: An animal model of osteoarthritis. Toxicol. Pathol. 2003, 31, 619–624. [Google Scholar] [CrossRef]
- Wilson, A.W.; Medhurst, S.J.; Dixon, C.I.; Bontoft, N.C.; Winyard, L.A.; Brackenborough, K.T.; De Alba, J.; Clarke, C.J.; Gunthorpe, M.J.; Hicks, G.A.; et al. An animal model of chronic inflammatory pain: Pharmacological and temporal differentiation from acute models. Eur. J. Pain 2006, 10, 537–549. [Google Scholar] [CrossRef]
- Pitcher, T.; Sousa-Valente, J.; Malcangio, M. The Monoiodoacetate Model of Osteoarthritis Pain in the Mouse. J. Vis. Exp. 2016, 111, 53746. [Google Scholar] [CrossRef]
- Ivanavicius, S.P.; Ball, A.D.; Heapy, C.G.; Westwood, R.F.; Murray, F.; Read, S.J. Structural pathology in a rodent model of osteoarthritis is associated with neuropathic pain: Increased expression of ATF-3 and pharmacological characterisation. Pain 2007, 128, 272–282. [Google Scholar] [CrossRef]
- Im, H.J.; Kim, J.S.; Li, X.; Kotwal, N.; Sumner, D.R.; van Wijnen, A.J.; Davis, F.J.; Yan, D.; Levine, B.; Henry, J.L.; et al. Alteration of sensory neurons and spinal response to an experimental osteoarthritis pain model. Arthritis Rheum. 2010, 10, 2995–3005. [Google Scholar] [CrossRef]
- Fu, W.; Vasylyev, D.; Bi, Y.; Zhang, M.; Sun, G.; Khleborodova, A.; Huang, G.; Zhao, L.; Zhou, R.; Li, Y.; et al. Nav1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature 2024, 625, 557–565. [Google Scholar] [CrossRef]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 2008, 12, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.A.; Zahra, F.T.; Martin, L.J. IUPHAR review: Navigating the role of preclinical models in pain research. Pharmacol. Res. 2024, 200, 107073. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.D.; Parekh, A.; Pancholi, Y.N. Evaluating pain behaviours: Widely used mechanical and thermal methods in rodents. Behav. Brain Res. 2023, 446, 114417. [Google Scholar] [CrossRef] [PubMed]
- Bradman, M.J.; Ferrini, F.; Salio, C.; Merighi, A. Practical mechanical threshold estimation in rodents using von Frey hairs/Semmes-Weinstein monofilaments: Towards a rational method. J. Neurosci. Methods 2015, 255, 92–103. [Google Scholar] [CrossRef]
- Campana, G.; Rimondini, R. Mechanical Nociception in Mice and Rats: Measurement with Automated von Frey Equipment. Methods Mol. Biol. 2021, 2201, 195–198. [Google Scholar] [CrossRef]
- Bannon, A.W.; Malmberg, A.B. Models of nociception: Hot-plate, tail-flick, and formalin tests in rodents. Curr. Protoc. Neurosci. 2007, 41, 8.9.1–8.9.16. [Google Scholar] [CrossRef]
- Yalcin, I.; Charlet, A.; Freund-Mercier, M.J.; Barrot, M.; Poisbeau, P. Differentiating thermal allodynia and hyperalgesia using dynamic hot and cold plate in rodents. J. Pain 2009, 10, 767–773. [Google Scholar] [CrossRef]
- Shields, S.D.; Deng, L.; Reese, R.M.; Dourado, M.; Tao, J.; Foreman, O.; Chang, J.H.; Hackos, D.H. Insensitivity to Pain upon Adult-Onset Deletion of Nav1.7 or Its Blockade with Selective Inhibitors. J. Neurosci. 2018, 38, 10180–10201. [Google Scholar] [CrossRef]
- Gingras, J.; Smith, S.; Matson, D.J.; Johnson, D.; Nye, K.; Couture, L.; Feric, E.; Yin, R.; Moyer, B.D.; Peterson, M.L.; et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 2014, 9, e105895. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3, 791. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Eijkelkamp, N.; Wood, J.N. Significant determinants of mouse pain behaviour. PLoS ONE 2014, 9, e104458. [Google Scholar] [CrossRef]
- Hockley, J.R.; González-Cano, R.; McMurray, S.; Tejada-Giraldez, M.A.; McGuire, C.; Torres, A.; Wilbrey, A.L.; Cibert-Goton, V.; Nieto, F.R.; Pitcher, T.; et al. Visceral and somatic pain modalities reveal NaV 1.7-independent visceral nociceptive pathways. J. Physiol. 2017, 595, 2661–2679. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Wook, Y.Y.; Sik, N.H.; Ho, K.S.; Mo, C.J. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 1994, 59, 369–376. [Google Scholar] [CrossRef]
