
Prevalence of Endocrinopathies in a Cohort of Patients with Rett Syndrome: A Two-Center Observational Study


 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Biochemical and Instrumental Evaluation
2.3. Statistical Analysis
3. Results
3.1. Main Clinical Features of the Study Population
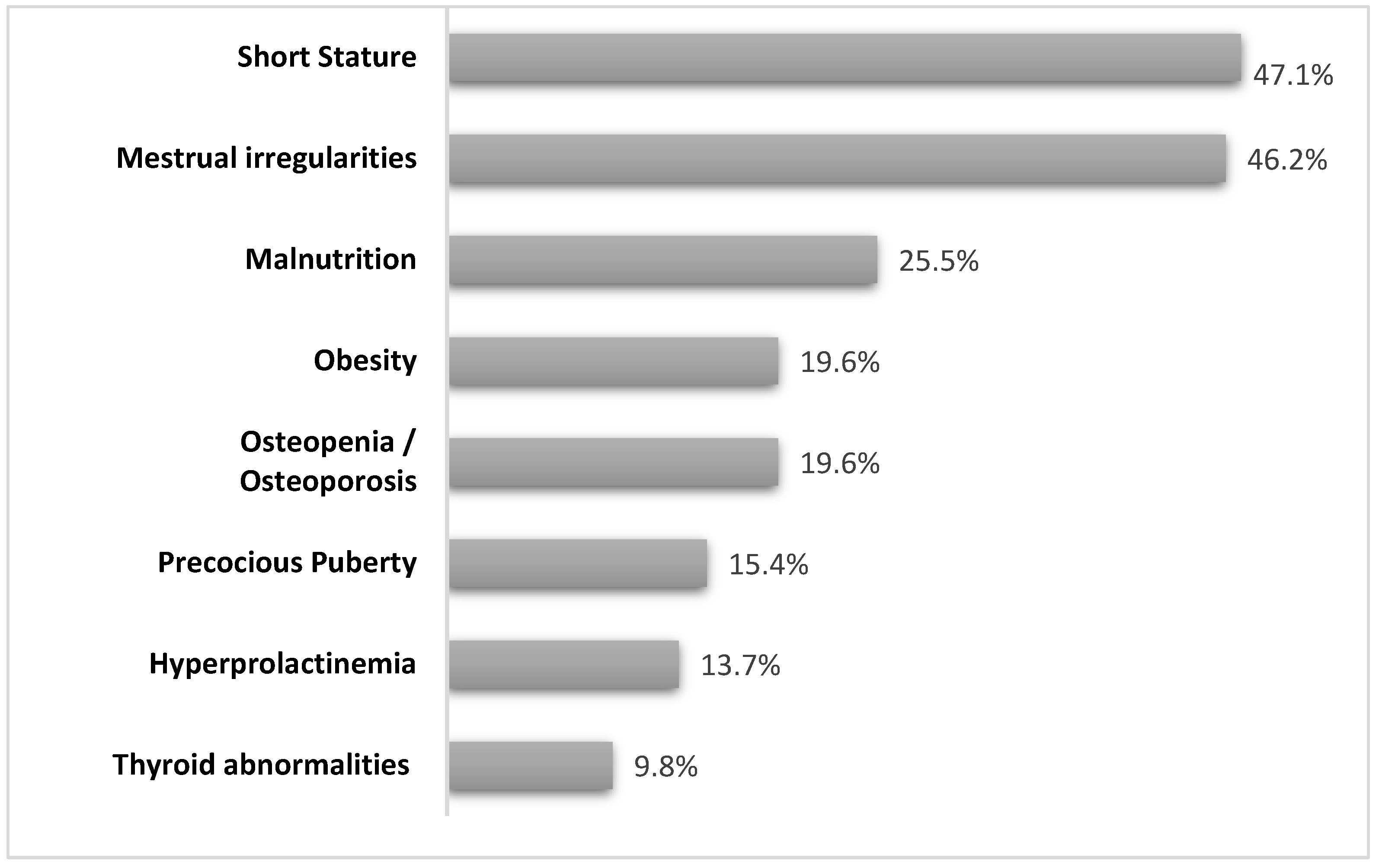
3.2. Endocrinological Report
3.2.1. Short Stature
3.2.2. Weight Disorders
3.2.3. Gonadal Function
3.2.4. Thyroid Disorders
3.2.5. Hyperprolactinemia
3.2.6. Bone Health and Orthopedic Issues
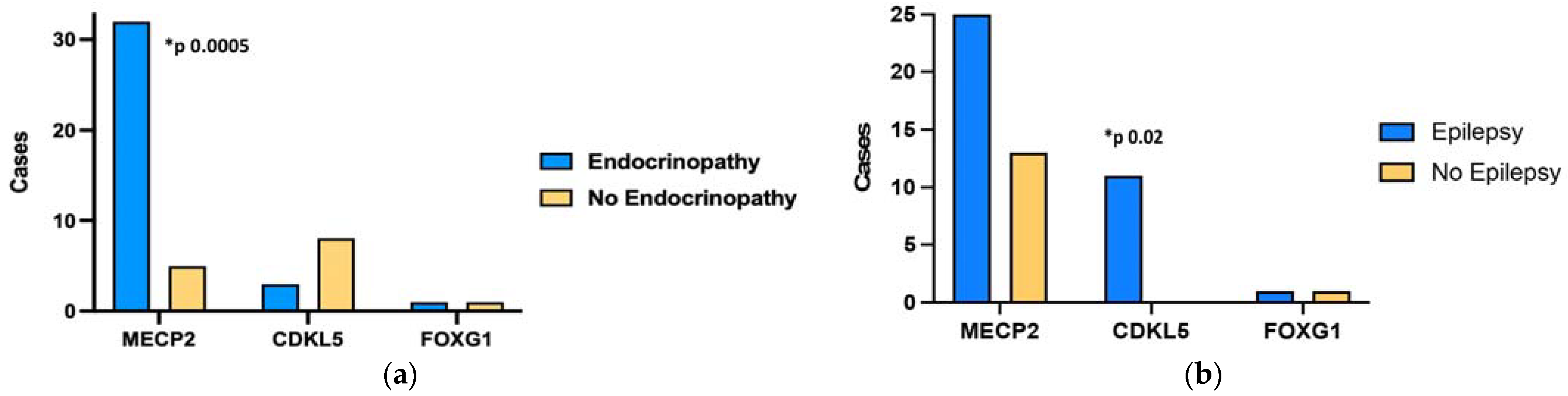
3.3. Genotype-Phenotype Correlation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iourov, I.Y.; Vorsanova, S.G.; Voinova, V.Y.; Kurinnaia, O.S.; Zelenova, M.A.; Demidova, I.A.; Yurov, Y.B. Xq28 (MECP2) Microdeletions Are Common in Mutation-Negative Females with Rett Syndrome and Cause Mild Subtypes of the Disease. Mol. Cytogenet. 2013, 6, 53. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M. Rett Syndrome: Revised Diagnostic Criteria and Nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Percy, A.K. Rett Syndrome: Current Status and New Vistas. Neurol. Clin. 2002, 20, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A Progressive Syndrome of Autism, Dementia, Ataxia, and Loss of Purposeful Hand Use in Girls: Rett’s Syndrome: Report of 35 Cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Halbach, N.S.J.; Smeets, E.E.J.; Steinbusch, C.; Maaskant, M.A.; Van Waardenburg, D.; Curfs, L.M.G. Aging in Rett Syndrome: A Longitudinal Study. Clin. Genet. 2013, 84, 223–229. [Google Scholar] [CrossRef]
- Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt, R.; Standridge, S.; Lane, J.; Dinkel, T.; Jones, M. Multisystem Comorbidities in Classic Rett Syndrome: A Scoping Review. BMJ Paediatr. Open 2020, 4, e000731. [Google Scholar] [CrossRef] [PubMed]
- Motil, K.J.; Ellis, K.J.; Barrish, J.O.; Caeg, E.; Glaze, D.G. Bone Mineral Content and Bone Mineral Density Are Lower in Older than in Younger Females with Rett Syndrome. Pediatr. Res. 2008, 64, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.T.; Lane, J.B.; Cutter, G.R.; Skinner, S.A.; Kaufmann, W.E.; Tarquinio, D.C.; Glaze, D.G.; Motil, K.J.; Neul, J.L.; Percy, A.K. Pubertal Development in Rett Syndrome Deviates from Typical Females. Pediatr. Neurol. 2014, 51, 769–775. [Google Scholar] [CrossRef]
- Thompson, C.C.; Potter, G.B. Thyroid Hormone Action in Neural Development. Cereb. Cortex 2000, 10, 939–945. [Google Scholar] [CrossRef]
