
Full-Length Transcriptome Sequencing and RNA-Seq Analysis Offer Insights into Terpenoid Biosynthesis in Blumea balsamifera (L.) DC.


 ,
,
Abstract
1. Introduction
2. Results
2.1. Terpenoids of the Essential Oils in B. balsamifera
2.2. Transcriptome Assembly from NGS and SMRT Sequencing of B. balsamifera
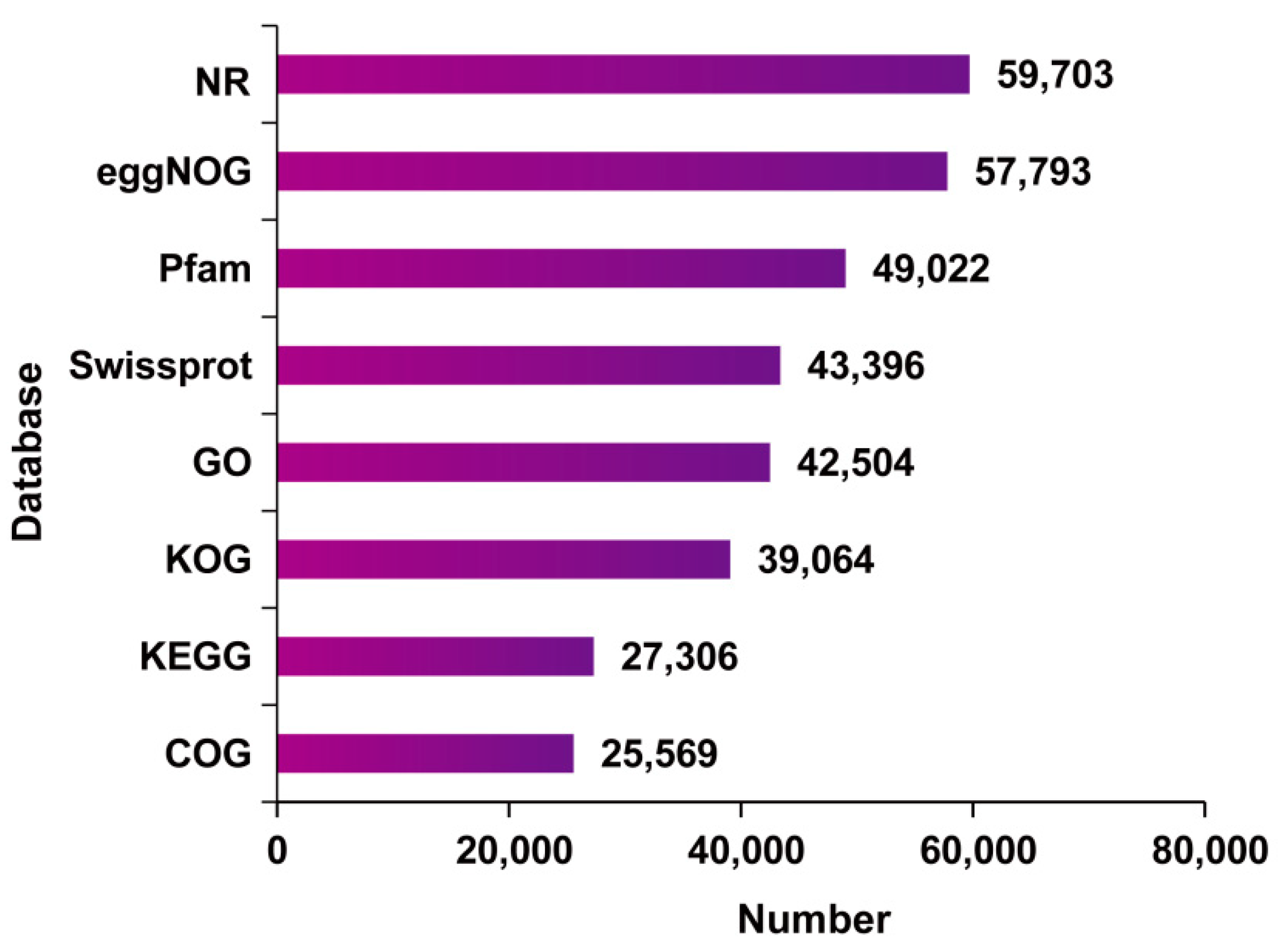
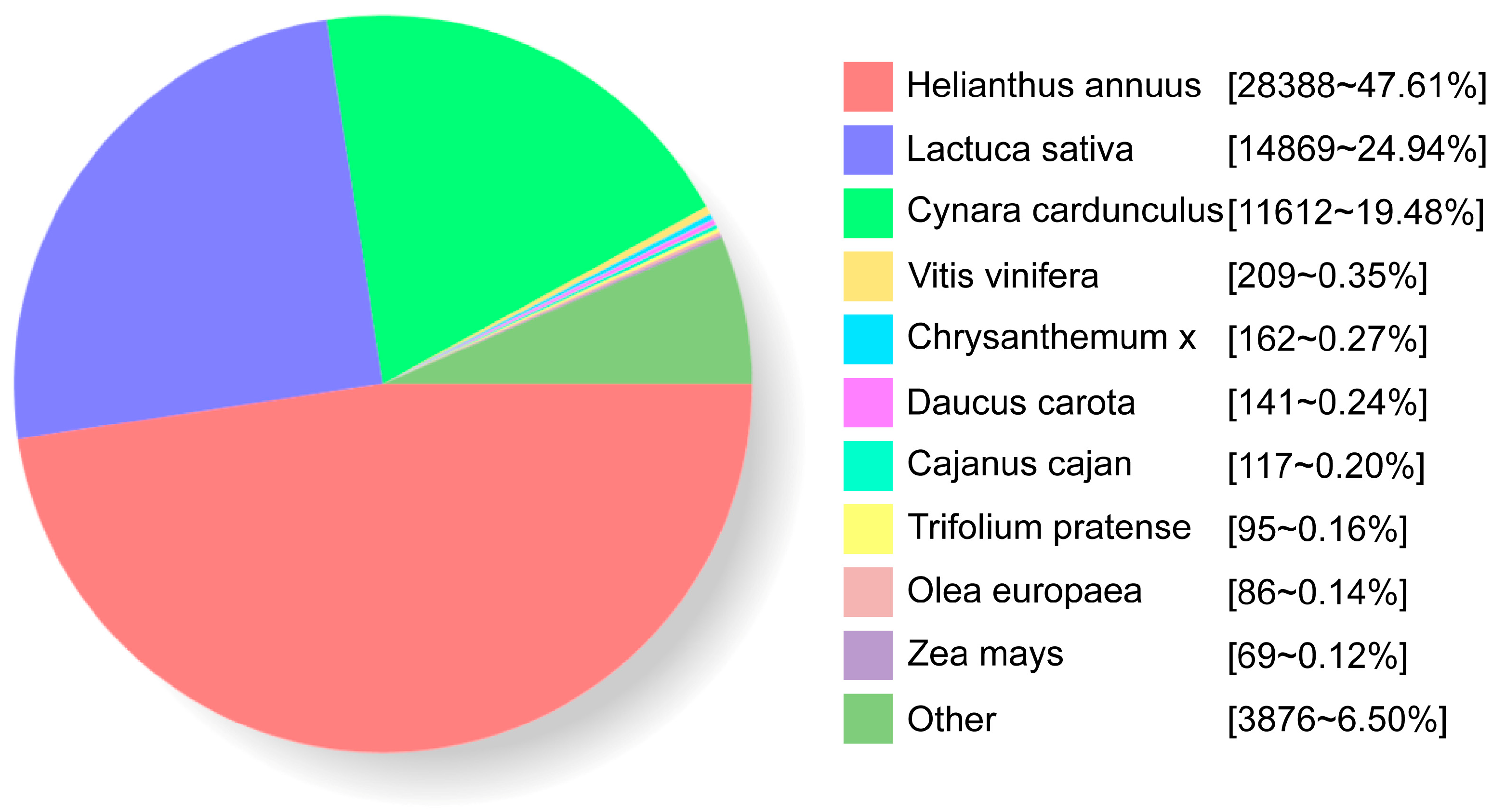
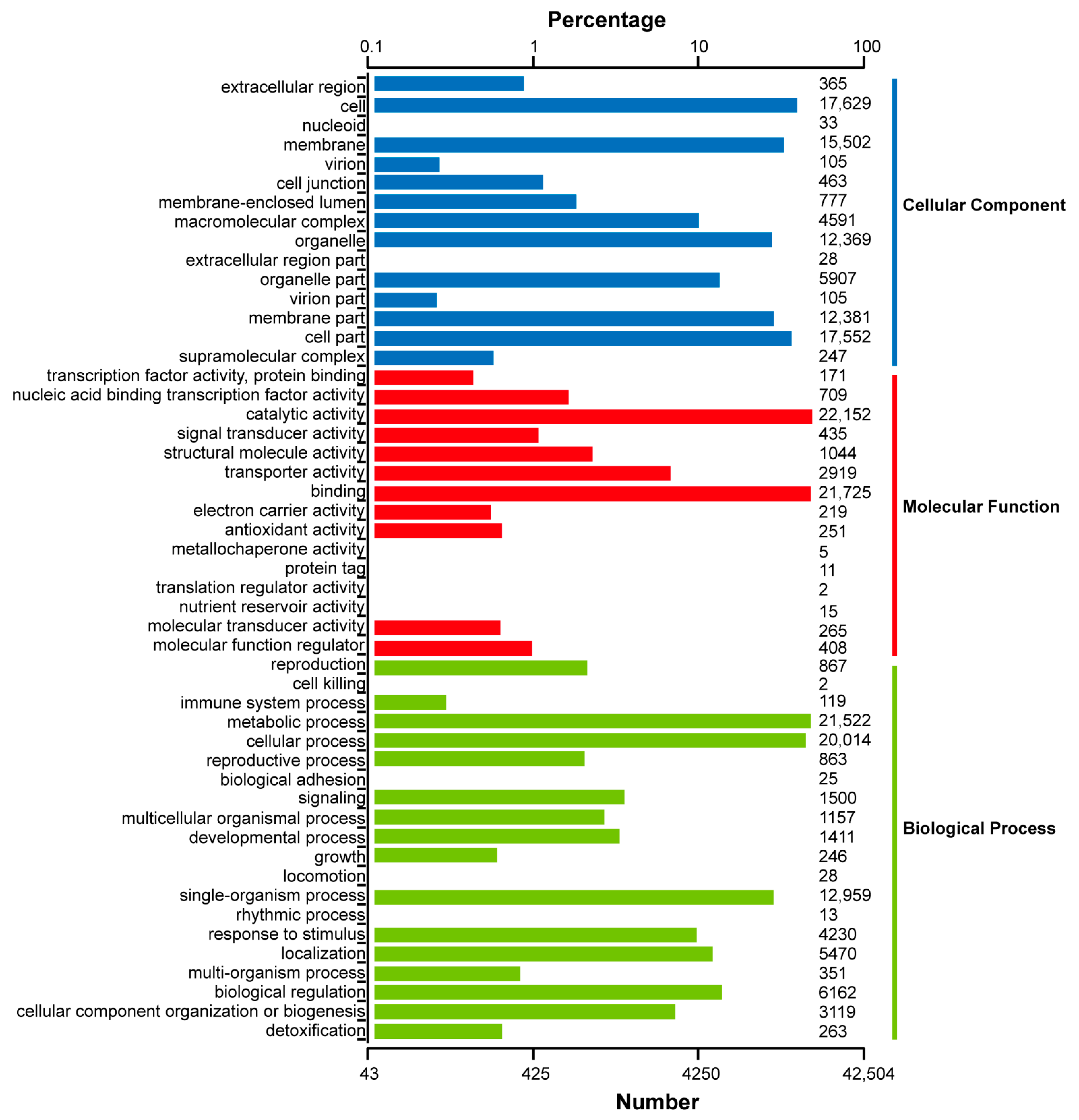
2.3. Functional Annotation of Full-Length Transcriptome
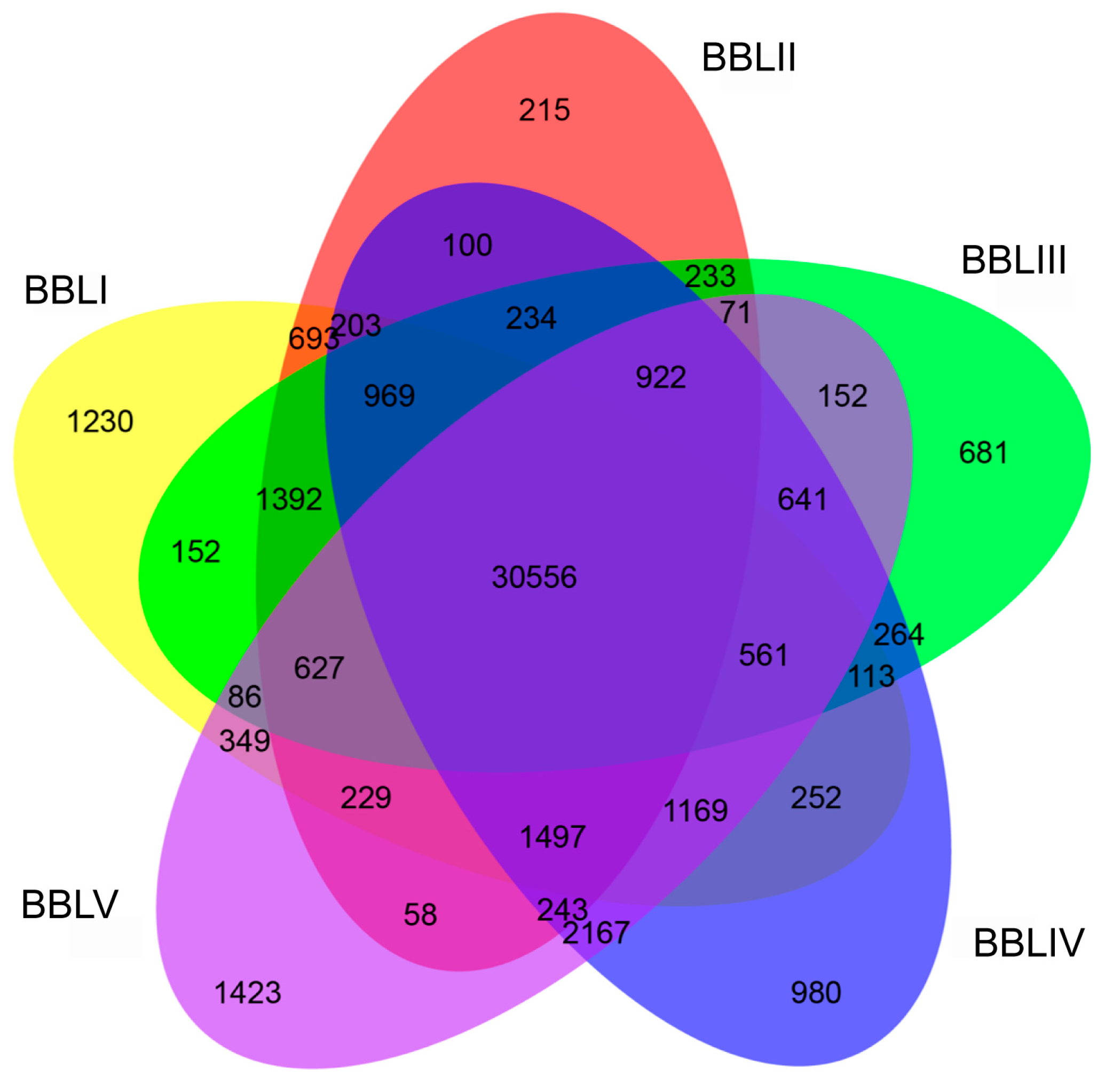
2.4. Identification of Differentially Expressed Transcripts (DETs)
2.5. Identification of TFs—1–2000
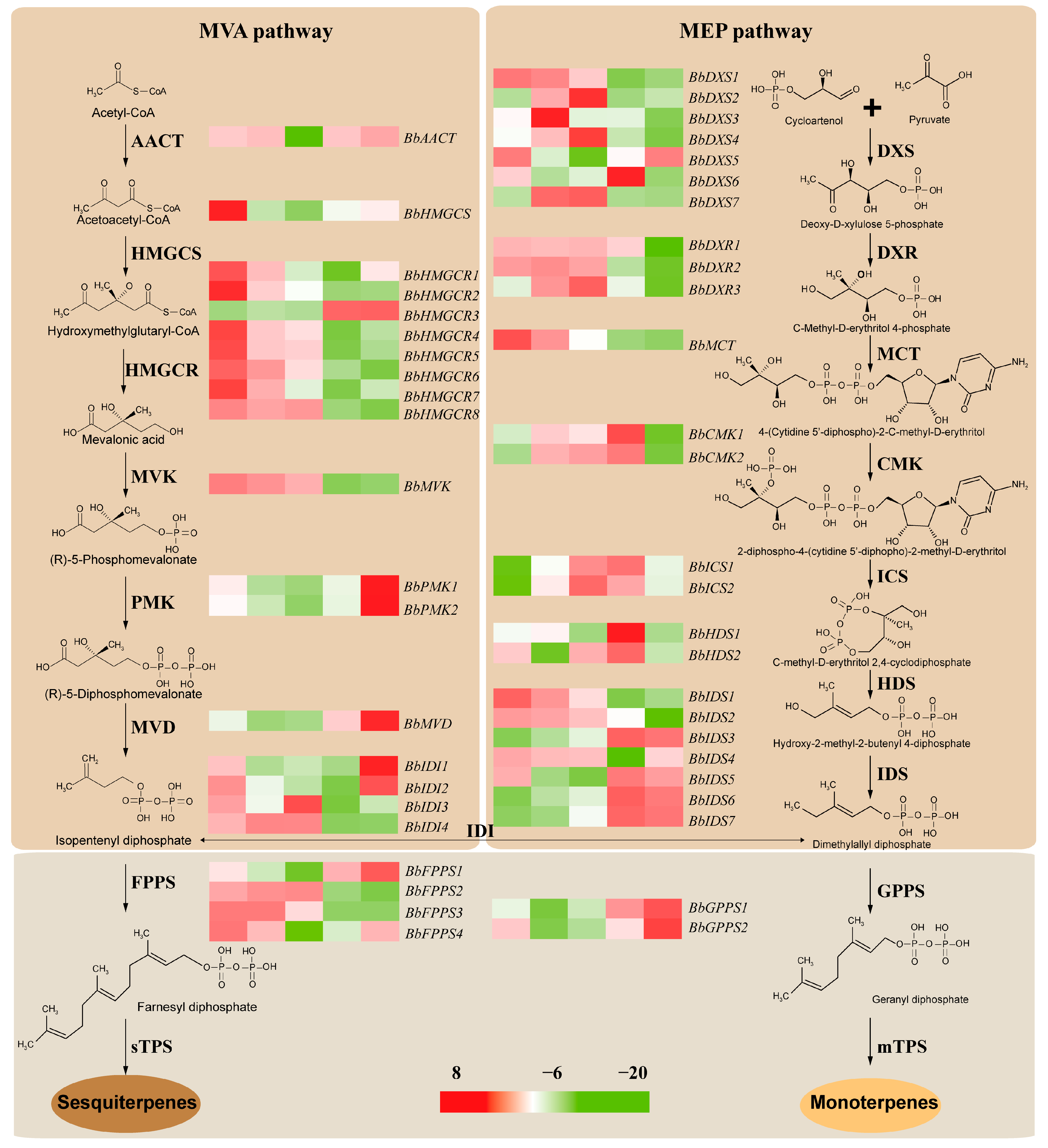
2.6. Identification of Hub Genes Involved in Terpenoid Biosynthesis
2.7. RT-qPCR Validation of DETs
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. GC–MS Analysis of B. balsamifera Essential Oils
4.3. RNA Extraction and Sequencing
4.4. Full-Length Transcriptome Analysis
4.5. Functional Annotation of Non-Redundant Isoforms
4.6. Identification of Differentially Expressed Transcripts (DETs)
4.7. Real Time-Quantitative PCR (RT-qPCR) Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pang, Y.; Wang, D.; Fan, Z.; Chen, X.; Yu, F.; Hu, X. Blumea balsamifera—A phytochemical and pharmacological review. Molecules 2014, 19, 9453–9477. [Google Scholar] [CrossRef]
- Yuan, Y.; Pang, Y.X.; Wang, W.Q.; Zhang, Y.B.; Yu, J.B. Investigation on the plant resources of Blumea balsamifera (L.) DC. in China. J. Trop. Biol. 2011, 2, 78–82. [Google Scholar]
- Guan, L.L.; Pang, Y.X.; Wang, D.; Zhang, Y.B.; Wu, K.Y. Research progress on Chinese Minority Medicine of Blumea balsamifera L. DC. J. Plant Genet. Res. 2012, 13, 695–698. [Google Scholar]
- Hasegawa, H.; Yamada, Y.; Komiyama, K.; Hayashi, M.; Ishibashi, M.; Yoshida, T.; Sakai, T.; Koyano, T.; Kam, T.S.; Murata, K.; et al. Dihydroflavonol BB-1, an extract of natural plant Blumea balsamifera, abrogates TRAIL resistance in leukemia cells. Blood 2006, 107, 679–688. [Google Scholar] [CrossRef]