- Jasmin, L.; Kohan, L.; Franssen, M.; Janni, G.; Goff, J.R. The cold plate as a test of nociceptive behaviors: Description and application to the study of chronic neuropathic and inflammatory pain models. Pain 1998, 75, 367–382. [Google Scholar] [CrossRef]
- Brenner, D.S.; Golden, J.P.; Gereau, R.W., 4th. A novel behavioral assay for measuring cold sensation in mice. PLoS ONE 2012, 7, e39765. [Google Scholar] [CrossRef]
- Allchorne, A.J.; Broom, D.C.; Woolf, C.J. Detection of cold pain, cold allodynia and cold hyperalgesia in freely behaving rats. Mol. Pain 2005, 1, 36. [Google Scholar] [CrossRef]
- Huerta, M.Á.; Cisneros, E.; Alique, M.; Roza, C. Strategies for measuring non-evoked pain in preclinical models of neuropathic pain: Systematic review. Neurosci. Biobehav. Rev. 2024, 163, 105761. [Google Scholar] [CrossRef]
- Djouhri, L.; Koutsikou, S.; Fang, X.; McMullan, S.; Lawson, S.N. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J. Neurosci. 2006, 26, 1281–1292. [Google Scholar] [CrossRef]
- Matson, D.J.; Hamamoto, D.T.; Bregman, H.; Cooke, M.; DiMauro, E.F.; Huang, L.; Johnson, D.; Li, X.; McDermott, J.; Morgan, C.; et al. Inhibition of Inactive States of Tetrodotoxin-Sensitive Sodium Channels Reduces Spontaneous Firing of C-Fiber Nociceptors and Produces Analgesia in Formalin and Complete Freund’s Adjuvant Models of Pain. PLoS ONE 2015, 10, e0138140. [Google Scholar] [CrossRef]
- Black, J.A.; Liu, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J., 3rd; England, J.D.; Soignier, R.D.; Nolan, P.; Minor, L.D.; Liu, Z.P.; Levinson, S.R.; Paul, D. Ibuprofen blocks changes in Na v 1.7 and 1.8 sodium channels associated with complete Freund’s adjuvant-induced inflammation in rat. J. Pain 2004, 5, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed]
- Yeomans, D.C.; Levinson, S.R.; Peters, M.C.; Koszowski, A.G.; Tzabazis, A.Z.; Gilly, W.F.; Wilson, S.P. Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Nav1.7 sodium channels in primary afferents. Hum. Gene Ther. 2005, 16, 271–277. [Google Scholar] [CrossRef]
- Moreno, A.M.; Alemán, F.; Catroli, G.F.; Hunt, M.; Hu, M.; Dailamy, A.; Pla, A.; Woller, S.A.; Palmer, N.; Parekh, U.; et al. Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice. Sci. Transl. Med. 2021, 13, eaay9056. [Google Scholar] [CrossRef]
- Li, S.; Jin, Y.; Li, M.; Yu, H. NAN-190, a 5-HT1A antagonist, alleviates inflammatory pain by targeting Nav1.7 sodium channels. Life Sci. 2023, 319, 121520. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, R.; Zhang, M.; Chen, D.; Zhang, Q.; Zhang, N.; Shi, Y.; Hu, X.; Li, N.; Fang, Q. NaV1.7 Channel Blocker [Ala5, Phe6, Leu26, Arg28]GpTx-1 Attenuates CFA-induced Inflammatory Hypersensitivity in Rats via Endogenous Enkephalin Mechanism. J. Pain 2023, 24, 840–859. [Google Scholar] [CrossRef]
- Martina, M.; Banderali, U.; Yogi, A.; Arbabi Ghahroudi, M.; Liu, H.; Sulea, T.; Durocher, Y.; Hussack, G.; van Faassen, H.; Chakravarty, B.; et al. A Novel Antigen Design Strategy to Isolate Single-Domain Antibodies that Target Human Nav1.7 and Reduce Pain in Animal Models. Adv. Sci. 2024, 11, e2405432. [Google Scholar] [CrossRef]
- Bankar, G.; Goodchild, S.J.; Howard, S.; Nelkenbrecher, K.; Waldbrook, M.; Dourado, M.; Shuart, N.G.; Lin, S.; Young, C.; Xie, Z.; et al. Selective NaV1.7 Antagonists with Long Residence Time Show Improved Efficacy against Inflammatory and Neuropathic Pain. Cell Rep. 2018, 24, 3133–3145. [Google Scholar] [CrossRef]
- Han, P.; Liu, S.; Zhang, M.; Zhao, J.; Wang, Y.; Wu, G.; Mi, W. Inhibition of Spinal Interlukin-33/ST2 Signaling and Downstream ERK and JNK Pathways in Electroacupuncture Analgesia in Formalin Mice. PLoS ONE 2015, 10, e0129576. [Google Scholar] [CrossRef] [PubMed]