- Tanner, J.M. Growth and Maturation during Adolescence. Nutr. Rev. 2009, 39, 43–55. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Committee on Adolescence; American College of Obstetricians and Gynecologists. Committee on Adolescent Health Care Menstruation in Girls and Adolescents: Using the Menstrual Cycle as a Vital Sign. Pediatrics 2006, 118, 2245–2250. [Google Scholar] [CrossRef]
- Borloz, E.; Villard, L.; Roux, J.-C. Rett Syndrome: Think Outside the (Skull) Box. Fac. Rev. 2021, 10, 59. [Google Scholar] [CrossRef]
- Stagi, S.; Giani, T.; Simonini, G.; Falcini, F. Thyroid Function, Autoimmune Thyroiditis and Coeliac Disease in Juvenile Idiopathic Arthritis. Rheumatology 2005, 44, 517–520. [Google Scholar] [CrossRef]
- Schultz, R.J.; Glaze, D.G.; Motil, K.J.; Armstrong, D.D.; del Junco, D.J.; Hubbard, C.R.; Percy, A.K. The Pattern of Growth Failure in Rett Syndrome. Am. J. Dis. Child 1993, 147, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Thommessen, M.; Kase, B.; Heiberg, A. Growth and Nutrition in 10 Girls with Rett Syndrome. Acta Paediatr. 1992, 81, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Cass, S.R. Hilary Growth and Nutrition in Rett Syndrome. Disabil. Rehabil. 2001, 23, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Roth, C.; Christen, H.; Brockmann, K.; Hanefeld, F. Endocrinological Study on Growth Retardation in Rett Syndrome. Acta Paediatr. 2001, 90, 1257–1261. [Google Scholar] [CrossRef]
- Hara, M.; Nishi, Y.; Yamashita, Y.; Hirata, R.; Takahashi, S.; Nagamitsu, S.; Hosoda, H.; Kangawa, K.; Kojima, M.; Matsuishi, T. Relation between Circulating Levels of GH, IGF-1, Ghrelin and Somatic Growth in Rett Syndrome. Brain Dev. 2014, 36, 794–800. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific Mutations in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Held, M.; Laccone, F.; Hanefeld, F. The Spectrum of Phenotypes in Females with Rett Syndrome. Brain Dev. 2003, 25, 346–351. [Google Scholar] [CrossRef]
- Oddy, W.H.; Webb, K.G.; Baikie, G.; Thompson, S.M.; Reilly, S.; Fyfe, S.D.; Young, D.; Anderson, A.M.; Leonard, H. Feeding Experiences and Growth Status in a Rett Syndrome Population. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 582–590. [Google Scholar] [CrossRef]
- Wong, L.C.; Chen, Y.; Tsai, S.; Lin, Y.; Hsu, C.; Wang, H.; Hu, S.; Shen, H.; Tsai, W.; Lee, W. Dietary Intake and Growth Deficits in Rett Syndrome—A Cross-section Study. Autism Res. 2021, 14, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Tarquinio, D.C. Growth Failure and Outcome in Rett Syndrome. Neurology 2012, 79, 1653–1661. [Google Scholar] [CrossRef]
