Abstract
Reynoutria japonica Houtt., a traditional medicine herb of the Polygonaceae family, has been used since ancient times in China due to its various pharmacological effects. Chloroplast genomes are conservative and play an essential role in population diversity analysis. However, there are few studies on the chloroplast genome of R. japonica. In this study, the complete chloroplast genomes of three R. japonica from different regions were performed by next-generation sequencing technology. The results revealed that the lengths of the three chloroplast genomes are between 163,371~163,372 bp, and they have a highly conserved structure with a pair of inverted repeat (IR) regions (31,121 bp), a large single-copy (LSC) region (87,571~87,572 bp), and a small single-copy (SSC) region (13,558 bp). In total, 132 genes were annotated, including 8 rRNA genes, 37 tRNA genes, and 87 protein-coding genes. The phylogenetic analysis strongly revealed that 13 populations of R. japonica form a monophyly, and Fallopia multiflora (Polygonaceae) is its closest species. The two species diverged at ~20.47 million years ago, and R. japonica in China could be further divided into two major groups based on genetic structure analysis. In addition, several potential loci with suitable polymorphism were identified as molecular markers. Our study provides important genetic resources for further development and utilization of R. japonica germplasm, as well as some new insights into the evolutionary characteristics of this medicinal plant.
1. Introduction
R. japonica Houtt., also known as Polygonum cuspidatum Sieb. et Zucc., Fallopia japonica (Japanese knotweed), and HuZhang, is a perennial herb in the Polygonaceae family and has been used as a traditional Chinese medicine for thousands of years [1]. R. japonica is rich in stilbenes, flavonoids, and anthraquinones [2], which play important roles in human health, functioning in anti-inflammation, anti-infection, and anticancer [1,3,4], and has been listed in the Pharmacopoeia of the People’s Republic of China since 1977. At present, it is in the recommendation of “Diagnosis and Treatment Protocol for COVID-19 (Trial Version 7).” As a medicinal plant with fast growth, easy adaptation, and high tolerance [5,6], R. japonica is considered an invasive species in Europe, causing enormous economic losses [7]. Meanwhile, this medicinal/invasive plant also has the ability to accumulate high levels of heavy metals [8]. The allelopathy effect of R. japonica may help it to rapidly establish an environment conducive to its own growth in new habitats and to interrupt the regeneration process in native plant species [9]. Moreover, the places with R. japonica usually form monospecific stands [10]. Hence, it is desirable to conduct a molecular-level analysis of the genetic diversity and variation range of R. japonica populations.
Chloroplast is a critical site for photosynthesis and has its own DNA and genetic system [11]. The chloroplast genomes are ubiquitous in plants and possess a typical quadripartite structure [12], including a large single-copy (LSC) and a small single-copy (SSC), as well as a pair of inverted repeats (IRs), more conserved than nuclear and mitochondrial genomes [13]. In addition, chloroplast genomes are small and, with a length of 120 kb to 160 kb [14], are easy to assemble and are of diverse value such as for elucidating phylogenetic relationships, resolving seemingly intractable problems in traditional taxonomy [15], clarifying interspecific and intraspecific variation [16], and identifying species [17]. With the advances in next-generation sequencing technology, more and more species of chloroplast genomes in the Polygonaceae family have been obtained, including Fagopyrum esculentum, Fagopyrum tataricum, Fagopyrum dibotrys [13,18,19,20], F. multiflora [21], and other species sequences published in public databases. In Flora of China, R. japonica was separated from Polygonum and considered to belong to a new genus, Reynoutria Houtt. Several studies using chloroplast genomes revealed that R. japonica and F. multiflora had a close genetic relationship [22,23]. However, the intraspecific and population structure among R. japonica has not yet been clarified till now. As a result, it is of great interest to provide its genomic data in terms of intraspecific and population structure of R. japonica.
Currently, most research into this herbal medicine has focused on pharmacological activities [24], biosynthesis, and regulation of active compounds [25,26,27]. Few studies are available to systematically compare the characteristics and phylogenetic relationships of R. japonica across different regions of China based on chloroplast genomes [23]. In this study, chloroplast genomes of R. japonica from three different Chinese regions were sequenced by us, and a total of 13 chloroplast genomes of R. japonica were obtained, together with other chloroplast genomes published in public databases. Then, a series of bioinformatics analyses was carried out, including identifying molecular markers that could be used to discriminate between different germplasm populations of R. japonica and exploring the evolutionary time and geographical distribution characteristics of R. japonica in China. The chloroplast genome resources assembled in this study could also serve as some material foundation for the further development and utilization of R. japonica germplasm.
2. Materials and Methods
2.1. Materials and DNA Extraction
The seeds of R. japonica were collected from Lichuan City (Rj. LiChuan), Hubei Province of China; Shandong Province of China (Rj. ShanDong); and Beijing (Rj. BeiJing), China. All sample seedings were propagated in plastic pots containing a soil/vermiculite mixture (1:1) and the growth environments, as described in a previous study [25]. The fresh leaves of R. japonica were quickly frozen with liquid nitrogen immediately after picking and cleaning. Total whole genomic DNA was extracted with Super Plant Genomic DNA Kit (polysaccharide- and polyphenolics-rich) (Tiangen, Beijing, China), and purity and integrity of the extracted total genomic DNA were detected by 1% agarose gel electrophoresis, and the total concentration was estimated by Qubit DNA Assay Kit in Qubit 3.0 Fluorometer (Invitrogen, Waltham, MA, USA). The qualified samples were selected for subsequent experiments.