- Stamboulian, S.; Choi, J.S.; Ahn, H.S.; Chang, Y.W.; Tyrrell, L.; Black, J.A.; Waxman, S.G.; Dib-Hajj, S.D. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J. Neurosci. 2010, 30, 1637–1647. [Google Scholar] [CrossRef]
- Zhao, Z.; Pan, T.; Chen, S.; Harvey, P.J.; Zhang, J.; Li, X.; Yang, M.; Huang, L.; Wang, S.; Craik, D.J.; et al. Design, synthesis, and mechanism of action of novel μ-conotoxin KIIIA analogues for inhibition of the voltage-gated sodium channel Nav1.7. J. Biol. Chem. 2023, 299, 103068. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Ou-Yang, X.S.; Zhou, P.; Dong, L.Y.; Shao, L.M.; Wang, K.W.; Liu, Y.N. Inhibition of TTX-S Na+ currents by a novel blocker QLS-278 for antinociception. J. Pharmacol. Exp. Ther. 2024, 392, 100030. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Maier, C.; Attal, N.; Binder, A.; Bouhassira, D.; Cruccu, G.; Finnerup, N.B.; Haanpää, M.; Hansson, P.; Hüllemann, P.; et al. Peripheral neuropathic pain: A mechanism-related organizing principle based on sensory profiles. Pain 2017, 158, 261–272. [Google Scholar] [CrossRef]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Front. Cell Neurosci. 2019, 13, 116. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Boakye, P.A.; Tang, S.J.; Smith, P.A. Mediators of Neuropathic Pain; Focus on Spinal Microglia, CSF-1, BDNF, CCL21, TNF-α, Wnt Ligands, and Interleukin 1β. Front. Pain Res. 2021, 2, 698157. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.J.; Yang, L.; Fang, X.L.; Hu, H.F.; Zhao, M.Y.; Li, L.; Guo, Y.Y.; Shao, J.P. Voltage-gated sodium channel 1.7 expression decreases in dorsal root ganglia in a spinal nerve ligation neuropathic pain model. Kaohsiung J. Med. Sci. 2019, 35, 493–500. [Google Scholar] [CrossRef]
- Liu, C.; Cao, J.; Ren, X.; Zang, W. Nav1.7 protein and mRNA expression in the dorsal root ganglia of rats with chronic neuropathic pain. Neural Regen. Res. 2012, 7, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Dong, W.; Zhang, L.; Yang, X. Activating Sirt1 by resveratrol suppresses Nav1.7 expression in DRG through miR-182 and alleviates neuropathic pain in rats. Channels 2020, 1, 69–78. [Google Scholar] [CrossRef]
- Luo, G.; Chen, L.; Easton, A.; Newton, A.; Bourin, C.; Shields, E.; Mosure, K.; Soars, M.G.; Knox, R.J.; Matchett, M.; et al. Discovery of Indole- and Indazole-acylsulfonamides as Potent and Selective NaV1.7 Inhibitors for the Treatment of Pain. J. Med. Chem. 2019, 62, 831–856. [Google Scholar] [CrossRef]
- Ghelardini, C.; Desaphy, J.F.; Muraglia, M.; Corbo, F.; Matucci, R.; Dipalma, A.; Bertucci, C.; Pistolozzi, M.; Nesi, M.; Norcini, M.; et al. Effects of a new potent analog of tocainide on hNav1.7 sodium channels and in vivo neuropathic pain models. Neuroscience 2010, 169, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stratton, H.J.; Lorca, S.A.; Grace, P.M.; Khanna, R. Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in chronic constriction injury (CCI) rats. Channels 2022, 16, 1–8. [Google Scholar] [CrossRef]
- Dustrude, E.T.; Wilson, S.M.; Ju, W.; Xiao, Y.; Khanna, R. CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 2013, 288, 24316–24331. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Moutal, A.; Yu, J.; Chew, L.A.; Isensee, J.; Chawla, R.; Gomez, K.; Luo, S.; Zhou, Y.; Chefdeville, A.; et al. Selective targeting of NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in rodents. Sci. Transl. Med. 2021, 13, eabh1314. [Google Scholar] [CrossRef]
- Dustrude, E.T.; Moutal, A.; Yang, X.; Wang, Y.; Khanna, M.; Khanna, R. Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc. Natl. Acad. Sci. USA 2016, 113, E8443–E8452. [Google Scholar] [CrossRef]
- Yin, J.B.; Liu, H.X.; Dong, Q.Q.; Wu, H.H.; Liang, Z.W.; Fu, J.T.; Zhao, W.J.; Hu, H.Q.; Guo, H.W.; Zhang, T.; et al. Correlative increasing expressions of KIF5b and Nav1.7 in DRG neurons of rats under neuropathic pain conditions. Physiol. Behav. 2023, 263, 114115. [Google Scholar] [CrossRef]