- Motil, K.J.; Caeg, E.; Barrish, J.O.; Geerts, S.; Lane, J.B.; Percy, A.K.; Annese, F.; McNair, L.; Skinner, S.A.; Lee, H.-S.; et al. Gastrointestinal and Nutritional Problems Occur Frequently throughout Life in Girls and Women with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Blardi, P.; De Lalla, A.; D’Ambrogio, T.; Zappella, M.; Cevenini, G.; Ceccatelli, L.; Auteri, A.; Hayek, J. Rett Syndrome and Plasma Leptin Levels. J. Pediatr. 2007, 150, 37–39. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Licinio, J.; Tauchnitz, R.; Engelmann, L.; Negrão, A.; Gold, P.; Chrousos, G.P. Plasma Leptin Levels Are Increased in Survivors of Acute Sepsis: Associated Loss of Diurnal Rhythm in Cortisol and Leptin Secretion. J. Clin. Endocrinol. Metab. 1998, 83, 280–283. [Google Scholar] [CrossRef]
- Wang, X.; Lacza, Z.; Sun, Y.E.; Han, W. Leptin Resistance and Obesity in Mice with Deletion of Methyl-CpG-Binding Protein 2 (MeCP2) in Hypothalamic pro-Opiomelanocortin (POMC) Neurons. Diabetologia 2014, 57, 236–245. [Google Scholar] [CrossRef]
- Quint, E.H. Menstrual Issues in Adolescents with Physical and Developmental Disabilities. Ann. N. Y. Acad. Sci. 2008, 1135, 230–236. [Google Scholar] [CrossRef]
- Humphrey, K.N.; Horn, P.S.; Olshavsky, L.; Reebals, L.; Standridge, S.M. Features of Menstruation and Menstruation Management in Individuals with Rett Syndrome. J. Pediatr. Adolesc. Gynecol. 2021, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.I.; Matoso, E.; Pinto, M.; Almeida, J.; Liehr, T.; Melo, J.B.; Carreira, I.M. X-Chromosome Terminal Deletion in a Female with Premature Ovarian Failure: Haploinsufficiency of X-Linked Genes as a Possible Explanation. Mol. Cytogenet. 2010, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Canton, A.P.M.; Tinano, F.R.; Guasti, L.; Montenegro, L.R.; Ryan, F.; Shears, D.; De Melo, M.E.; Gomes, L.G.; Piana, M.P.; Brauner, R.; et al. Rare Variants in the MECP2 Gene in Girls with Central Precocious Puberty: A Translational Cohort Study. Lancet Diabetes Endocrinol. 2023, 11, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Gong, Y.; Liu, H.; Zhao, J.; Wang, J.; Lu, W. Expression and Distribution Pattern of DNMT1 and MeCP2 and Their Relationship with GnRH and Kisspeptin in the Hypothalamus during Puberty Onset in Ewes. IJAR 2021. [Google Scholar] [CrossRef]