2.2. Chloroplast Genome Assembly and Annotation
The raw sequencing reads were obtained by the company (Novogene, Beijing, China). A total amount of 0.2 μg DNA per sample was used as input material for the DNA library preparations. Sequencing library was generated using NEB Next Ultra DNA Library Prep Kit for Illumina (San Diego, CA, USA) following manufacturer’s recommendations, and index codes were added to each sample. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using Illumina PE Cluster Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. After cluster generation, the DNA libraries were sequenced on Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA), and 150 bp paired-end reads were generated. The raw data contained adapter contamination, low-quality nucleotides, and unrecognizable nucleotide (N). We used fastp v0.19.7 [28] to obtain high-quality reads. The specific process was as follows: (1) Discard a pair of reads if either one read contains adapter contamination. (2) Discard a pair of reads if more than 10% of bases are uncertain in either one read. (3) Discard a pair of reads if the proportion of low-quality (Phred quality < 5) bases is over 50% in either one read. Then, the high-quality reads were used to assemble the whole chloroplast genome with GetOrganelle [29]. WTDBG [30] was used to polish the chloroplast genome with clean reads. BWA v0.7.17-r1188 [31] was used to map the clean data to assemble chloroplast genome, and samtools v1.9 [32] was used to calculate the sequencing depth of each locus. The complete chloroplast genome sequences of three R. japonica were annotated via GeSeq [33]. All three annotation results were manually curated. Finally, the resulting plastid genome maps were drawn with OGDRAW [34].
2.3. Chloroplast Genomes Feature Analysis
We compared the boundaries of the LSC, IR, and SSC regions of R. japonica chloroplast genomes by IRscope [35]. All the shared genes of R. japonica were extracted and aligned with MAFFT v7.503 [36], including 75 protein-coding genes, 4 rRNA, and 28 tRNA. Then, the alignments of each gene were used to evaluate nucleotide diversity (Pi) using DnaSP v6.0 [37]. The mVISTA program [38] in Shuffle-LAGAN mode [39] was used to analyze the intraspecific variation with Rj. BeiJing acting as the reference genome. For the protein-coding genes codon usage, the relative synonymous codon usage (RSCU), codon adaption index (CAI), codon bias index (CBI), frequency of optimal codons (FOP), GC contents of silent third codon position (GC3s), and effective codon number (ENC) were determined by CodonW v1.4.4 (available online: https://codonw.sourceforge.net/, accessed on 1 September 2022).
2.4. Chloroplast Phylogenetic and Divergence Time Estimation
In addition to the chloroplast genomes of R. japonica from three Chinese regions obtained by us, five samples of R. japonica were provided by [23], and another five chloroplast genomes data were obtained from the NCBI (Accession: NC_059800.1, MW411186.1, MT955361.1, MK381448.1, and MT301955.1). F. multiflora (MK330002.1), Fagopyrum dibotrys (MF491390.1), Fagopyrum tataricum (KM201427.1), Fagopyrum esculentum (EU254477.1), Fallopia convolvulus (OK0409957.1), Polygonum aviculare (NC_058892.1), and Vitis vinifera (NC_007957.1) were used as an outgroup to root the phylogenetic tree.
Phylogenetic analysis based on the shared protein-coding genes (PCGs) and MAFFT v7.503 [36] was used to align the shared PCGs under default parameters. A total of 74 PCGs were used in the concatenated dataset, which were: atpA, atpB, atpE, atpF, atpH, atpI, ccsA, cemA, clpP, infA, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK, petA, petB, petD, petG, petL, petN, psaA, psaB, psaC, psaI, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ, rbcL, rpl14, rpl16, rpl2, rpl20, rpl22, rpl32, rpl33, rpl36, rpoA, rpoB, rpoC1, rpoC2, rps11, rps12, rps14, rps15, rps16, rps18, rps19, rps2, rps3, rps4, rps7, rps8, ycf1, ycf2, ycf3, ycf4. Nucleotide substitution model selection was estimated with ModelTest-NG [40]. The GTR + G4 model was chosen as the best-fit model based on Bayesian information criterion. Maximum likelihood (ML) phylogenetic inference tree was performed by RAxML-NG v0.9.0 [41] with 1000 bootstrap replicates. The Bayesian inference (BI) tree was generated in MrBayes v3.2.7a [42], running for 2,000,000 generations, and the parameters were sampled every 1000 generations. The best-fit model (GTR + I+G) was selected by Akaike information criterion in MrModeltest v2.4 (available online: https://github.com/nylander/MrModeltest2, accessed on 1 September 2022).
