
Identification of Superior Haplotypes and Haplotype Combinations for Grain Size- and Weight-Related Genes for Breeding Applications in Rice (Oryza sativa L.)
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Field Experiment and Agronomic Trait Investigation
2.3. DNA Extraction and Library Construction
2.4. Probe Design and Targeted Enrichment Assay
2.5. Reference Genome Alignment and Variation Analysis
2.6. Phylogenetic and Population Genetics Analyses
2.7. Candidate Gene-Based Association Analysis
2.8. Identification of Superior Haplotypes and Haplotype Combinations for the Associated Genes
3. Results
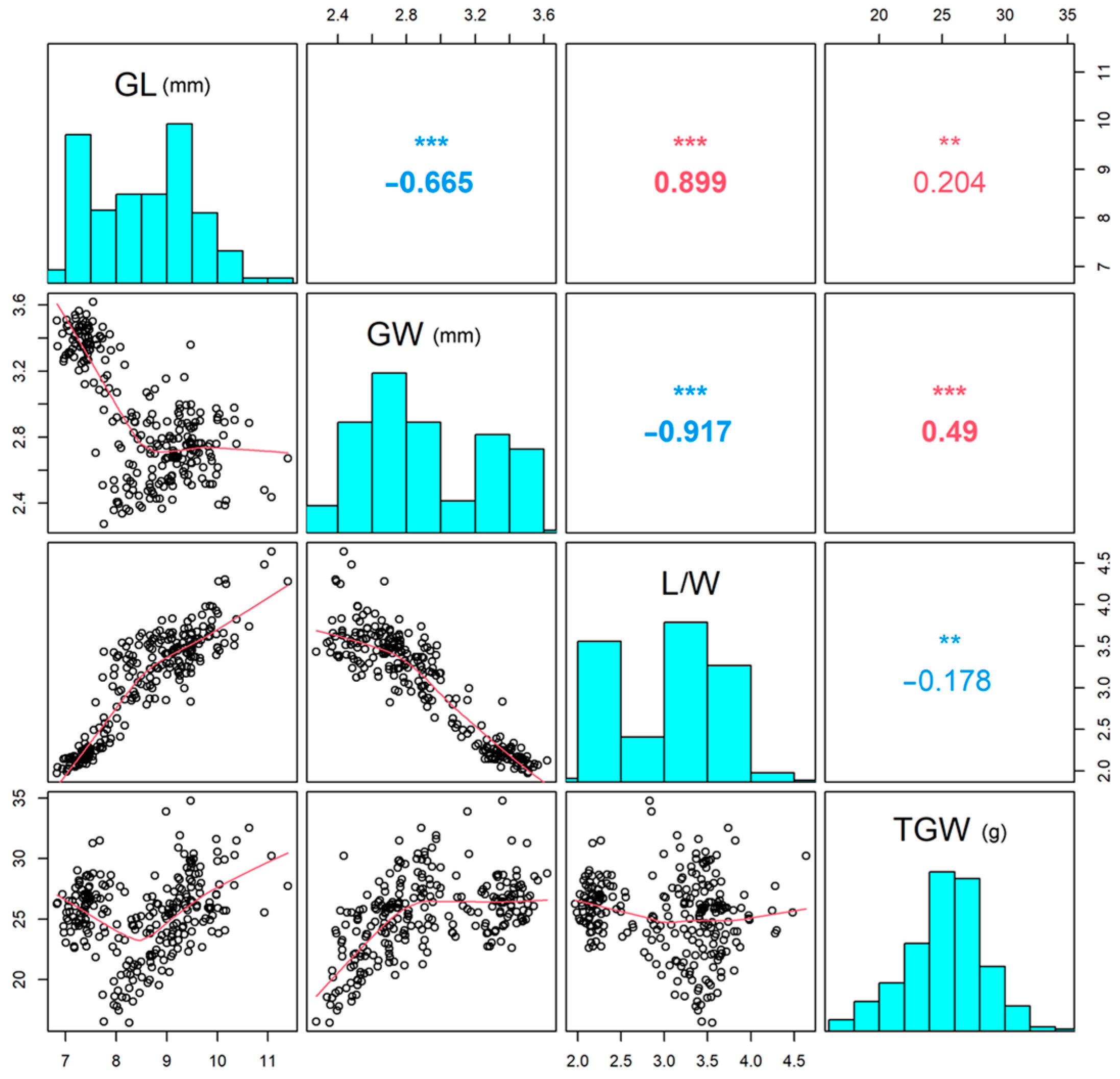
3.1. Phenotypic Variation Analysis
3.2. Targeted Enrichment Analysis and Sequence Variation of the 42 Genes in the Population
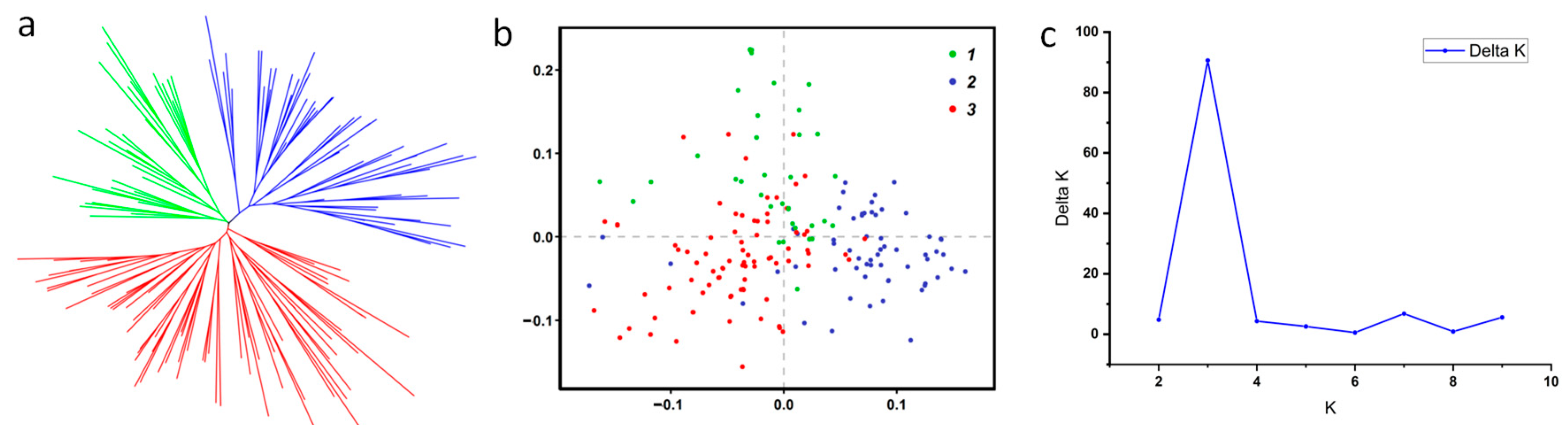
3.3. Population Structure
3.4. Association Analysis
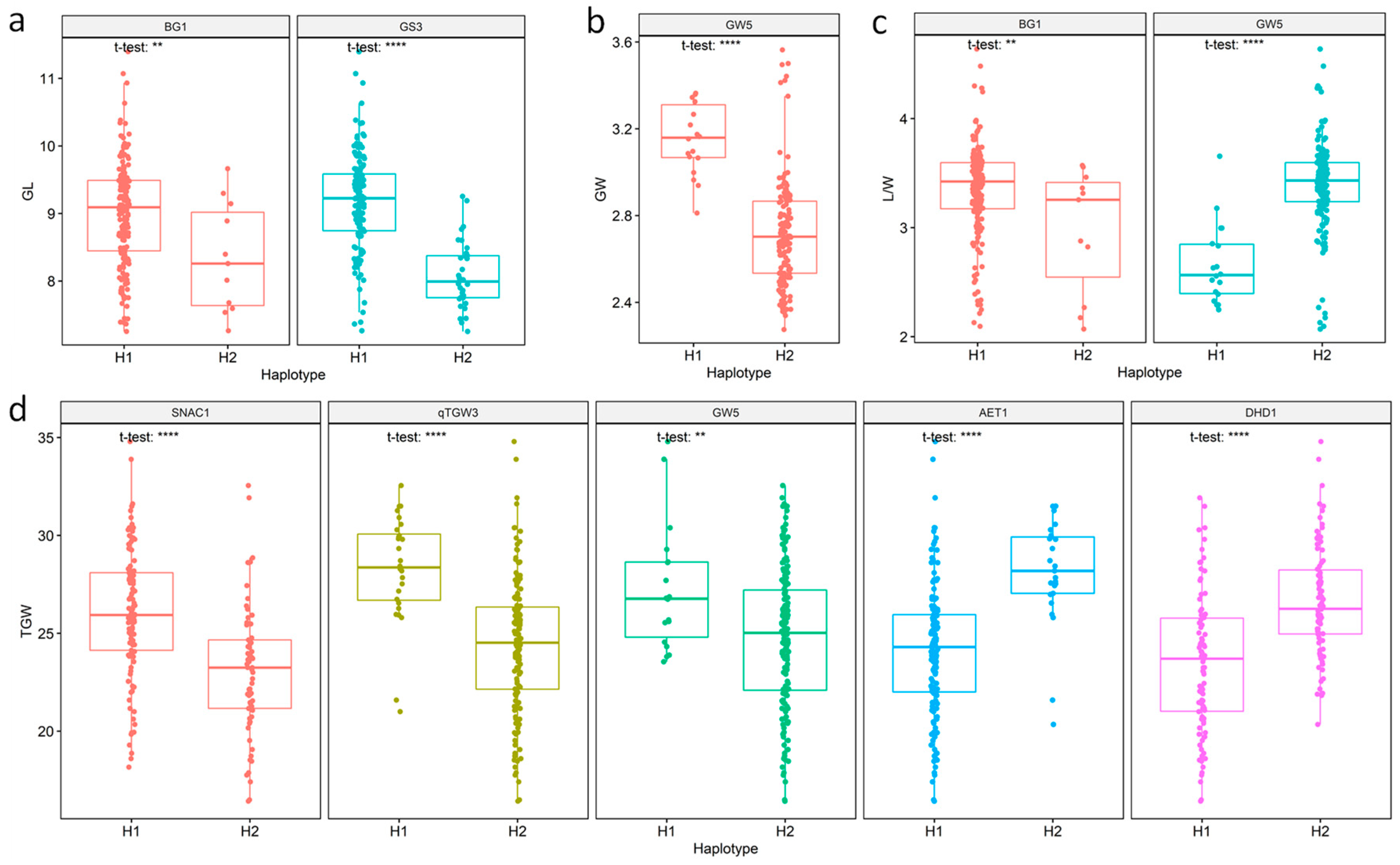
3.5. Haplotype Analysis and Identification of Superior Haplotypes
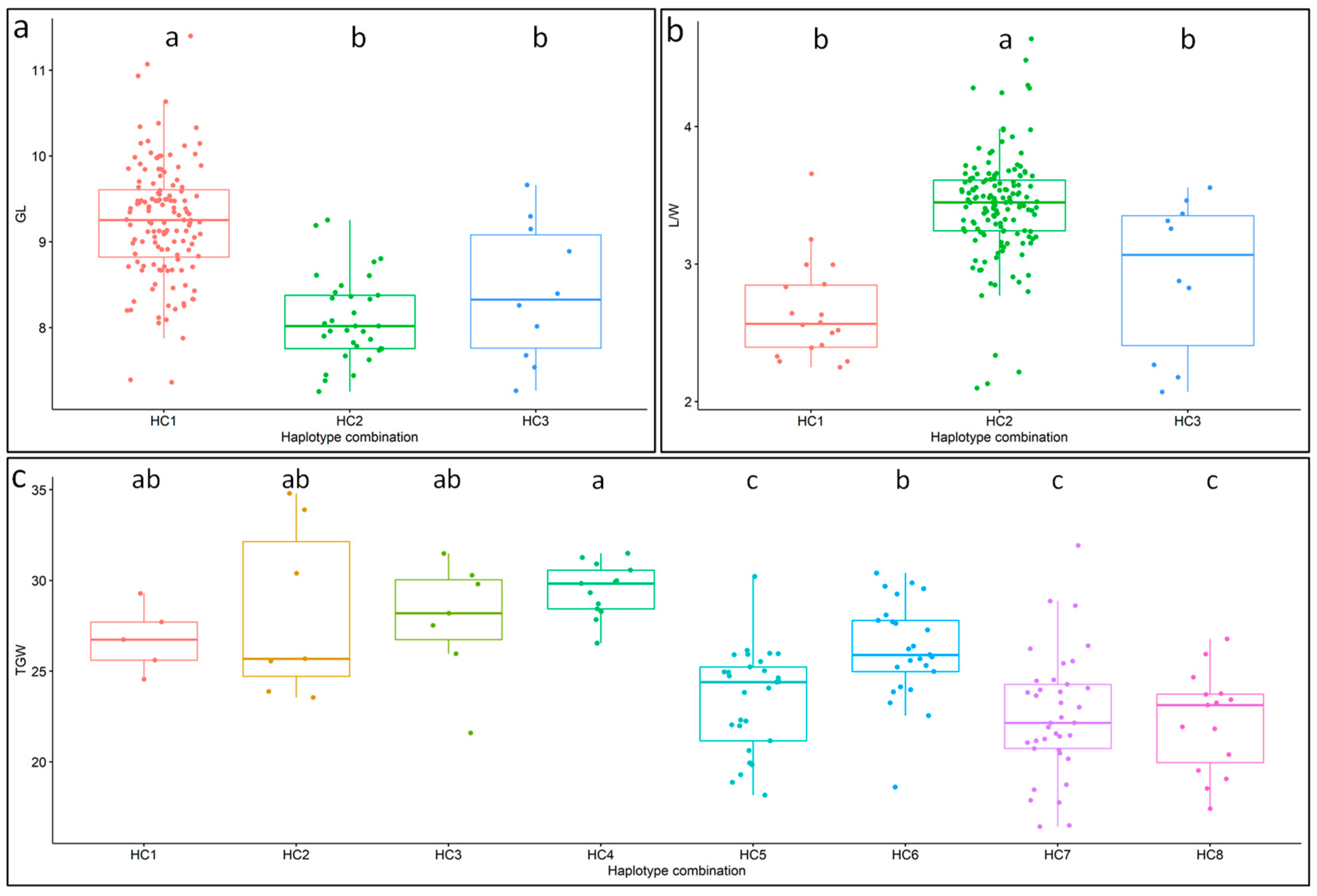
3.6. Identification of Superior Haplotype Combinations
3.7. Tracing of the Superior Haplotype Combination forThousand-Grain Weight in Past Breeding Practices
4. Discussion
4.1. The Balance of Grain Length and Grain Weight Is Important for Rice Breeding
4.2. Candidate Gene-Based Association Analysis Is Useful for Identifying New Functions and Favorable Alleles of Old Genes
4.3. Selection during Breeding May Lead to an Imbalanced Distribution of Different Haplotypes and Haplotype Combinations