- Garcia-Rudaz, C.; Deng, V.; Matagne, V.; Ronnekleiv, O.K.; Bosch, M.; Han, V.; Percy, A.K.; Ojeda, S.R. FXYD1, a Modulator of Na+, K+ -ATPase Activity, Facilitates Female Sexual Development by Maintaining Gonadotrophin-Releasing Hormone Neuronal Excitability. J. Neuroendocrinol. 2009, 21, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, M.; Yu, R.; Hu, R.; Xiong, F.; Li, J. A Case Report of Precocious Puberty Related to Rett Syndrome and a Literature Review. Pharmazie 2021, 75, 559–561. [Google Scholar] [CrossRef]
- Casto, C.; Pepe, G.; Li Pomi, A.; Corica, D.; Aversa, T.; Wasniewska, M. Hashimoto’s Thyroiditis and Graves’ Disease in Genetic Syndromes in Pediatric Age. Genes 2021, 12, 222. [Google Scholar] [CrossRef]
- Kron, M.; Howell, C.J.; Adams, I.T.; Ransbottom, M.; Christian, D.; Ogier, M.; Katz, D.M. Brain Activity Mapping in Mecp2 Mutant Mice Reveals Functional Deficits in Forebrain Circuits, Including Key Nodes in the Default Mode Network, That Are Reversed with Ketamine Treatment. J. Neurosci. 2012, 32, 13860–13872. [Google Scholar] [CrossRef]
- Bunker, S.K.; Dandapat, J.; Chainy, G.B.N.; Sahoo, S.K.; Nayak, P.K. Neonatal Exposure to 6-n-Propyl-Thiouracil, an Anti-Thyroid Drug, Alters Expression of Hepatic DNA Methyltransferases, Methyl CpG-Binding Proteins, Gadd45a, P53, and PCNA in Adult Male Rats. Eur. Thyroid. J. 2017, 6, 281–291. [Google Scholar] [CrossRef]
- Leonard, H.; Thomson, M.; Bower, C.; Fyfe, S.; Constantinou, J. Skeletal Abnormalities in Rett Syndrome: Increasing Evidence for Dysmorphogenetic Defects. Am. J. Med. Genet. 1995, 58, 282–285. [Google Scholar] [CrossRef]
- Motil, K.J.; Barrish, J.O.; Lane, J.; Geerts, S.P.; Annese, F.; McNair, L.; Percy, A.K.; Skinner, S.A.; Neul, J.L.; Glaze, D.G. Vitamin D Deficiency Is Prevalent in Girls and Women with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 569–574. [Google Scholar] [CrossRef]
- Jefferson, A.L.; Woodhead, H.J.; Fyfe, S.; Briody, J.; Bebbington, A.; Strauss, B.J.; Jacoby, P.; Leonard, H. Bone Mineral Content and Density in Rett Syndrome and Their Contributing Factors. Pediatr. Res. 2011, 69, 293–298. [Google Scholar] [CrossRef]
- O’Connor, R.D.; Zayzafoon, M.; Farach-Carson, M.C.; Schanen, N.C. Mecp2 Deficiency Decreases Bone Formation and Reduces Bone Volume in a Rodent Model of Rett Syndrome. Bone 2009, 45, 346–356. [Google Scholar] [CrossRef]