Species divergence times were estimated by mcmctree program in PAML v4.8a [43] based on the maximum likelihood phylogenetic inference tree, the 74 PCGs, and calibrated time from TimeTree [44]. Specifically, pairwise divergence time for R. japonica and F. multiflora was 14.6~43.9 million years ago (MYA), 25.5~55.2 MYA for R. japonica and F. tataricum, and 112.4~125.0 MYA for R. japonica and V. vinifera. The results were treated with FigTree v1.4.4.
2.5. Genetic Diversity and Population Differentiation of R. japonica
The whole chloroplast genomes of the 13 R. japonica were aligned by MAFFT v7.503 [36]. The 13 whole chloroplast genomes had an aligned length of 163,725 bp. A total of 245 mutation sites were screened in MEGA v7.0 [45], including 142 singleton and 103 parsimony informative sites, which were used to analyze population structure with an admixture model-based clustering method implemented in STRUCTURE v2.3.4 [46]. We conducted 20 independent runs for each value from K = 1~10 to estimate the “true” number of clusters in 200,000 Markov chain Monte Carlo cycles following a burn-in step of 500,000 iterations. The most likely number of clusters was determined via the online website STRUCTURE HARVESTER [47]. Then, CLUMPP v1.1.2 and the greedy algorithm were used to align multiple runs of STRUCTURE for the best value of K [48]. In addition, the 245 mutation sites were also used to perform principal component analysis (PCA) to assess genetic structure.
3. Results
3.1. Chloroplast Genome Assembly and Annotation of R. japonica
The whole chloroplast genome of the three sequenced R. japonica comprised a typical covalently closed and double-stranded circular molecule (Figure 1). The results indicated that the complete chloroplast genome had a length of 163,371 bp (Rj. BeiJing), 163,371 bp (Rj. LiChuan), and 163,372 bp (Rj. ShanDong), respectively. The structure of the chloroplast genome included a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions. The coverage of sequencing depth ranged from 2677.20× to 3740.61× (Table S1). The length of the LSC region was 87,571 bp (Rj. BeiJing and Rj. LiChuan) and 87,571 bp (Rj. ShanDong), respectively. The length of the SSC and IR regions in three R. japonica were 13,558 bp and 31,121 bp, respectively (Table 1). The total GC content of the three plastomes ranged from 37.52% to 37.53%, and the three plastomes encoded 79 protein-coding genes, 30 tRNA genes, and 4 rRNA genes (Table 1). In addition, 19 genes contained introns, 17 genes had a single intron, and only ycf3 and clpP genes had 2 introns. The intron in trnK-UUU contained matK (Figure 1). Two genes, psbL and ndhD, were transcribed with ACG because of RNA editing. The obtained whole chloroplast genome sequences of the three R. japonica were submitted to the NCBI database with the GenBank accession numbers OP583946 (Rj. BeiJing), OP583947 (Rj. LiChuan), and OP583948 (Rj. ShanDong).
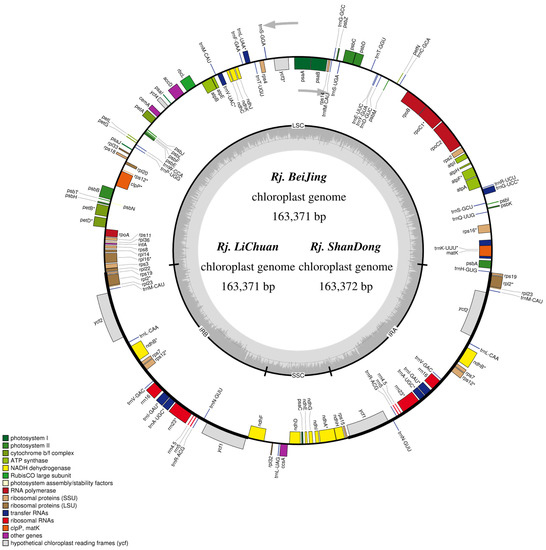
Figure 1.
Gene map of the three R. japonica chloroplast genomes. Genes drawn inside the circle are transcribed clockwise, and genes outside are transcribed counter-clockwise. Different colors encode genes belonging to different functional groups. The area in darker gray and lighter gray in the inner circle corresponds to GC content and AT content, respectively.

Table 1.
Chloroplast genome features of the three R. japonica.
3.2. Features of the R. japonica Chloroplast Genomes
The chloroplast genome length of 13 R. japonica ranged from 163,183 (Rj. ZheJiangZYY) to 163,425 bp (Rj. ZheJiangU) (Table S2), with a GC content of 37.52~37.54%. The LSC region varied the most in length (87594.92 ± 90.65 bp, mean ± standard deviation), but the GC content varied little (35.63 ± 0.0060%). The SSC region was the second most variable in length at 31105.92 ± 71.99 bp, and the difference in GC content variation was at 41.22 ± 0.016%. Compared with the LSC and IRs regions, the SSC region showed the smallest range of length variation (13559.77 ± 4.59 bp), while the GC content showed the greatest variation (32.76 ± 0.26%) (Table S2). The border positions of JLB (junction of LSC and IRb), JSB (junction of SSC and IRb), JSA (junction of SSC and IRa), and JLA (junction of LSC and IRa) among the 13 R. japonica samples were compared. The above results revealed the length of IR regions with some contraction or expansion ranging from 30,859 to 31,150 bp (Figure 2). Some notable differences were found in the JLB junction point; for example, the JLBs of the 12 R. japonica populations were located between rpl22 and rps19, except for Rj. ZheJiangZYY, whose JLB was located in rps19, and the length of rps19 in IRb was 41 bp. There was only one copy of the rps19 gene in Rj. ZheJiangZYY, while there were two copies in the other samples. It is consistent in R. japonica that the ndhF gene stretched over JSB, with 62 bp in the IRb region and 2182 bp in the SSC region. The JSA junction point was located between rps15 and ycf1; the JLA junction point was located between rps19 and trnH (Figure 2). The mVISTA results revealed that the whole chloroplast genomes of the 13 R. japonica samples were relatively similar (Figure S1), indicating that the chloroplast genome in R. japonica is highly conserved. The mVISTA results also indicated that the variations in these 13 samples could be divided into two groups, one with less variation than Rj. BeiJing, and the other with the opposite pattern (Figure S1).