4.4. Haplotype Combination 4 forThousand-Grain Weight Can Be Used to Improve Tgw by Mas
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xing, Y.; Zhang, Q. Genetic and molecular bases of rice yield. Annu. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.A.; McCouch, S.R.; Hall, R.D. Not just a grain of rice: The quest for quality. Trends Plant Sci. 2009, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Takano-Kai, N.; Hui, J.; Kubo, T.; Sweeney, M.; Matsumoto, T.; Kanamori, H.; Padhukasahasram, B.; Bustamante, C.; Yoshimura, A.; Doi, K.; et al. Evolutionary history of GS3, a gene conferring grain length in rice. Genetics 2009, 182, 1323–1334. [Google Scholar] [CrossRef]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef]
- Mao, H.; Sun, S.; Yao, J.; Wang, C.; Yu, S.; Xu, C.; Li, X.; Zhang, Q. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 19579–19584. [Google Scholar] [CrossRef] [PubMed]
- Shomura, A.; Izawa, T.; Ebana, K.; Ebitani, T.; Kanegae, H.; Konishi, S.; Yano, M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 2008, 40, 1023–1028. [Google Scholar] [CrossRef]
- Weng, J.; Gu, S.; Wan, X.; Gao, H.; Guo, T.; Su, N.; Lei, C.; Zhang, X.; Cheng, Z.; Guo, X.; et al. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008, 18, 1199–1209. [Google Scholar] [CrossRef]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J.; et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef]