- Metwalley, K.A.; Farghaly, H.S. Endocrinal Dysfunction in Children with Down Syndrome. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Levy-Shraga, Y.; Gothelf, D.; Pinchevski-Kadir, S.; Katz, U.; Modan-Moses, D. Endocrine Manifestations in Children with Williams–Beuren Syndrome. Acta Paediatr. 2018, 107, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Heksch, R.; Kamboj, M.; Anglin, K.; Obrynba, K. Review of Prader-Willi Syndrome: The Endocrine Approach. Transl. Pediatr. 2017, 6, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Artuso, R.; Mencarelli, M.A.; Polli, R.; Sartori, S.; Ariani, F.; Pollazzon, M.; Marozza, A.; Cilio, M.R.; Specchio, N.; Vigevano, F.; et al. Early-Onset Seizure Variant of Rett Syndrome: Definition of the Clinical Diagnostic Criteria. Brain Dev. 2010, 32, 17–24. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Organ/System | Disorders/Abnormalities |
|---|---|
| Respiratory system | Breathing arrhythmias (apneas) Breath-holding |
| Cardiovascular system | Rythm defects QT prolonged interval No structural changes |
| Digestive system | Gastrointestinal dysmotility (Dysphagia) Altered microbiota |
| Metabolic system | Severe dyslipidemia Fatty liver disease Metabolic syndrome Insulin resistance Alters energy homeostasis |
| Skeletal system | Scoliosis Fractures |
| Endocrine system | Low-bone mineral content Delayed menarche Thyroid hormones |
| Muscular system | Mild hypotonia Muscle atrophy |
| Urinary system | Urinary tract infection Kidney stones Urine retention |
| Patients (n) | 51 |
| Males, n (%) | 4 (7.8%) |
| Females, n (%) | 47 (92.2%) |
| Age at visit (mean, SDS) | 9.65 ± 5.9 |
| Age at diagnosis (mean, SDS) | 3.22 ± 2.4 |
| Height (mean, SDS) | −1.76 ± 1.84 |
| BMI (mean, SDS) | −0.77 ± 2.53 |
| Prepubertal, n (%) | 25 (49%) |
| Pubertal, n (%) | 26 (51%) |
| Genotype | |
| MeCP2 | 38 (74.5%) |
| CDKL5 | 11 (21.6%) |
| FOXG1 | 2 (3.9%) |
| Epilepsy | 37 (72.5%) |
| Drug-resistant epilepsy | 10 (27%) |
| Epileptic encephalopathy | 8 (21.6%) |
| Patient n. | Gene | Mutation/Deletion | Endocrine Disease |
|---|---|---|---|
| 1 | MeCP2 | P322A | Obesity, amenorrhea (POF), osteopenia, vitamin D deficiency |
| 2 | MeCP2 | P322S | None |
| 3 | MeCP2 | R133C | Short stature, obesity, amenorrhea (POF), thyroid nodules, hyperprolactinemia, osteopenia |
| 4 | MeCP2 | R133C | Obesity |
| 5 | MeCP2 | R168X | Short stature, obesity, menstrual irregularities, central hypothyroidism, hyperprolactinemia, osteoporosis |
| 6 | MeCP2 | R168X | Short stature, precocious puberty |
| 7 | MeCP2 | R255X | Short stature, malnutrition |
| 8 | MeCP2 | R255X | Short stature, malnutrition, menstrual irregularities, hyperprolactinemia, central hypothyroidism, thyroid nodules |
| 9 | MeCP2 | R255X | Short stature, precocious puberty |
| 10 | MeCP2 | R270X | Short stature, osteoporosis, vitamin D deficiency |