- Saewan, N.; Koysomboon, S.; Chantrapromma, K. Anti-tyrosinase and anti-cancer activities of flavonoids from Blumea balsamifera DC. J. Med. Plants Res. 2011, 5, 1018–1025. [Google Scholar]
- Chen, X.; Su, H.Z.Z. Detailed studies on the anticancer action of rosmarinic acid in human Hep-G2 liver carcinoma cells: Evaluating its effects on cellular apoptosis, caspase activation and suppression of cell migration and invasion. Off. J. Balk. Union Oncol. 2020, 25, 2011–2016. [Google Scholar]
- Li, J.; Zhao, G.Z.; Chen, H.H.; Wang, H.B.; Qin, S.; Zhu, W.Y.; Xu, L.H.; Jiang, C.L.; Li, W.J. Antitumour and antimicrobial activities of endophytic streptomycetes from pharmaceutical plants in rainforest. Lett. Appl. Microbiol. 2008, 47, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Ragasa, C.Y.; Co, A.L.K.C.; Rideout, J.A. Antifungal metabolites from Blumea balsamifera. Nat. Prod. Res. 2005, 19, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Nessa, F.; Ismail, Z.; Mohamed, N.; Haris, M.R.H.M. Free radical-scavenging activity of organic extracts and of pure flavonoids of Blumea balsamifera DC leaves. Food Chem. 2004, 88, 243–252. [Google Scholar] [CrossRef]
- Xiong, Y.; Yi, P.; Li, Y.; Gao, R.; Chen, J.; Hu, Z.; Lou, H.; Du, C.; Zhang, J.; Zhang, Y.; et al. New sesquiterpeniod esters form Blumea balsamifera (L.) DC. and their anti-influenza virus activity. Nat. Prod. Res. 2022, 36, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. Adv. Biochem. Eng. Biotechnol. 2015, 148, 63–106. [Google Scholar]