- Liu, L.; Tong, H.; Xiao, Y.; Che, R.; Xu, F.; Hu, B.; Liang, C.; Chu, J.; Li, J.; Chu, C. Activation of Big Grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 11102–11107. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.Z.; Ma, M.; Bai, C.; Huang, X.H.; Liu, J.L.; Fan, Y.Y.; Song, X.J. TGW3, a Major QTL that Negatively Modulates Grain Length and Weight in Rice. Mol. Plant 2018, 11, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Zhou, H.; Liu, R.; Dan, W.; Li, P.; Wu, B.; Chen, J.; Wang, L.; Gao, G.; Zhang, Q.; et al. GL3.3, a novel QTL encoding a GSK3/SHAGGY-like kinase, epistatically interacts with GS3 to produce extra-long grains in rice. Mol. Plant 2018, 11, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lu, S.J.; Wang, M.J.; He, H.; Sun, L.; Wang, H.; Liu, X.H.; Jiang, L.; Sun, J.L.; Xin, X.; et al. A novel QTL qTGW3 encodes the GSK3/SHAGGY-Like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol. Plant 2018, 11, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Ren, Y.; Liu, L.; Wang, F.; Zhang, H.; Tian, P.; Pan, T.; Wang, Y.; Jing, R.; Liu, T.; et al. Ubiquitin specific protease 15 has an important role in regulating grain width and size in rice. Plant Physiol. 2019, 180, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genomics-assisted breeding for crop improvement. Trends Plant Sci. 2005, 10, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196. [Google Scholar] [CrossRef]
- Bhat, J.A.; Yu, D.; Bohra, A.; Ganie, S.A.; Varshney, R.K. Features and applications of haplotypes in crop breeding. Commun. Biol. 2021, 4, 1266. [Google Scholar] [CrossRef]
- Bevan, M.W.; Uauy, C.; Wulff, B.B.H.; Zhou, J.; Krasileva, K.; Clark, M.D. Genomic innovation for crop improvement. Nature 2017, 543, 346–354. [Google Scholar] [CrossRef]