| 11 | MeCP2 | R270X | Short stature, malnutrition, menstrual irregularities |
| 12 | MeCP2 | R270X | Premature pubarche, vitamin D deficiency |
| 13 | MeCP2 | R294X | vitamin D deficiency |
| 14 | MeCP2 | R294X | Short stature, vitamin D deficiency |
| 15 | MeCP2 | R294X | Malnutrition, osteopenia |
| 16 | MeCP2 | R294X | Malnutrition |
| 17 | MeCP2 | R306C | Short stature, obesity, menstrual irregularities, Hashimoto’s thyroiditis, hyperprolactinemia |
| 18 | MeCP2 | R306C | Short stature, malnutrition, menstrual irregularities, hyperprolactinemia |
| 19 | MeCP2 | R306C | Short stature, hyperprolactinemia |
| 20 | MeCP2 | R306X | Short stature, precocious puberty |
| 21 | MeCP2 | S134C | Short stature, obesity, menstrual irregularities, subclinical hypothyroidism, vitamin D deficiency |
| 22 | MeCP2 | T158M | Short stature, overweight, osteopenia |
| 23 | MeCP2 | T158M | Short stature, malnutrition, menstrual irregularities, osteoporosis, vitamin D deficiency |
| 24 | MeCP2 | T158M | Malnutrition |
| 25 | MeCP2 | T158P | Osteoporosis |
| 26 | MeCP2 | T158M | Short stature, malnutrition, menstrual irregularities, central hypothyroidism, vitamin D deficiency |
| 27 | MeCP2 | c.749_752dup(p.Gly252Profs * 8) | Obesity |
| 28 | MeCP2 | c.763C>T | Short stature, osteoporosis |
| 29 | MeCP2 | c.880C>T | Short stature, osteoporosis |
| 30 | MeCP2 | c.880>T | Short stature, malnutrition |
| 31 | MeCP2 | c.880>T p, (Arg294 *) | Short stature |
| 32 | MeCP2 | c.316C>T (p.Arg106Trp) | Short stature, vitamin D deficiency |
| 33 | MeCP2 | c.915G>T (p.Lys305Asn) | Obesity, menstrual irregularities |
| 34 | MeCP2 | c.917G>A | Obesity |
| 35 | MECP2 | c.1061_1062del p.(Arg354Glnfs * 38) | Short stature, malnutrition |
| 36 | MeCP2 | 1097 delinsCC; 1130_1190delinG * | Short stature |
| 37 | MeCP2 | c.1164_1207del [p.(Pro389 *) | Obesity, menstrual irregularities, hyperprolactinemia, vitamin D deficiency |
| 38 | MeCP2 | c.1164_1207del [p.(Pro389 *) | None |
| 39 | CDKL5 | whole gene del | None |
| 40 | CDKL5 | whole gene del | Precocious puberty |
| 41 | CDKL5 | c.119 C>T (p.Ala40Val) | None |
| 42 | CDKL5 | c.119 C>T (p.Ala40Val) | Malnutrition, vitamin D deficiency |
| 43 | CDKL5 | c.380A>G [p.his127Arg] | None |
| 44 | CDKL5 | c628G>A p (Gly228Arg) | None |
| 45 | CDKL5 | Exon 1 | None |
| 46 | CDKL5 | c.855A>T; p Arg 285 ser | None |
| 47 | CDKL5 | c.1648C>T p.(Arg550 *) | None |
| 48 | CDKL5 | c.2217dup p.(Pro740Thrfs * 24) | None |
| 49 | CDKL5 | c.2713+19G>A e c.2732G>A | None |
| 50 | FOXG1 | C.256delC | None |