- Zhou, F.; Pichersky, E. More is better: The diversity of terpene metabolism in plants. Curr. Opin. Plant Biol. 2020, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.D.; Chappll, J. Isoprenoid biosynthesis in plant: Carbon partitioning within the cytoplasmic pathway. Crit. Rev. Biochem. Mol. Biol. 1999, 34, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Ward, B.L.; Bostock, R.M. Differential induction and suppression of potato 3-hydroxy-3-methylglutaryl coenzyme A reductase genes in response to Phytophthora infestans and to its elicitor arachidonic acid. Plant Cell 1992, 4, 1333–1344. [Google Scholar] [PubMed]
- Zhao, S.; Wang, L.; Liu, L.; Liang, Y.; Sun, Y.; Wu, J. Both the mevalonate and the non-mevalonate pathways are involved in ginsenoside biosynthesis. Plant Cell Rep. 2014, 33, 393–400. [Google Scholar] [CrossRef]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.C.; Croteau, R.B. Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics 2001, 158, 811–832. [Google Scholar] [CrossRef]
- Zhu, F.Y.; Chen, M.X.; Ye, N.H.; Qiao, W.M.; Gao, B.; Law, W.K.; Tian, Y.; Zhang, D.; Zhang, D.; Liu, T.Y.; et al. Comparative performance of the BGISEQ-500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant Methods 2018, 14, 69. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Cui, Y.; Gao, X.; Wang, J.; Shang, Z.; Zhang, Z.; Zhou, Z.; Zhang, K. Full-Length Transcriptome Analysis Reveals Candidate Genes Involved in terpenoids Biosynthesis in Artemisia argyi. Front. Genet. 2021, 12, 659962. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, Z.; Yang, Y.; Liu, X.; Lv, H.; Song, B.H. Transcriptome profiling reveals the spatial-temporal dynamics of gene expression essential for soybean seed development. BMC Genom. 2021, 22, 453. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, C.; Zhao, S.; Zhang, S.; Wang, W.; Fu, H. De novo transcriptome and phytochemical analyses reveal differentially expressed genes and characteristic secondary metabolites in the original oolong tea (Camellia sinensis) cultivar ‘Tieguanyin’ compared with cultivar ‘Benshan’. BMC Genom. 