- Sinha, P.; Singh, V.K.; Saxena, R.K.; Khan, A.W.; Abbai, R.; Chitikineni, A.; Desai, A.; Molla, J.; Upadhyaya, H.D.; Kumar, A.; et al. Superior haplotypes for haplotype-based breeding for drought tolerance in pigeonpea (Cajanus cajan L.). Plant Biotechnol. J. 2020, 18, 2482–2490. [Google Scholar] [CrossRef]
- Selvaraj, R.; Singh, A.K.; Singh, V.K.; Abbai, R.; Habde, S.V.; Singh, U.M.; Kumar, A. Superior haplotypes towards development of low glycemic index rice with preferred grain and cooking quality. Sci. Rep. 2021, 11, 10082. [Google Scholar] [CrossRef]
- Liu, J.; Zhan, J.; Chen, J.; Lu, X.; Zhi, S.; Ye, G. Validation of genes affecting rice grain zinc content through candidate gene-based association analysis. Front. Genet. 2021, 12, 701658. [Google Scholar] [CrossRef]
- Abbai, R.; Singh, V.K.; Nachimuthu, V.V.; Sinha, P.; Selvaraj, R.; Vipparla, A.K.; Singh, A.K.; Singh, U.M.; Varshney, R.K.; Kumar, A. Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol. J. 2019, 17, 1612–1622. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 2, 359–361. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. FastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Lu, J.; Hou, J.; Ouyang, Y.; Luo, H.; Zhao, J.; Mao, C.; Han, M.; Wang, L.; Xiao, J.; Yang, Y.; et al. A direct PCR–based SNP marker–assisted selection system (D-MAS) for different crops. Mol. Breed. 2020, 40, 9. [Google Scholar] [CrossRef]