Figure 2.
Comparison of the borders of large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among the 13 R. japonica chloroplast genomes. Genes are denoted by colored boxes, and the gaps between the genes and boundaries are proportional to the distances in bps.
3.3. Molecular Marker Identification
To investigate the genetic diversity of various R. japonica and exploit potential polymorphic genes for identifying the specificity of R. japonica in different regions, gene nucleotide diversity (Pi) values of the 13 samples in R. japonica were calculated (Figure 3). In the protein-coding and nonprotein-coding genes, the Pi value ranged from 0 to 0.08381 (Figure 3A,B). In addition, the sliding window method was used to calculate the Pi values in each chloroplast genomes region using a window size of 600 bp (Figure 3C). The results indicated that most variations in the chloroplast genomes occurred in the LSC region. Gene nucleotide diversity was lowest in the IR region. Therefore, the IR region was more conserved than the other two regions. Six peaks were identified in the 13 R. japonica, with Pi values > 0.0025 (Figure 3C). All peaks were located in intergenic regions. Among them, trnS-GCU/trnG-UCC, trnG-UCC/trnR-UCU, psbM/trnD-GUC, and petG/trnW-CCA were located in the LSC region, and ndhF/rpl32 and rpl32/trnL-UAG were located in the SSC region (Figure 3C). The results showed that the 13 R. japonica samples manifested, in general, a lower degree of nucleotide diversity.
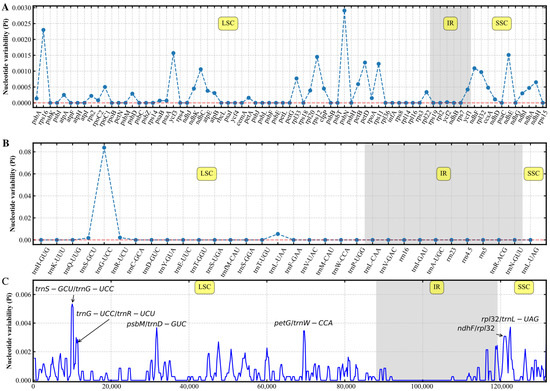
Figure 3.
Gene nucleotide diversity (Pi) values of the 13 R. japonica. The 75 protein-coding genes (A) and 32 nonprotein-coding genes (B). X-axis, gene name; Y-axis, Pi value. (C) Six regions with the highest Pi values were masked based on alignment. Window length, 600 bp; step size, 200 bp. LSC, large single copy; IR, inverted repeats; SSC, small single copy.
3.4. Indices of Codon Usage
In order to analyze the codon usage of R. japonica chloroplast genomes, 75 shared protein-coding genes were extracted and connected as a concatenated dataset for each chloroplast genome. These sequences ranged from 64,275 bp to 64,665 bp, encoding 21,425~21,555 codons. The Leu was encoded by the highest number of codons (2275~2285), while the Cys was by the lowest (228~235). The relative synonymous codon usage (RSCU) values of all the codons ranged from 0.36 to 1.92 (Figure 4, Table S3). The four most frequently used codons with a usage count of more than 800 were AUU (Ile), GAA (Glu), AAA (Lys), and UUU (Phe). The results indicated that the CAI, CBI, FOP, GC3s, and ENC values were similar among the 13 R. japonica samples (Table S4).
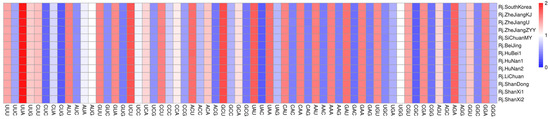
Figure 4.
The RSCU values of all the concatenated protein-coding genes for the 13 R. japonica chloroplast genomes.