2019, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Shen, Q.; Huang, H.; Xie, L.; Hao, X.; Kayani, S.I.; Liu, H.; Qin, W.; Chen, T.; Pan, Q.; Liu, P.; et al. Basic Helix-Loop-Helix Transcription Factors AabHLH2 and AabHLH3 Function Antagonistically with AaMYC2 and Are Negative Regulators in Artemisinin Biosynthesis. Front. Plant Sci. 2022, 13, 885622. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.J.; Xue, X.Y.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 2012, 24, 2635–4268. [Google Scholar] [CrossRef]
- Matías-Hernández, L.; Jiang, W.; Yang, K.; Tang, K.; Brodelius, P.E.; Pelaz, S. AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J. 2017, 90, 520–534. [Google Scholar] [CrossRef]
- Paul, P.; Singh, S.K.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoids indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2017, 213, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Suttipanta, N.; Pattanaik, S.; Kulshrestha, M.; Patra, B.; Singh, S.K.; Yuan, L. The transcription factor CrWRKY1 positively regulates the terpenoids indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 2011, 157, 2081–2093. [Google Scholar] [CrossRef]
- Zhang, F.; Xiang, L.; Yu, Q.; Zhang, H.; Zhang, T.; Zeng, J. Artemisinin biosynthesis promoting kinase 1 positively regulates artemisinin biosynthesis through phosphorylating AabZIP1. J. Exp. Bot. 2018, 69, 1109–1123. [Google Scholar] [CrossRef]
- Hu, X.; Wang, K.; Yu, F.L.; Wang, D.; Xie, X.L.; Pang, Y.X.; Chen, H.F. Composition and Antibacterial Activity of Blumea balsamifera Extracts. J. Fujian Agric. 2021, 36, 1131–1138. [Google Scholar]
- Wang, Y.H.; Wang, H.X.; Tian, H.Y.; Ma, C.Y.; Chen, T.; Zou, C.L.; Wang, X. Headspace Solid-Phase Microextraction Coupled with GC-MS for Analysis of Aromatic Components in Leaves of Blumea balsamifera (L.) DC. in Different Seasons. Food Sci. 2012, 33, 166–170. [Google Scholar]