- Liu, J.; Chen, J.; Zheng, X.; Wu, F.; Lin, Q.; Heng, Y.; Tian, P.; Cheng, Z.J.; Yu, X.; Zhou, K.; et al. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 2017, 3, 17043. [Google Scholar] [CrossRef]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef]
- Redillas, M.C.F.R.; Jeong, J.S.; Kim, Y.S.; Jung, H.; Bang, S.W.; Choi, Y.D.; Ha, S.H.; Reuzeau, C.; Kim, J.K. The overexpression of OsNAC9 alters the root architecture of rice plants enhancing drought resistance and grain yield under field conditions. Plant Biotechnol. J. 2012, 10, 792–805. [Google Scholar] [CrossRef]
- Chen, K.; Guo, T.; Li, X.M.; Zhang, Y.M.; Yang, Y.B.; Ye, W.W.; Dong, N.Q.; Shi, C.L.; Kan, Y.; Xiang, Y.H.; et al. Translational regulation of plant response to high temperature by a dual-function tRNAHis guanylyltransferase in rice. Mol. Plant 2019, 12, 1123–1142. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, S.; Liu, T.; Wang, C.; Cheng, Z.; Zhang, X.; Chen, L.; Sheng, P.; Cai, M.; Li, C.; et al. DELAYED HEADING DATE1 interacts with OsHAP5C/D, delays flowering time and enhances yield in rice. Plant Biotechnol. J. 2019, 17, 531–539. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Gene | Locus ID | Haplotype | Percent of Accessions (%) | Mean | R2 (%) | p Value |
|---|---|---|---|---|---|---|---|
| GL | BG1 | LOC_Os03g07920 | H1 | 95.00 | 9.01 | 3.24 | 1.62 × 10−2 |
| (mm) | H2 | 5.00 | 8.34 | ||||
| GS3 | Os03g0407400 | H1 | 81.11 | 9.19 | 27.66 | 4.04 × 10−12 | |
| H2 | 18.89 | 8.08 | |||||
| GW | GW5 | LOC_Os05g09520 | H1 | 10.00 | 3.15 | 25.19 | 4.90 × 10−11 |
| (mm) | H2 | 88.33 | 2.72 | ||||