- Berta, T.; Poirot, O.; Pertin, M.; Ji, R.R.; Kellenberger, S.; Decosterd, I. Transcriptional and functional profiles of voltage-gated Na(+) channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell Neurosci. 2008, 37, 196–208. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, J.; Zhang, Y.; Xun, X.; Tang, D.; Peng, D.; Yi, J.; Liu, Z.; Shi, X. Synthesis and analgesic effects of μ-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins 2014, 6, 2363–2378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shu, J.; Yang, H.; Hong, K.; Yang, X.; Guo, W.; Fang, J.; Li, F.; Liu, T.; Shan, Z.; et al. Nav1.7 Modulator Bearing a 3-Hydroxyindole Backbone Holds the Potential to Reverse Neuropathic Pain. ACS Chem. Neurosci. 2024, 15, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Dustrude, E.T.; Largent-Milnes, T.M.; Vanderah, T.W.; Khanna, M.; Khanna, R. Blocking CRMP2 SUMOylation reverses neuropathic pain. Mol. Psychiatry 2018, 23, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.; Stratton, H.J.; Duran, P.; Loya, S.; Tang, C.; Calderon-Rivera, A.; François-Moutal, L.; Khanna, M.; Madura, C.L.; Luo, S.; et al. Identification and targeting of a unique NaV1.7 domain driving chronic pain. Proc. Natl. Acad. Sci. USA 2023, 120, e2217800120. [Google Scholar] [CrossRef]
- Fukuoka, T.; Noguchi, K. Contribution of the spared primary afferent neurons to the pathomechanisms of neuropathic pain. Mol. Neurobiol. 2002, 26, 57–67. [Google Scholar] [CrossRef]
- Shim, B.; Ringkamp, M.; Lambrinos, G.L.; Hartke, T.V.; Griffin, J.W.; Meyer, R.A. Activity-dependent slowing of conduction velocity in uninjured L4 C fibers increases after an L5 spinal nerve injury in the rat. Pain 2007, 128, 40–51. [Google Scholar] [CrossRef]
- Fukuoka, T.; Miyoshi, K.; Noguchi, K. De novo expression of Nav1.7 in injured putative proprioceptive afferents: Multiple tetrodotoxin-sensitive sodium channels are retained in the rat dorsal root after spinal nerve ligation. Neuroscience 2015, 284, 693–706. [Google Scholar] [CrossRef]
- Nassar, M.A.; Levato, A.; Stirling, L.C.; Wood, J.N. Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol. Pain 2005, 1, 24. [Google Scholar] [CrossRef]
- Grubinska, B.; Chen, L.; Alsaloum, M.; Rampal, N.; Matson, D.J.; Yang, C.; Taborn, K.; Zhang, M.; Youngblood, B.; Liu, D.; et al. Rat NaV1.7 loss-of-function genetic model: Deficient nociceptive and neuropathic pain behavior with retained olfactory function and intra-epidermal nerve fibers. Mol. Pain 2019, 15, 1744806919881846. [Google Scholar] [CrossRef]
- Hildebrand, M.E.; Smith, P.L.; Bladen, C.; Eduljee, C.; Xie, J.Y.; Chen, L.; Fee-Maki, M.; Doering, C.J.; Mezeyova, J.; Zhu, Y.; et al. A novel slow-inactivation-specific ion channel modulator attenuates neuropathic pain. Pain 2011, 152, 833–843. [Google Scholar] [CrossRef]
- Yang, S.W.; Ho, G.D.; Tulshian, D.; Bercovici, A.; Tan, Z.; Hanisak, J.; Brumfield, S.; Matasi, J.; Sun, X.; Sakwa, S.A.; et al. Bioavailable pyrrolo-benzo-1,4-diazines as Na(v)1.7 sodium channel blockers for the treatment of pain. Bioorg. Med. Chem. Lett. 2014, 24, 4958–4962. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Wu, W.; Li, Z.; Yang, G.; Qin, J.; Wang, Y.; Hu, Z.; Dong, H.; Hou, L.; Hu, G.; et al. Discovery of aminocyclohexene analogues as selective and orally bioavailable hNav1.7 inhibitors for analgesia. Bioorg. Med. Chem. Lett. 2017, 27, 4979–4984. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, L.; Zhu, F.; Xia, W.; Wen, T.; Xia, R.; Yu, X.; Xu, D.; Peng, C. Ectopic expression of Nav1.7 in spinal dorsal horn neurons induced by NGF contributes to neuropathic pain in a mouse spinal cord injury model. Front. Mol. Neurosci. 2023, 16, 1091096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dougherty, P.M. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 2014, 120, 1463–1475. [Google Scholar] [CrossRef]