- Xie, X.L.; Chen, Z.X.; Yu, F.L.; Pang, Y.X.; Hu, X.; Chen, H.F. Discussion on the Industry Development Status of Blumea balsamifera Based on Patent Analysis. China Pharm. 2021, 32, 1158–1164. [Google Scholar]
- Yuan, C.; Wang, H.F.; Hu, X.; Pang, Y.X. Rapid Identification on Chemical Constituents of Antimicrobial Fractions from Blumea balsamifera (L.) DC. by UPLC-Q-TOF-MSE. Nat. Prod. Res. Dev. 2018, 30, 1904–1912. [Google Scholar]
- Guan, L.L.; Xia, Q.F.; Pang, Y.X.; Zhao, Z.; Hu, Y.P.; Bai, L.; Wang, H.F.; Liu, H.C. Analysis of metabolic pathway of terpenoids in Blumea balsamifera. China J. Chin. Mater. Medica 2016, 41, 1585–1591. [Google Scholar]
- Chuang, Y.C.; Hung, Y.C.; Tsai, W.C.; Chen, W.H.; Chen, H.H. PbbHLH4 regulates floral monoterpene biosynthesis in Phalaenopsis orchids. J. Exp. Bot. 2018, 69, 4363–4377. [Google Scholar] [CrossRef]
- Chen, M.; Yan, T.; Shen, Q.; Lu, X.; Pan, Q.; Huang, Y.; Tang, Y.; Fu, X.; Liu, M.; Jiang, W.; et al. Glandular trichome-specific WRKY 1 promotes artemisinin biosynthesis in Artemisia annua. New Phytol. 2017, 214, 304–316. [Google Scholar] [CrossRef]
- Reddy, V.A.; Wang, Q.; Dhar, N.; Kumar, N.; Venkatesh, P.N.; Rajan, C.; Panicker, D.; Sridhar, V.; Mao, H.Z.; Sarojam, R. Spearmint R2R3-MYB transcription factor MsMYB negatively regulates monoterpene production and suppresses the expression of geranyl diphosphate synthase large subunit (MsGPPS.LSU). Plant Biotech. J. 2017, 15, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, D.; Zhang, Q.; Chai, J.; Peng, Y.; Cai, X. Identification and cytochemical immunolocalization of acetyl-CoA acetyltransferase involved in the terpenoids mevalonate pathway in Euphorbia helioscopia laticifers. Bot. Stud. 2017, 58, 62. [Google Scholar] [CrossRef] [PubMed]