| 51 | FOXG1 | c.681C>G | Malnutrition |
| Endocrinological Evaluation | Infancy | Childhood-Adolescence | Adulthood |
|---|---|---|---|
| Growth assessment | |||
| Height | √ | √ | NA |
| Growth velocity | √ | √ | NA |
| Weight, BMI | √ | √ | √ |
| IGF-1 and GH secretion | in case of short stature and/or growth deceleration | in case of short stature and/or growth deceleration | NA |
| Bone age | √ | √ | NA |
| Thyroid function | |||
| FT4, TSH, TPO-AB, TG-AB | √ | √ | √ |
| Thyroid ultrasound | if thyroid alterations, goitre or family history | if thyroid alterations, goitre or family history | if thyroid alterations, goitre or family history |
| Metabolic assessment | |||
| Blood pressure | √ | √ | √ |
| Waist circumference | if BMI > 2 SDS | if BMI > 2 SDS | if BMI > 2 SDS |
| Blood glucose, Hba1c | √ | √ | √ |
| Insulin | if BMI > 2 SDS | if BMI > 2 SDS | if BMI > 2 SDS |
| Lipid profile | √ | √ | √ |
| Gonadal function | |||
| Gonadotropins | NA | in case of pubertal alterations or menstrual irregularities | in case of menstrual irregularities |
| Sex steroids | NA | in case of pubertal alterations or menstrual irregularities | in case of menstrual irregularities |
| Bone health | |||
| Calcium-phosphorus metabolism | √ | √ | √ |
| DEXA | NA | √ | √ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepe, G.; Coco, R.; Corica, D.; Di Rosa, G.; Bossowski, F.; Skorupska, M.; Aversa, T.; Stagi, S.; Wasniewska, M. Prevalence of Endocrinopathies in a Cohort of Patients with Rett Syndrome: A Two-Center Observational Study. Genes 2024, 15, 287. https://doi.org/10.3390/genes15030287
Pepe G, Coco R, Corica D, Di Rosa G, Bossowski F, Skorupska M, Aversa T, Stagi S, Wasniewska M. Prevalence of Endocrinopathies in a Cohort of Patients with Rett Syndrome: A Two-Center Observational Study. Genes. 2024; 15(3):287. https://doi.org/10.3390/genes15030287
Chicago/Turabian StylePepe, Giorgia, Roberto Coco, Domenico Corica, Gabriella Di Rosa, Filip Bossowski, Magdalena Skorupska, Tommaso Aversa, Stefano Stagi, and Malgorzata Wasniewska. 2024. "Prevalence of Endocrinopathies in a Cohort of Patients with Rett Syndrome: A Two-Center Observational Study" Genes 15, no. 3: 287. https://doi.org/10.3390/genes15030287
APA StylePepe, G., Coco, R., Corica, D., Di Rosa, G., Bossowski, F., Skorupska, M., Aversa, T., Stagi, S., & Wasniewska, M. (2024). Prevalence of Endocrinopathies in a Cohort of Patients with Rett Syndrome: A Two-Center Observational Study. Genes, 15(3), 287. https://doi.org/10.3390/genes15030287