- Xia, Z.; Xiao, Y.; Wu, Y.; Zhao, B. Sodium channel Nav1.7 expression is upregulated in the dorsal root ganglia in a rat model of paclitaxel-induced peripheral neuropathy. SpringerPlus 2016, 5, 1738. [Google Scholar] [CrossRef]
- Li, Y.; North, R.Y.; Rhines, L.D.; Tatsui, C.E.; Rao, G.; Edwards, D.D.; Cassidy, R.M.; Harrison, D.S.; Johansson, C.A.; Zhang, H.; et al. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. J. Neurosci. 2018, 38, 1124–1136. [Google Scholar] [CrossRef]
- Wang, G.J.; Zhang, X.; Huang, L.D.; Xiao, Y. Involvement of the Sodium Channel Nav1.7 in Paclitaxel-induced Peripheral Neuropathy through ERK1/2 Signaling in Rats. Curr. Neurovasc. Res. 2020, 17, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Akin, E.J.; Alsaloum, M.; Higerd, G.P.; Liu, S.; Zhao, P.; Dib-Hajj, F.B.; Waxman, S.G.; Dib-Hajj, S.D. Paclitaxel increases axonal localization and vesicular trafficking of Nav1.7. Brain 2021, 144, 1727–1737. [Google Scholar] [CrossRef]
- Chandra, S.; Wang, Z.; Tao, X.; Chen, O.; Luo, X.; Ji, R.R.; Bortsov, A.V. Computer-aided Discovery of a New Nav1.7 Inhibitor for Treatment of Pain and Itch. Anesthesiology 2020, 133, 611–627. [Google Scholar] [CrossRef]
- Misset, J.L.; Bleiberg, H.; Sutherland, W.; Bekradda, M.; Cvitkovic, E. Oxaliplatin clinical activity: A review. Crit. Rev. Oncol. Hematol. 2000, 35, 75–93. [Google Scholar] [CrossRef]
- Beijers, A.J.; Mols, F.; Vreugdenhil, G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support. Care Cancer 2014, 22, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Sereno, M.; Gutiérrez-Gutiérrez, G.; Rubio, J.M.; Apellániz-Ruiz, M.; Sánchez-Barroso, L.; Casado, E.; Falagan, S.; López-Gómez, M.; Merino, M.; Gómez-Raposo, C.; et al. Genetic polymorphisms of SCN9A are associated with oxaliplatin-induced neuropathy. BMC Cancer 2017, 17, 63. [Google Scholar] [CrossRef]
- Xu, L.J.; Wang, J.; Li, Y.D.; Shen, K.F.; He, X.H.; Wu, W.; Liu, C.C. Reduction of SIRT1-Mediated Epigenetic Upregulation of Nav1.7 Contributes to Oxaliplatin-Induced Neuropathic Pain. Pain Physician 2023, 26, E213–E222. [Google Scholar]
- Braden, K.; Stratton, H.J.; Salvemini, D.; Khanna, R. Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces and prevents pain chronification in a mouse model of oxaliplatin-induced neuropathic pain. Neurobiol. Pain 2021, 11, 100082. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Liu, X.; Tang, Q.; Deng, Y. Pain-related mediators underlie incision-induced mechanical nociception in the dorsal root ganglia. Neural Regen. Res. 2013, 8, 3325–3333. [Google Scholar] [CrossRef]
- Sun, J.; Li, N.; Duan, G.; Liu, Y.; Guo, S.; Wang, C.; Zhu, C.; Zhang, X. Increased Nav1.7 expression in the dorsal root ganglion contributes to pain hypersensitivity after plantar incision in rats. Mol. Pain 2018, 14, 1744806918782323. [Google Scholar] [CrossRef]
- Barbosa Neto, J.O.; Garcia, J.B.S.; Cartágenes, M.D.S.S.; Amaral, A.G.; Onuchic, L.F.; Ashmawi, H.A. Influence of androgenic blockade with flutamide on pain behaviour and expression of the genes that encode the NaV1.7 and NaV1.8 voltage-dependent sodium channels in a rat model of postoperative pain. J. Transl. Med. 2019, 17, 287. [Google Scholar] [CrossRef]
- Liu, B.W.; Zhang, J.; Hong, Y.S.; Li, N.B.; Liu, Y.; Zhang, M.; Wu, W.Y.; Zheng, H.; Lampert, A.; Zhang, X.W. NGF-Induced Nav1.7 Upregulation Contributes to Chronic Post-surgical Pain by Activating SGK1-Dependent Nedd4-2 Phosphorylation. Mol. Neurobiol. 2021, 58, 964–982. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Xiang, G.; Zhang, X.; Yuan, R.; Zhan, H.; Qi, D. A single-nucleotide polymorphism in SCN9A may decrease postoperative pain sensitivity in the general population. Anesthesiology 2013, 118, 436–442. [Google Scholar] [CrossRef]