- Pathak, G.; Dudhagi, S.S.; Raizada, S.; Singh, R.K.; Sane, A.P.; Sane, V.A. Phosphomevalonate kinase regulates the MVA/MEP pathway in mango during ripening. Plant Physiol. Bioch. 2023, 196, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Berasategui, A.; Paetz, C.; Gershenzon, J.; Schmidt, A. Overexpression of an isoprenyl diphosphate synthase in spruce leads to unexpected terpene diversion products that function in plant defense. Plant Physiol. 2014, 164, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Suenaga-Hiromori, M.; Mogi, D.; Kikuchi, Y.; Tong, J.; Kurisu, N.; Aoki, Y.; Amano, H.; Furutani, M.; Shimoyama, T.; Waki, T.; et al. Comprehensive identification of terpene synthase genes and organ-dependent accumulation of terpenoids volatiles in a traditional medicinal plant Angelica archangelica L. Plant Biotechnol. 2022, 39, 391–404. [Google Scholar] [CrossRef]
- Felipe, A.S.; Robert, M.W.; Panagiotis, I.; Evgenia, V.K.; Evgeny, M.Z. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein family database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Wang, L.G.; Park, H.J.; Dasari, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression mode. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated NR Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Name of Compound | Formula | Retention Index | Count Per Second |
|---|---|---|---|---|
| 1 | L(-)-Borneol | C10H18O | 1.17 × 103 | 5186.23 |
| 2 | (-)-Terpinen-4-ol | C10H18O | 1.18 × 103 | 86.64 |
| 3 | Linalool | C10H18O | 1.10 × 103 | 23.27 |
| 4 | (-)-Orthodene | C10H16 | 1.04 × 103 | 14.13 |
| 5 | Camphene | C10H16 | 9.49 × 102 | 12.40 |
| 6 | (-)-Bornyl acetate | C12H20O2 | 1.29 × 103 | 8.60 |
| 7 | (+)-2-Bornanone | C10H16O | 1.15 × 103 | 6.84 |
| 8 | 1,3-Cyclohexadiene | C10H16 | 1.04 × 103 | 5.86 |
| 9 | L-α-Terpineol | C10H18O | 1.20 × 103 | 1.80 |
| 10 | p-Mentha-1,5,8-triene | C10H14 | 1.13 × 103 | 1.69 |
| 11 | Pinocarvone | C10H14O | 1.16 × 103 | 0.54 |
| 12 | 1-(2-Ethyl-3-cyclohexenyl)ethanol | C10H18O | 1.14 × 103 | 0.19 |
| 13 | Allo-ocimene | C10H16 | 1.14 × 103 | 0.19 |