3.5. Plastid Phylogenomics of R. japonica
Phylogenetic analysis was carried out based on alignment of the concatenated nucleotide sequences of all the 20 chloroplast genomes through ML and BI (Figure 5). The topologies of the ML and BI trees were consistent, and most nodes had strong support values. Both trees indicated that R. japonica was sister to F. multiflora and formed an alone clade, which is consistent with traditional taxonomic classifications in Flora of China. In addition, the molecular data showed that the divergence time between R. japonica and F. multiflora was about 20.47 MYA (95% HPD = 13.82~34.46 MYA) (Figure 6). It could be inferred from the current data that R. japonica appeared earlier in South Korea than in China. At least before 2.22 MYA (95% HPD = 0.84~5.02 MYA), R. japonica had been delivered to China (Figure 6). The results also showed that all R. japonica clustered together with high resolution and were clearly divided into two major subclades, indicating that they may have relatively different homology and genetic relationships. Among them, one subclade is distributed in Zhejiang Province (Rj. ZheJiangKJ, Rj. ZheJiangZYY, and Rj. ZheJiangU), while the other is relatively scattered (Figure 5 and Figure 6).
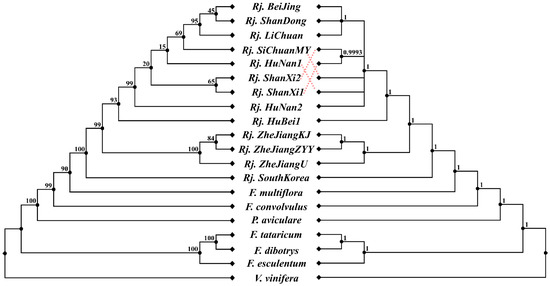
Figure 5.
Phylogenetic trees based on protein-coding genes. Maximum likelihood (ML) phylogenetic inference tree (left) and Bayesian inference (BI) tree (right); ML bootstrap support values/Bayesian posterior probabilities are shown at each node; F. convolvulus, Fallopia convolvulus; F. tataricum, Fagopyrum tataricum; F. dibotrys, Fagopyrum dibotrys; F. esculentum, Fagopyrum esculentum.
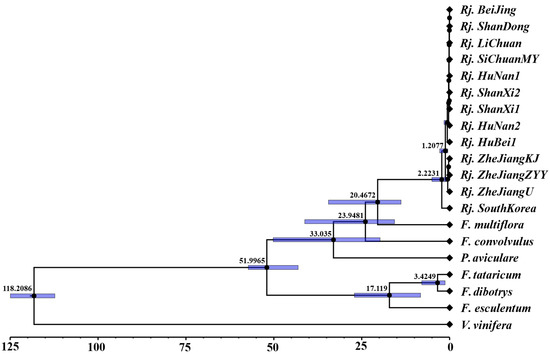
Figure 6.
Divergence times of R. japonica and other plants based on shared protein-coding genes with three priors. The mean divergence times of the nodes are shown next to the nodes, and the blue bars correspond to the 95% highest posterior density (HPD); X-axis unit, million years ago (MYA).
3.6. Intraspecific Diversity and Genetic Structure of R. japonica
To further understand the possible group of R. japonica in China, molecular phylogeography was carried out. The samples were clearly divided into two clades according to phylogenetic relationships (Figure 7A), STRUCTURE analysis (Figure 7B), and PCA (Figure 7C). The results also showed that R. japonica in South Korea and Zhejiang Province in China had closer consanguinity. In addition, Rj. HuBei1 processes 52.15% consanguinity, similar to that in South Korea and Zhejiang Province, and 47.85%, similar to other provinces and cities in China. During the propagation of R. japonica in China, the essential transition zone was Hubei Province, about 0.65 MYA (95% HPD = 0.24~1.59 MYA) (Figure 7A,B). This was followed by penetration into other regions of China, including Hunan, Shanxi, Sichuan, Beijing, Shandong, and so on. The two clades showed remarkable genetic differences and diverged at 1.21 MYA (95% HPD = 0.51~2.81 MYA) (Figure 7A). R. japonica appeared in several provinces in China within a similar period of time, reflecting its great adaptability and rapid invasion capacity.
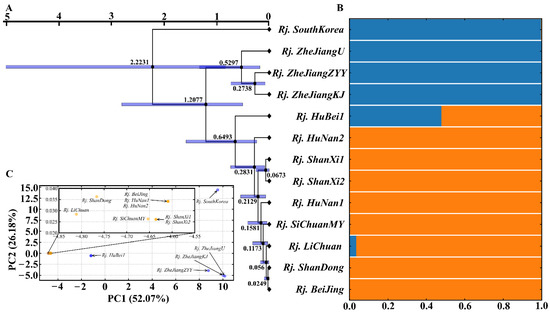
Figure 7.
Intraspecific diversity and genetic structure of R. japonica based on whole chloroplast genomes: (A) phylogenetic tree; (B) population structure analysis with K = 2; (C) principal component analysis.
4. Discussion
4.1. Comparison of Chloroplast Genomes in R. japonica
In this study, we sequenced and assembled three chloroplast genomes of R. japonica and performed a series of comprehensive comparative analyses with other published data. All the chloroplast genomes in R. japonica presented a typically quadripartite structure [22,23]. The chloroplast genome structure, codon bias, and gene order were conserved, consistent with buckwheat [20]. A slight variation was observed in chloroplast genome size and GC content (Table S2). However, some evident diversities in R. japonica were also detected. IR regions were considered the most conserved regions in the chloroplast genome [49], and its contraction or expansion was the main cause of chloroplast genome size variation [50]. In Rj. ZheJiangZYY, the whole chloroplast length was the shortest in the 13 accessions of R. japonica—it was only 163,183 bp (Figure 2). As 238 bp of rps19 were located in the LSC region, it appeared that the IR region had contracted. In addition, the genes near the boundary of IR and LSC were rpl2 and trnH rather than rps19 and trnH in the other 12 R. japonica. We also found that the accD and matK genes were present as pseudogenes in Rj. SiChuanMY, which produce acetyl-CoA carboxylase subunit β and maturase K, respectively. The accD gene is also a pseudogene in some species [51,52] according to incomplete sequence and annotation.