- Beckley, J.T.; Pajouhesh, H.; Luu, G.; Klas, S.; Delwig, A.; Monteleone, D.; Zhou, X.; Giuvelis, D.; Meng, I.D.; Yeomans, D.C.; et al. Antinociceptive properties of an isoform-selective inhibitor of Nav1.7 derived from saxitoxin in mouse models of pain. Pain 2021, 162, 1250–1261. [Google Scholar] [CrossRef]
- Staunton, C.A.; Lewis, R.; Barrett-Jolley, R. Ion channels and osteoarthritic pain: Potential for novel analgesics. Curr. Pain Headache Rep. 2013, 17, 378. [Google Scholar] [CrossRef]
- Reimann, F.; Cox, J.J.; Belfer, I.; Diatchenko, L.; Zaykin, D.V.; McHale, D.P.; Drenth, J.P.; Dai, F.; Wheeler, J.; Sanders, F.; et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc. Natl. Acad. Sci. USA 2010, 107, 5148–5153. [Google Scholar] [CrossRef] [PubMed]
- Rahman, W.; Dickenson, A.H. Osteoarthritis-dependent changes in antinociceptive action of Nav1.7 and Nav1.8 sodium channel blockers: An in vivo electrophysiological study in the rat. Neuroscience 2015, 295, 103–116. [Google Scholar] [CrossRef]
- Hestehave, S.; Allen, H.N.; Gomez, K.; Duran, P.; Calderon-Rivera, A.; Loya-López, S.; Rodríguez-Palma, E.J.; Khanna, R. Small molecule targeting NaV1.7 via inhibition of CRMP2-Ubc9 interaction reduces pain-related outcomes in a rodent osteoarthritic model. Pain 2024, 166, 99–111. [Google Scholar] [CrossRef]
- Abboud, C.; Duveau, A.; Bouali-Benazzouz, R.; Massé, K.; Mattar, J.; Brochoire, L.; Fossat, P.; Boué-Grabot, E.; Hleihel, W.; Landry, M. Animal models of pain: Diversity and benefits. J. Neurosci. Methods 2021, 348, 108997. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xie, Y.F.; Smith, R.; Ratté, S.; Prescott, S.A. Discordance between preclinical and clinical testing of Na V 1.7-selective inhibitors for pain. Pain 2025, 166, 481–501. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, N.; Fahmideh, F.; Pascale, A.; Allegri, M.; Govoni, S. Neuropathic Pain in Aged People: An Unresolved Issue Open to Novel Drug Approaches, Focusing on Painful Diabetic Neuropathy. Curr. Neuropharmacol. 2024, 22, 53–64. [Google Scholar] [CrossRef]
- Ruiz-Medina, J.; Baulies, A.; Bura, S.A.; Valverde, O. Paclitaxel-induced neuropathic pain is age dependent and devolves on glial response. Eur. J. Pain 2013, 17, 75–85. [Google Scholar] [CrossRef]
- Mogil, J.S.; Wilson, S.G.; Bon, K.; Lee, S.E.; Chung, K.; Raber, P.; Pieper, J.O.; Hain, H.S.; Belknap, J.K.; Hubert, L.; et al. Heritability of nociception I: Responses of 11 inbred mouse strains on 12 measures of nociception. Pain 1999, 80, 67–82. [Google Scholar] [CrossRef]
- Mogil, J.S. Qualitative sex differences in pain processing: Emerging evidence of a biased literature. Nat. Rev. Neurosci. 2020, 21, 353–365. [Google Scholar] [CrossRef]
- Shansky, R.M.; Murphy, A.Z. Considering sex as a biological variable will require a global shift in science culture. Nat. Neurosci. 2021, 24, 457–464. [Google Scholar] [CrossRef]
- Sadler, K.E.; Mogil, J.S.; Stucky, C.L. Innovations and advances in modelling and measuring pain in animals. Nat. Rev. Neurosci. 2022, 23, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.; Bohnert, T.; Damian-Iordache, V.; Gibson, C.; Hsu, C.P.; Krishnatry, A.S.; Liederer, B.M.; Lin, J.; Lu, Q.; Mettetal, J.T.; et al. Translational pharmacokinetic-pharmacodynamic analysis in the pharmaceutical industry: An IQ Consortium PK-PD Discussion Group perspective. Drug Discov. Today 2017, 22, 1447–1459. [Google Scholar] [CrossRef]
- Ballard, J.E.; Pall, P.S.; Vardigan, J.; Zhao, F.; Holahan, M.A.; Zhou, X.; Jochnowitz, N.; Kraus, R.L.; Klein, R.M.; Henze, D.A.; et al. Translational Pharmacokinetic-Pharmacodynamic Modeling of NaV1.7 Inhibitor MK-2075 to Inform Human Efficacious Dose. Front. Pharmacol. 2021, 12, 786078. [Google Scholar] [CrossRef]
- Jansson-Löfmark, R.; Hjorth, S.; Gabrielsson, J. Does In Vitro Potency Predict Clinically Efficacious Concentrations? Clin. Pharmacol. Ther. 2020, 108, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Frézel, N.; Dib-Hajj, S.D.; Waxman, S.G. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 2012, 8, 82. [Google Scholar] [CrossRef]