| 14 | α-Thujene | C10H16 | 9.25 × 102 | 0.19 |
| 15 | cis-Sabinenhydrate | C10H18O | 9.77 × 102 | 0.07 |
| 16 | 2-Bornene | C10H16 | 9.73 × 102 | 0.05 |
| 17 | 2-methylene-4,8,8-trimethyl-4-vinylbicyclo[5.2.0]nonane | C15H24 | 1.43 × 103 | 1272.53 |
| 18 | 1R,4R,7R,11R-1,3,4,7-Tetramethyltricyclo[5.3.1.0(4,11)]undec-2-ene | C15H24 | 1.35 × 103 | 942.58 |
| 19 | β-Copaene | C15H24 | 1.43 × 103 | 661.04 |
| 20 | (-)-α-Gurjunene | C15H24 | 1.44 × 103 | 231.35 |
| 21 | (+)-epi-Bicyclosesquiphellandrene | C15H24 | 1.49 × 103 | 56.64 |
| 22 | β-Maaliene | C15H24 | 1.41 × 103 | 29.59 |
| 23 | γ-Cadinene | C15H24 | 1.52 × 103 | 14.22 |
| 24 | α-Calacorene | C15H20 | 1.55 × 103 | 8.52 |
| 25 | Cadina-3,5-diene | C15H24 | 1.38 × 103 | 8.17 |
| 26 | Epizonarene | C15H24 | 1.51 × 103 | 7.33 |
| 27 | γ-Selinene | C15H24 | 1.50 × 103 | 6.82 |
| 28 | β-Cadinene | C15H24 | 1.49 × 103 | 5.62 |
| 29 | Rosifoliol | C15H26O | 1.61 × 103 | 5.04 |
| 30 | β-Elemen | C15H24 | 1.39 × 103 | 4.64 |
| 31 | γ-Muurolene | C15H24 | 1.37 × 103 | 2.48 |
| 32 | (-)-Clovene | C15H24 | 1.37 × 103 | 2.00 |
| 33 | α-Corocalene | C15H20 | 1.62 × 103 | 1.24 |
| 34 | 6-Isopropyl-1,4-dimethylnaphthalene | C15H18 | 1.78 × 103 | 0.53 |
| 35 | β-Guaiene | C15H24 | 1.56 × 103 | 0.36 |
| 36 | Kessane | C15H26O | 1.54 × 103 | 0.26 |
| cDNA Size | Number of Unpolished Consensus Isoforms | Mean Unpolished Consensus Isoform Read Length | Number of Polished HQ Isoforms | Percent of Polished HQ Isoforms (%) |
|---|---|---|---|---|
| 1–6 K | 116,656 | 2244 | 116,639 | 99.99 |
| All | 116,656 | 2244 | 116,639 | 99.99 |
| Order Number | Name of TF Family | Number of Transcripts | Number of TFs with Highest Expression (FPKM > 1) | ||||
|---|---|---|---|---|---|---|---|
| BBLI | BBLII | BBLIII | BBLIV | BBLV | |||
| 1 | C3H | 119 | 34 | 2 | 3 | 16 | 64 |
| 2 | bZIP | 110 | 20 | 9 | 3 | 23 | 55 |
| 3 | bHLH | 109 | 24 | 14 | 14 | 24 | 33 |