| L/W | BG1 | LOC_Os03g07920 | H1 | 95.00 | 3.35 | 3.21 | 1.66 × 10−2 |
| H2 | 5.00 | 2.98 | |||||
| GW5 | LOC_Os05g09520 | H1 | 10.00 | 2.66 | 21.1 | 2.58 × 10−9 | |
| H2 | 88.33 | 3.39 | |||||
| TGW | SNAC1 | LOC_Os03g60080 | H1 | 60.56 | 26.05 | 14.78 | 8.36 × 10−7 |
| (g) | H2 | 37.78 | 23.09 | ||||
| qTGW3 | LOC_Os03g62500 | H1 | 15.56 | 28.28 | 13.48 | 2.63 × 10−6 | |
| H2 | 83.33 | 24.39 | |||||
| GW5 | LOC_Os05g09520 | H1 | 10.00 | 27.26 | 5.88 | 1.73 × 10−3 | |
| H2 | 88.33 | 24.78 | |||||
| AET1 | LOC_Os05g45890 | H1 | 78.89 | 24.17 | 15.01 | 6.83 × 10−7 | |
| H2 | 20.00 | 28.06 | |||||
| DHD1 | LOC_Os11g47920 | H1 | 51.11 | 23.45 | 10.78 | 2.73 × 10−5 | |
| H2 | 46.67 | 26.65 |
| Trait | Haplotype Combination | Single-Gene Haplotype | Percentage (%) | Mean | R2 (%) | p Value |
|---|---|---|---|---|---|---|
| GL | HC1 | BG1-H1, GS3-H1 | 75.56 | 9.24 | 25.38 | 3.96 × 10−13 |
| (mm) | HC2 | BG1-H1, GS3-H2 | 18.33 | 8.09 | ||
| HC3 | BG1-H2, GS3-H1 | 5.56 | 8.41 | |||
| L/W | HC1 | BG1-H1, GW5-H1 | 10 | 2.66 | 5.5 | 9.79 × 10−4 |
| HC2 | BG1-H1, GW5-H2 | 82.78 | 3.42 | |||
| HC3 | BG1-H2, GW5-H2 | 5.56 | 2.91 | |||
| TGW (g) | HC1 | SNAC1-H1, qTGW3-H2, GW5-H1, AET1-H1, DHD1-H1 | 2.78 | 26.78 | 22.49 | 2.53 × 10−9 |
| HC2 | SNAC1-H1, qTGW3-H2, GW5-H1, AET1-H1, DHD1-H2 | 3.89 | 28.25 | |||
| HC3 | SNAC1-H1, qTGW3-H1, GW5-H2, AET1-H2, DHD1-H1 | 3.33 | 27.84 | |||
| HC4 | SNAC1-H1, qTGW3-H1, GW5-H2, AET1-H2, DHD1-H2 | 6.67 | 29.47 | |||
| HC5 | SNAC1-H1, qTGW3-H2, GW5-H2, AET1-H1, DHD1-H1 | 16.11 | 23.39 | |||
| HC6 | SNAC1-H1, qTGW3-H2, GW5-H2, AET1-H1, DHD1-H2 | 13.89 | 26.19 | |||