- Kraus, R.L.; Zhao, F.; Pall, P.S.; Zhou, D.; Vardigan, J.D.; Danziger, A.; Li, Y.; Daley, C.; Ballard, J.E.; Clements, M.K.; et al. Na(v)1.7 target modulation and efficacy can be measured in nonhuman primate assays. Sci. Transl. Med. 2021, 13, eaay1050. [Google Scholar] [CrossRef] [PubMed]
- Morinville, A.; Fundin, B.; Meury, L.; Juréus, A.; Sandberg, K.; Krupp, J.; Ahmad, S.; O’Donnell, D. Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: Expression in the autonomic and endocrine systems. J. Comp. Neurol. 2007, 504, 680–689. [Google Scholar] [CrossRef]
- Kim, J.S.; Meeker, S.; Ru, F.; Tran, M.; Zabka, T.S.; Hackos, D.; Undem, B.J. Role of NaV1.7 in postganglionic sympathetic nerve function in human and guinea-pig arteries. J. Physiol. 2024, 602, 3505–3518. [Google Scholar] [CrossRef]
- Kollarik, M.; Ru, F.; Pavelkova, N.; Mulcahy, J.; Hunter, J.; Undem, B.J. Role of NaV 1.7 in action potential conduction along human bronchial vagal afferent C-fibres. Br. J. Pharmacol. 2022, 179, 242–251. [Google Scholar] [CrossRef]
- Rush, A.M.; Dib-Hajj, S.D.; Liu, S.; Cummins, T.R.; Black, J.A.; Waxman, S.G. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. USA 2006, 103, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Dourado, M.; Reese, R.M.; Huang, K.; Shields, S.D.; Stark, K.L.; Maksymetz, J.; Lin, H.; Kaminker, J.S.; Jung, M.; et al. Nav1.7 is essential for nociceptor action potentials in the mouse in a manner independent of endogenous opioids. Neuron 2023, 111, 2642–2659. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Tagen, M.; Chang, J.H.; Boyce-Rustay, J.; Friesenhahn, M.; Hackos, D.H.; Hains, A.; Sutherlin, D.; Ward, M.; Cho, W. Safety, Tolerability, and Pharmacokinetics of GDC-0276, a Novel NaV1.7 Inhibitor, in a First-in-Human, Single- and Multiple-Dose Study in Healthy Volunteers. Clin. Drug Investig. 2019, 39, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, J.V.; Beckley, J.T.; Klas, S.D.; Odink, D.A.; Delwig, A.; Pajouhesh, H.; Monteleone, D.; Zhou, X.; Du Bois, J.; Yeomans, D.C.; et al. ST-2560, a selective inhibitor of the NaV1.7 sodium channel, affects nocifensive and cardiovascular reflexes in non-human primates. Br. J. Pharmacol. 2024, 181, 3160–3171. [Google Scholar] [CrossRef] [PubMed]
- Regan, C.P.; Morissette, P.; Kraus, R.L.; Wang, E.; Arrington, L.; Vavrek, M.; de Hoon, J.; Depre, M.; Lodeweyck, T.; Demeyer, I.; et al. Autonomic Dysfunction Linked to Inhibition of the Nav1.7 Sodium Channel. Circulation 2024, 149, 1394–1396. [Google Scholar] [CrossRef]
- Martina, M.; Perkins, M. Small molecules versus biologics: The quest for the ideal Nav1.7 inhibitor. Drug Target. Rev. 2015, 2, 34–37. [Google Scholar]
- Osteen, J.D.; Immani, S.; Tapley, T.L.; Indersmitten, T.; Hurst, N.W.; Healey, T.; Aertgeerts, K.; Negulescu, P.A.; Lechner, S.M. Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain. Pain Ther. 2025, 14, 655–674. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Wen, J.; Yang, W.; Wang, C.; Gao, L.; Zheng, L.H.; Wang, T.; Ran, K.; Li, Y.; Li, X.; et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 2013, 93, 957–966. [Google Scholar] [CrossRef]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; López García, J.A. Potassium channels in neuropathic pain: Advances, challenges, and emerging ideas. Pain 2016, 157 (Suppl. S1), S7–S14. [Google Scholar] [CrossRef]
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium channel modulation as a target in chronic pain control. Br. J. Pharmacol. 2018, 175, 2173–2184. [Google Scholar] [CrossRef]