| 4 | AP2 | 105 | 28 | 6 | 10 | 21 | 40 |
| 5 | MYB-related | 102 | 18 | 2 | 13 | 27 | 42 |
| 6 | C2H2 | 100 | 22 | 6 | 3 | 24 | 45 |
| 7 | NAC | 94 | 10 | 3 | 2 | 13 | 66 |
| 8 | MYB | 88 | 39 | 4 | 7 | 16 | 22 |
| 9 | GRAS | 84 | 16 | 4 | 5 | 9 | 50 |
| 10 | WRKY | 81 | 11 | 1 | 1 | 19 | 49 |
| 11 | HB-HD-ZIP | 69 | 34 | 1 | 0 | 8 | 26 |
| 12 | B3-ARF | 68 | 48 | 4 | 1 | 12 | 3 |
| 13 | Trihelix | 68 | 24 | 5 | 3 | 11 | 25 |
| 14 | RWP-RK | 56 | 11 | 0 | 5 | 22 | 18 |
| 15 | GARP-G2-like | 49 | 9 | 0 | 7 | 11 | 22 |
| 16 | HSF | 46 | 10 | 0 | 1 | 20 | 15 |
| 17 | SBP | 37 | 17 | 2 | 1 | 2 | 15 |
| 18 | TCP | 37 | 9 | 7 | 3 | 10 | 8 |
| 19 | TUB | 32 | 5 | 2 | 2 | 11 | 12 |
| 20 | C2C2-Dof | 32 | 5 | 5 | 4 | 7 | 11 |
| 21 | C2C2-GATA | 31 | 16 | 3 | 5 | 2 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, Z.; Liang, L.; Zheng, Y.; Shi, H.; Zhao, W.; Sun, W.; Pang, Y. Full-Length Transcriptome Sequencing and RNA-Seq Analysis Offer Insights into Terpenoid Biosynthesis in Blumea balsamifera (L.) DC. Genes 2024, 15, 285. https://doi.org/10.3390/genes15030285
Ju Z, Liang L, Zheng Y, Shi H, Zhao W, Sun W, Pang Y. Full-Length Transcriptome Sequencing and RNA-Seq Analysis Offer Insights into Terpenoid Biosynthesis in Blumea balsamifera (L.) DC. Genes. 2024; 15(3):285. https://doi.org/10.3390/genes15030285
Chicago/Turabian StyleJu, Zhigang, Lin Liang, Yaqiang Zheng, Hongxi Shi, Wenxuan Zhao, Wei Sun, and Yuxin Pang. 2024. "Full-Length Transcriptome Sequencing and RNA-Seq Analysis Offer Insights into Terpenoid Biosynthesis in Blumea balsamifera (L.) DC." Genes 15, no. 3: 285. https://doi.org/10.3390/genes15030285
APA StyleJu, Z., Liang, L., Zheng, Y., Shi, H., Zhao, W., Sun, W., & Pang, Y. (2024). Full-Length Transcriptome Sequencing and RNA-Seq Analysis Offer Insights into Terpenoid Biosynthesis in Blumea balsamifera (L.) DC. Genes, 15(3), 285. https://doi.org/10.3390/genes15030285