4.2. Potential DNA Barcodes
Developing DNA barcodes based on differences in chloroplast genome sequences are widely used in species identification [20,53]. The comparison of whole chloroplast genomes by mVISTA indicated that the sequences of R. japonica were highly similar (Figure S1) but not as rich in variable sites as buckwheat [20]. The results showed that the chloroplast genomes of 13 R. japonica samples could be divided into two main categories. We further investigated the nucleotide diversities of these R. japonica (Figure 3). Based on the sequence variations, five protein-coding genes (rps16, ycf3, rps12, psbN, ndhE) and one nonprotein-coding gene (trnG-UCC) were selected, which were potentially useful for the population identification in R. japonica (Figure 3A,B). Moreover, six intergenic regions with high nucleotide diversity were found (Figure 3C). The intergenic regions of trnS-GCU/trnG-UCC and ndhF/rpl32 were also identified in other angiosperms [16,53]. These results suggested that this DNA barcode could be used to identify R. japonica and conduct a phylogenetic tree. The results also indicated that mutation sites appeared clustered and mainly concentrated in the LSC region (Figure 3C). These polymorphic loci might be helpful for phylogenetic inference and population genetic studies of R. japonica.
4.3. Evolutionary Relationships
At present, a systematic study of genetic diversity and biogeographic evolution of R. japonica is still lacking. It is both desirable and helpful to investigate the systematical evolution and biogeographic relationships of this species. Although R. japonica was introduced to Europe, North America, and Australia with ornamental purposes in the 19th century and is now considered an invasive plant [54], it is native and widely distributed in China, Japan, and Korea [1]. Moreover, the medicinal use of R. japonica in China has a long history, first appearing in Mingyi Bielu, a famous medicinal book, 2000 years ago. In Japan, the R. japonica chloroplast genome region from the rbcL to the accD gene was obtained [55], and the intraspecific sequence variation in chloroplast regions reflected the species variety and geographical distribution. However, comparative analyses based on the whole chloroplast genome could provide a more comprehensive interpretation of intraspecific genetic structure and phylogenetic relationships than using only a few fragments [16].
The main aim of this study was to explore the characteristics of chloroplast genome divergence in R. japonica. The phylogenetic trees in our study showed the independent evolution of R. japonica species (Figure 5), which corroborates a separate taxonomic status for R. japonica in the Polygonaceae. The differentiation time of R. japonica and V. vinifera estimated based on the chloroplast genome was similar to that of the nuclear genome [56]. The results also revealed that R. japonica was divided into two subclades with significant genetic divergence (Figure 7B) and with Hubei Province as a transition zone. Located in the middle of the Yangtze River, an essential transport hub region in China, Hubei Province may serve as a crucial hub for R. japonica to further spread to other regions due to the frequent movement of personnel. The seeds of Rj. LiChuan were collected from Hubei Province and planted in our lab, and Rj. LiChuan and Rj. HuBei1 had partial consanguinity from population genetic structure analysis (Figure 7B) with a relatively distant genetic distance (Figure 7A). This might be caused by the adaption of R. japonica or the different differentiation times in other regions of Hubei Province. In addition, the current data showed that R. japonica occurred earlier in South Korea than in China (Figure 7A), contrary to the notion that R. japonica originated in China [57]. Considering the wide availability of R. japonica across diverse regions of China, the debate on the origin of this medicinal plant is evidently not conclusive at this stage. From the tight timescale deduced from the chloroplast genomic data (Figure 7A), it is safe to say that this plant is indeed highly invasive [9].
5. Conclusions
R. japonica. is an essential medicinal plant. A large amount of genetic information was contained in the chloroplast genome. At present, systematic research into R. japonica evolution based on the chloroplast genome is still lacking. This study seems the first attempt to comprehensively investigate the chloroplast genome features and infer phylogeny for R. japonica. We assembled three complete chloroplast genomes, together with 10 other currently available data, to elucidate the evolutionary relationship of R. japonica. Our comparative analyses showed that chloroplast genomes of R. japonica are well conserved in terms of genome structure and codon usage, and there are two main subclades of R. japonica in China, with Hubei Province probably acting as the dispersal center region. The collected genomes and obtained genetic relationship in this study could contribute to further studies on population genetics, phylogeny, and conservation biology in Polygonaceae.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13111979/s1, Figure S1: comparison of 13 R. japonica chloroplast genomes using mVISTA, with Rj. BeiJing as the reference. The Y-axis represents the percent identity within 50~100%. Gray arrows indicate the direction of gene transcription. Purple bars represent exons, blue bars represent untranslated regions (UTR), pink bars indicate conserved noncoding sequences (CNS), gray bars represent mRNA, and white peaks represent differences of genomics, Table S1: the sequencing depth of three R. japonica, Table S2: comparison of the complete chloroplast genomes for 13 R. japonica, Table S3: the RSCU values of 13 R. japonica, Table S4: the codon usage of 13 R. japonica.