- Dulai, J.S.; Smith, E.S.J.; Rahman, T. Acid-sensing ion channel 3: An analgesic target. Channels 2021, 15, 94–127. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carvajal, A.; González-Muñiz, R.; Fernández-Ballester, G.; Ferrer-Montiel, A. Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Expert Opin. Investig. Drugs 2020, 29, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Jo, Y.Y.; Kim, Y.H.; Park, C.K. Current insights and therapeutic strategies for targeting TRPV1 in neuropathic pain management. Life Sci. 2024, 355, 122954. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Pain Model | Test Compound | Class | Genetic Model | Reference |
|---|---|---|---|---|
| Inflammatory | ||||
| CFA | - | - | Nav1.7 cKO, Nav1.7 gKO Nav1.7R−/− | [91,92,105] |
| Compound 52 | Aminotriazine | - | [102] | |
| GX-201, GX-201, GX-585 | Acylsulfonamide | - | [91,111] | |
| NAN-190 | 5-HT1A antagonist | - | [108] | |
| GpTx-1-71 | Venom peptide | - | [109] | |
| VHH DI-D | SdAb | [110] | ||
| Carrageenan | - | - | Nav1.7R−/− | [105] |
| ZFP-KRAB | Epigenetic platform | - | [146] | |
| Formalin | - | - | Nav1.7 gKO, Nav1.7R−/− | [92,105] |
| Compound 52 | Aminotriazine | - | [102] | |
| VHH DI-D | SdAb | - | [110] | |
| μ-conotoxin KIIIA analogues | Venom peptide | - | [114] | |
| QLS-278 | Acylsulfonamide derivative | - | [115] | |
| Neuropathic | ||||
| CCI | - | - | Nav1.7Advill, Nav1.7Nav1.8, Nav1.7Wnt1 | [35] |
| Compound 29 | Acylsulfonamide derivative | - | [124] | |
| NeP1 | Tocainide congener | - | [125] | |
| Compound 194 | CRMP2 SUMOylation inhibitor | - | [126] | |
| SNI | - | - | Nav1.7 cKO | [91] |
| miR-182 agomir | MicroRNA | - | [51] | |
| VHH DI-D | SdAb | - | [110] | |
| QLS-278 | Acylsulfonamide derivative | - | [115] | |
| μ-TRTX-Hhn1b | Venom peptide | - | [132] | |
| Compound 3 g | 3-Hydroxyindole backbone | - | [133] | |
| Compound 194 | CRMP2 SUMOylation inhibitor | - | [128] | |
| Myr-TAT-Nav1.7-CRS | CRMP2/Nav1.7 interaction inhibitor | - | [135] | |
| SNL | - | - | Nav1.7Wnt1, Nav1.7Advill (no effect), nociceptor-specific Nav1.7 KO, HOM-KI rats | [93,139,140] |
| Compound 194 | CRMP2 SUMOylation inhibitor | - | [128] | |
| Z123212 | Piperazine derivative | - | [141] | |
| Compound 33 | Quinoline amide | - | [142] | |
| Compound 9 | Aminocyclohexene analogue | - | [143] | |
| SCI | PF-05089771 | Arylsulfonamide | - | [144] |
| GNE-0439 | VSD4 domain | - | [144] | |
| CIPN (Paclitaxel) | ZFP-KRAB, KRAB-dCas9 | Epigenetic platform | - | [146] |
| DA-0218 | Propanamide, methoxyphenyl, benzyl indolyl derivative | - | [150] | |
| Myr-TAT-Nav1.7-CRS | CRMP2/Nav1.7 interaction inhibitor | - | [135] | |
| CIPN (Oxaliplatin) | - | - | Nav1.7Advill, Nav1.7Wnt1 (no effect) | [35] |
| ProTx II | Venom peptide | - | [154] | |
| Compound 194 | CRMP2 SUMOylation inhibitor | - | [155] | |
| Post operative | ||||
| Plantar incision | - | - | Nav1.7 cKO (effect on thermal but not mechanical response) | [91] |
| SCN9A-RNAi-LV | siRNA lentivirus | - | [157] | |
| ST-2530 | Saxitoxin analog | - | [161] | |
| Osteoarthritis | ||||
| MIA | - | - | Nav1.7DRG;chondrocyte | [83] |
| PF-04756264 | Arylsulfonamide | - | [83] | |
| ProTx II | Venom peptide | - | [164] | |
| Compound 194 | CRMP2 SUMOylation inhibitor | - | [165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yogi, A.; Banderali, U.; Moreno, M.J.; Martina, M. Preclinical Animal Models to Investigate the Role of Nav1.7 Ion Channels in Pain. Life 2025, 15, 640. https://doi.org/10.3390/life15040640
Yogi A, Banderali U, Moreno MJ, Martina M. Preclinical Animal Models to Investigate the Role of Nav1.7 Ion Channels in Pain. Life. 2025; 15(4):640. https://doi.org/10.3390/life15040640
Chicago/Turabian StyleYogi, Alvaro, Umberto Banderali, Maria J. Moreno, and Marzia Martina. 2025. "Preclinical Animal Models to Investigate the Role of Nav1.7 Ion Channels in Pain" Life 15, no. 4: 640. https://doi.org/10.3390/life15040640
APA StyleYogi, A., Banderali, U., Moreno, M. J., & Martina, M. (2025). Preclinical Animal Models to Investigate the Role of Nav1.7 Ion Channels in Pain. Life, 15(4), 640. https://doi.org/10.3390/life15040640