| HC7 | SNAC1-H2, qTGW3-H2, GW5-H2, AET1-H1, DHD1-H1 | 20.56 | 22.6 | |||
| HC8 | SNAC1-H2, qTGW3-H2, GW5-H2, AET1-H1, DHD1-H2 | 7.78 | 22.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Qiu, D.; Lu, Y.; Wu, Y.; Han, X.; Jiao, Y.; Wang, T.; Yang, J.; You, A.; Chen, J.; et al. Identification of Superior Haplotypes and Haplotype Combinations for Grain Size- and Weight-Related Genes for Breeding Applications in Rice (Oryza sativa L.). Genes 2023, 14, 2201. https://doi.org/10.3390/genes14122201
Liu G, Qiu D, Lu Y, Wu Y, Han X, Jiao Y, Wang T, Yang J, You A, Chen J, et al. Identification of Superior Haplotypes and Haplotype Combinations for Grain Size- and Weight-Related Genes for Breeding Applications in Rice (Oryza sativa L.). Genes. 2023; 14(12):2201. https://doi.org/10.3390/genes14122201
Chicago/Turabian StyleLiu, Gang, Dongfeng Qiu, Yuxia Lu, Yan Wu, Xuesong Han, Yaru Jiao, Tingbao Wang, Jinsong Yang, Aiqing You, Jianguo Chen, and et al. 2023. "Identification of Superior Haplotypes and Haplotype Combinations for Grain Size- and Weight-Related Genes for Breeding Applications in Rice (Oryza sativa L.)" Genes 14, no. 12: 2201. https://doi.org/10.3390/genes14122201
APA StyleLiu, G., Qiu, D., Lu, Y., Wu, Y., Han, X., Jiao, Y., Wang, T., Yang, J., You, A., Chen, J., & Zhang, Z. (2023). Identification of Superior Haplotypes and Haplotype Combinations for Grain Size- and Weight-Related Genes for Breeding Applications in Rice (Oryza sativa L.). Genes, 14(12), 2201. https://doi.org/10.3390/genes14122201