Author Contributions
Conceptualization, J.C. (Jianhui Chen) and H.W.; methodology, J.C. (Jianhui Chen) and H.W.; software, J.C. (Jianhui Chen); validation, T.H., L.M. and H.W.; formal analysis, J.C. (Jianhui Chen) and T.H.; investigation, T.H., H.F., F.L., H.M. and J.C. (Jie Cao); resources, T.H., J.C. (Jie Cao), T.C. and L.M.; data curation, J.C. (Jianhui Chen); writing—original draft preparation, J.C. (Jianhui Chen) and T.H.; writing—review and editing, T.H. and H.W.; visualization, J.C. (Jianhui Chen); supervision, T.C., L.M. and H.W.; project administration, T.C., L.M. and H.W.; funding acquisition, T.C. and H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 61972374, 61672489) and the Fundamental Research Funds for the Central Universities (Grant No. E1E40605).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used in our study have been submitted to NCBI GenBank (Accession Number: OP583946-OP583948).
Acknowledgments
The authors would like to thank Zhuqing Zhang (College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China) for her computing resource assistance during the data analysis of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peng, W.; Qin, R.X.; Li, X.L.; Zhou, H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: A review. J. Ethnopharmacol. 2013, 148, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Wang, X.W.; Chen, M.; Hu, H.Y.; Cao, J.; Chai, T.Y.; Wang, H. A study on tissue-specific metabolite variations in Polygonum cuspidatum by high-resolution mass spectrometry-based metabolic profiling. Molecules 2019, 24, 1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Kwok, S.T.; Zhang, Q.W.; Chan, S.W. A review of the pharmacological effects of the dried root of Polygonum cuspidatum (Hu Zhang) and its constituents. Evid.-Based Complement. Altern. Med. 2013, 2013, 208349. [Google Scholar] [CrossRef]
- Lee, C.C.; Chen, Y.T.; Chiu, C.C.; Liao, W.T.; Liu, Y.C.; David Wang, H.M. Polygonum cuspidatum extracts as bioactive antioxidaion, anti-tyrosinase, immune stimulation and anticancer agents. J. Biosci. Bioeng. 2015, 119, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.L.; Walls, R.L.; Bailey, J.P.; Parameswaran, R.; George, T.; Pigliucci, M. Plasticity in salt tolerance traits allows for invasion of novel habitat by Japanese knotweed s. l. (Fallopia japonica and F.xbohemica, Polygonaceae). Am. J. Bot. 2008, 95, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Michalet, S.; Rouifed, S.; Pellassa-Simon, T.; Fusade-Boyer, M.; Meiffren, G.; Nazaret, S.; Piola, F. Tolerance of Japanese knotweed s.l. to soil artificial polymetallic pollution: Early metabolic responses and performance during vegetative multiplication. Environ. Sci. Pollut. Res. Int. 2017, 24, 20897–20907. [Google Scholar] [CrossRef]
- Nentwig, W.; Bacher, S.; Kumschick, S.; Pyšek, P.; Vilà, M. More than "100 worst" alien species in Europe. Biol. Invasions 2018, 20, 1611–1621. [Google Scholar] [CrossRef]
- Rahmonov, O.; Czylok, A.; Orczewska, A.; Majgier, L.; Parusel, T. Chemical composition of the leaves of Reynoutria japonica Houtt. and soil features in polluted areas. Cent. Eur. J. Biol. 2014, 9, 320–330. [Google Scholar] [CrossRef]
- Kato-Noguchi, H. Allelopathy of knotweeds as invasive plants. Plants 2021, 11, 3. [Google Scholar] [CrossRef]
- Bailey, J.P.; Bimova, K.; Mandak, B. Asexual spread versus sexual reproduction and evolution in Japanese knotweed s.l. sets the stage for the "Battle of the Clones". Biol. Invasions 2009, 11, 1189–1203. [Google Scholar] [CrossRef]
- Allen, J.F.; de Paula, W.B.; Puthiyaveetil, S.; Nield, J. A structural phylogenetic map for chloroplast photosynthesis. Trends Plant Sci. 2011, 16, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.J. Circular chloroplast chromosomes: The grand illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhou, T.; Bai, G.Q.; Zhao, Y.M. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Wang, S.; Wang, Y.H.; Wang, R.S.; Liu, K.J.; Li, E.Z.; Qiao, P.; Shi, L.Y.; Dong, W.P.; Huang, L.Q.; et al. Phylogenomics and genetic diversity of Arnebiae Radix and its allies (Arnebia, Boraginaceae) in China. Front. Plant Sci. 2022, 13, 920826. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Sun, J.H.; Liu, Y.L.; Xu, C.; Wang, Y.H.; Suo, Z.L.; Zhou, S.L.; Zhang, Z.X.; Wen, J. Phylogenomic relationships and species identification of the olive genus Olea (Oleaceae). J. Syst. Evol. 2021. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Samigullin, T.H.; Dhingra, A.; Penin, A.A. Comparative chloroplast genomics and phylogenetics of Fagopyrum esculentum ssp. ancestrale—A wild ancestor of cultivated buckwheat. BMC Plant Biol. 2008, 8, 59. [Google Scholar] [CrossRef]
- Cho, K.S.; Yun, B.K.; Yoon, Y.H.; Hong, S.Y.; Mekapogu, M.; Kim, K.H.; Yang, T.J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef]
- Fan, Y.; Jin, Y.N.; Ding, M.Q.; Tang, Y.; Cheng, J.P.; Zhang, K.X.; Zhou, M.L. The complete chloroplast genome sequences of eight Fagopyrum species: Insights into genome evolution and phylogenetic relationships. Front Plant Sci. 2021, 12, 799904. [Google Scholar] [CrossRef]
- Yao, G.; Jin, J.J.; Li, H.T.; Yang, J.B.; Mandala, V.S.; Croley, M.; Mostow, R.; Douglas, N.A.; Chase, M.W.; Christenhusz, M.J.M.; et al. Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet. Evol. 2019, 134, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.F.; Lin, J.G.; Zhou, M.J.; He, X.Y.; Yan, M.X.; Cheng, R.B. The complete chloroplast genome of the medicinal plant Polygonum cuspidatum (Polygonaceae) and its phylogenetic implications within the subfamily Polygonoideae. Mitochondrial DNA B Resour. 2021, 6, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.T.; Zhang, J.X.; Huang, T.R.; Yang, M.F.; Ma, L.Q.; Duan, L.S. Genome structure and variation of Reynoutria japonica Houtt. chloroplast genome. Chin. J. Biotechnol. 2022, 38, 1953–1964. [Google Scholar]
- Zhang, X.; Liu, F.; Feng, Z.M.; Yang, Y.N.; Jiang, J.S.; Zhang, P.C. Bioactive phenylpropanoid esters of sucrose and anthraquinones from Polygonum cuspidatum. Fitoterapia 2020, 146, 104673. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Hu, H.Y.; Wu, Z.J.; Fan, H.L.; Wang, G.W.; Chai, T.Y.; Wang, H. Tissue-specific transcriptome analyses reveal candidate genes for stilbene, flavonoid and anthraquinone biosynthesis in the medicinal plant Polygonum cuspidatum. BMC Genome 2021, 22, 353. [Google Scholar] [CrossRef]
- Zheng, L.L.; Zhou, C.; Li, T.H.; Yuan, Z.; Zhou, H.L.; Tamada, Y.; Zhao, Y.H.; Wang, J.; Zheng, Q.; Hao, X.C.; et al. Global transcriptome analysis reveals dynamic gene expression profiling and provides insights into biosynthesis of resveratrol and anthraquinones in a medicinal plant Polygonum cuspidatum. Ind. Crops Prod. 2021, 171, 113919. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Xu, J.X.; Wu, X.; Wang, Y.Y.; Lin, Y.L.; Wu, D.Y.; Zhang, H.J.; Qin, J.B. Molecular analysis of UV-C induced resveratrol accumulation in Polygonum cuspidatum leaves. Int. J. Mol. Sci. 2019, 20, 6185. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19 (Suppl. 1), i54–i62. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Liu, L.X.; Wang, Y.W.; He, P.Z.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C.X. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef]
- Fan, W.B.; Wu, Y.; Yang, J.; Shahzad, K.; Li, Z.H. Comparative chloroplast genomics of Dipsacales species: Insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 2018, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wuyun, T.N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Li, Y.T.; Dong, Y.; Liu, Y.C.; Yu, X.Y.; Yang, M.S.; Huang, Y.R. Comparative analyses of Euonymus chloroplast genomes: Genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within celastrineae. Front Plant Sci. 2020, 11, 593984. [Google Scholar] [CrossRef] [PubMed]
- Fennell, M.; Wade, M.; Bacon, K.L. Japanese knotweed (Fallopia japonica): An analysis of capacity to cause structural damage (compared to other plants) and typical rhizome extension. PeerJ 2018, 6, e5246. [Google Scholar] [CrossRef]
- Inamura, A.; Ohashi, Y.; Sato, E.; Yoda, Y.; Masuzawa, T.; Ito, M.; Yoshinaga, K. Intraspecific sequence variation of chloroplast DNA reflecting variety and geographical distribution of Polygonum cuspidatum (Polygonaceae) in Japan. J. Plant Res. 2000, 113, 419–426. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zheng, L.L.; Zheng, Y.; Zhou, C.; Huang, P.; Xiao, X.; Zhao, Y.H.; Hao, X.C.; Hu, Z.B.; Chen, Q.H.; et al. Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatum. Front. Plant Sci. 2019, 10, 1274. [Google Scholar] [CrossRef]
- Fan, P.H.; Hostettmann, K.; Lou, H.X. Allelochemicals of the invasive neophyte Polygonum cuspidatum Sieb. & Zucc. (Polygonaceae). Chemoecology 2010, 20, 223–227. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).