Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis

Abstract
:1. Introduction
2. Methods
2.1. Plant Materials
2.2. RNA Isolation, Library Construction, and RNA-Seq Analysis
2.3. Differentially Expressed Genes Analysis and Comparison of Differentially Expressed Genes Expression Patterns
2.4. Construction of Gene Co-Expression Networks and Screening of Hub Genes
3. Results
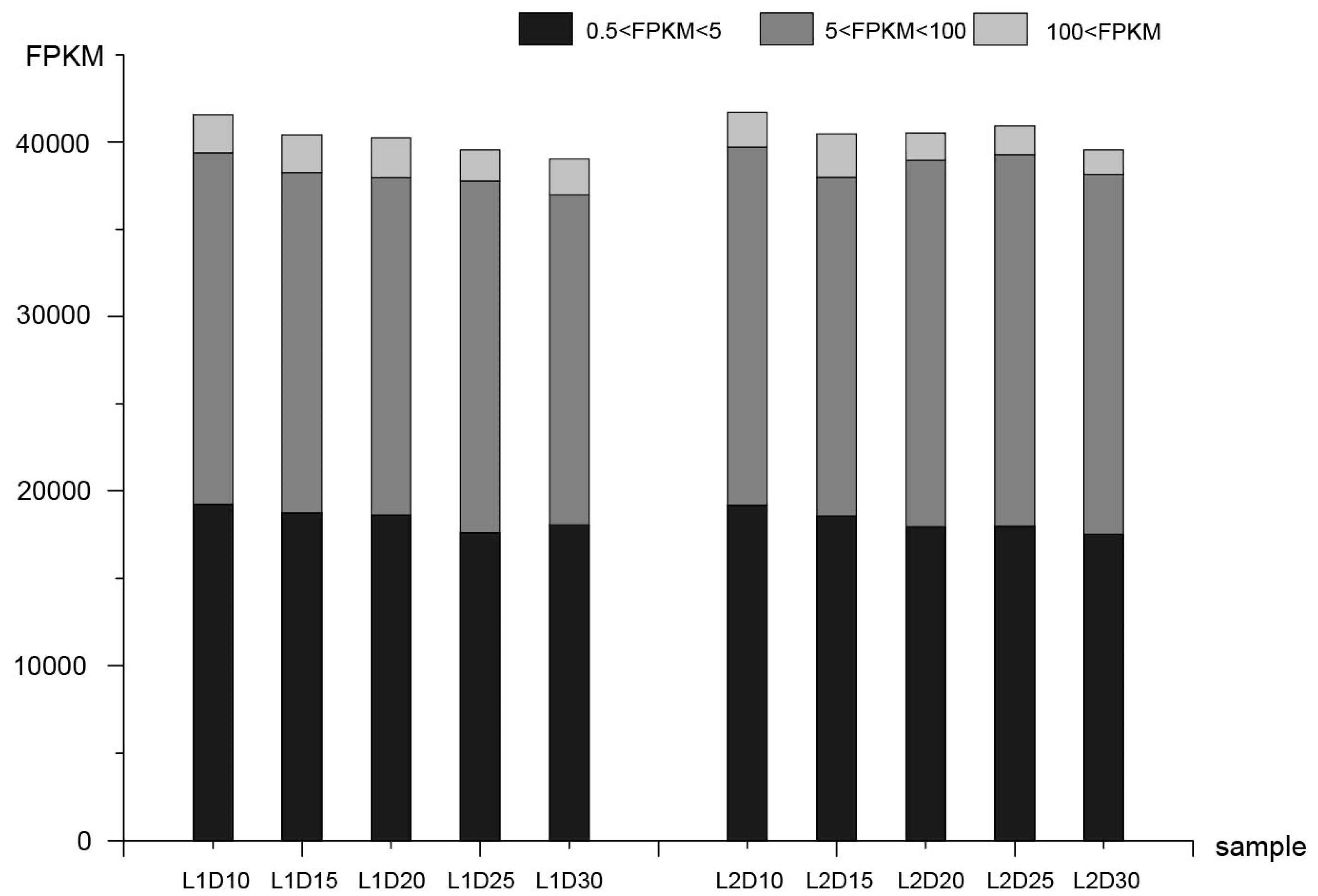
3.1. Transcriptome Sequencing Analysis and Correlation of Replicate Samples
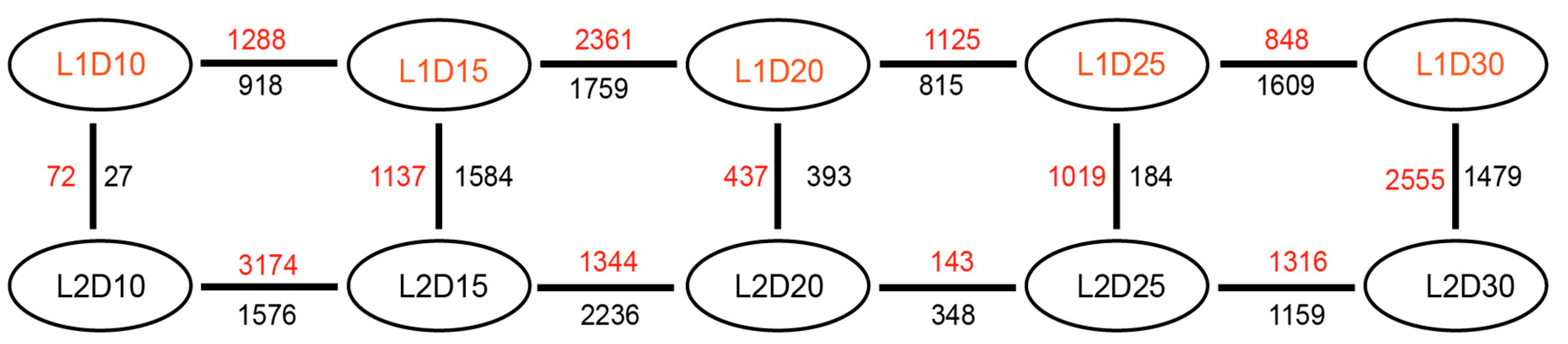
3.2. Differential Gene Expression Analysis
3.3. Congruence Analysis with the Previous Report
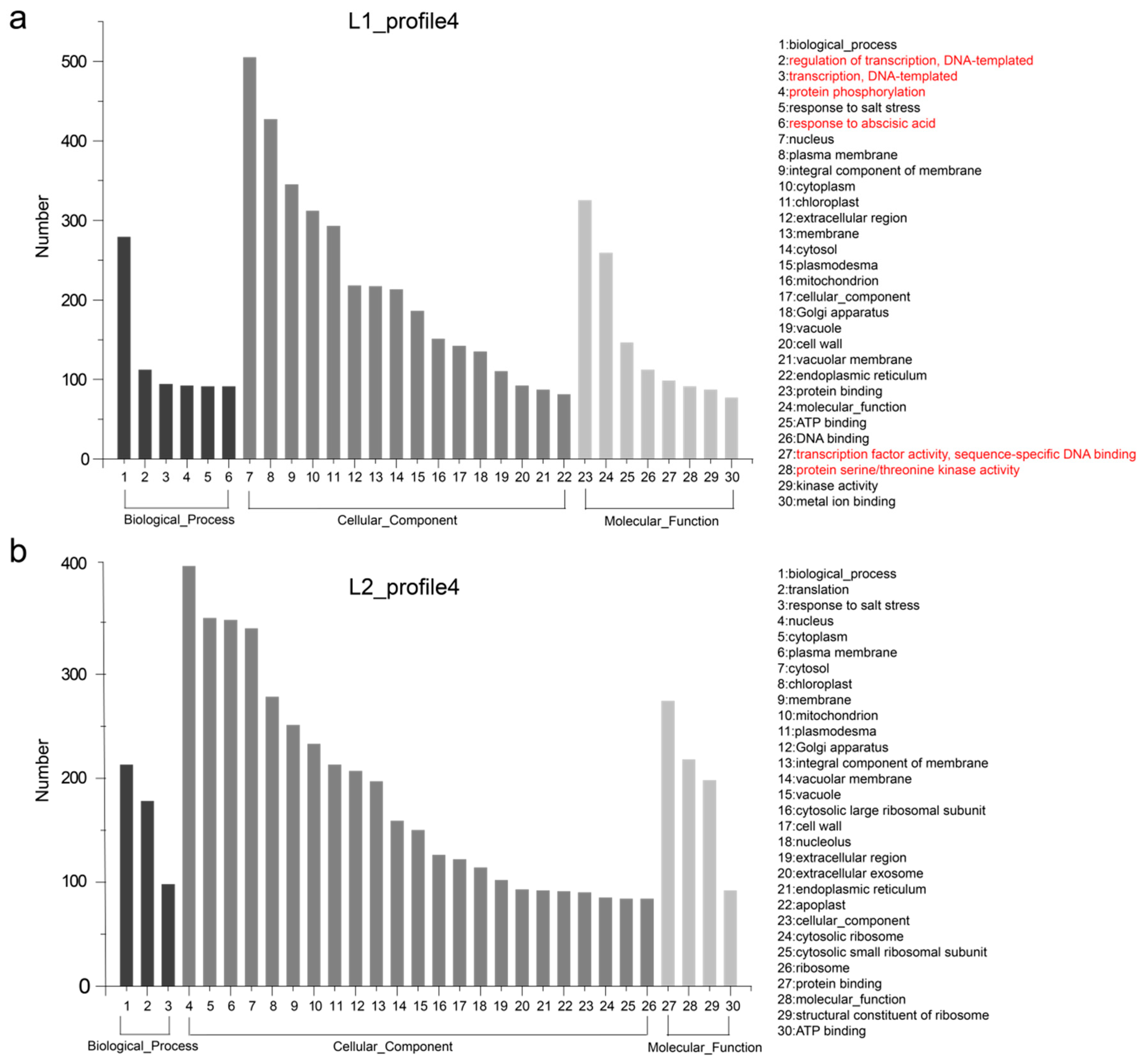
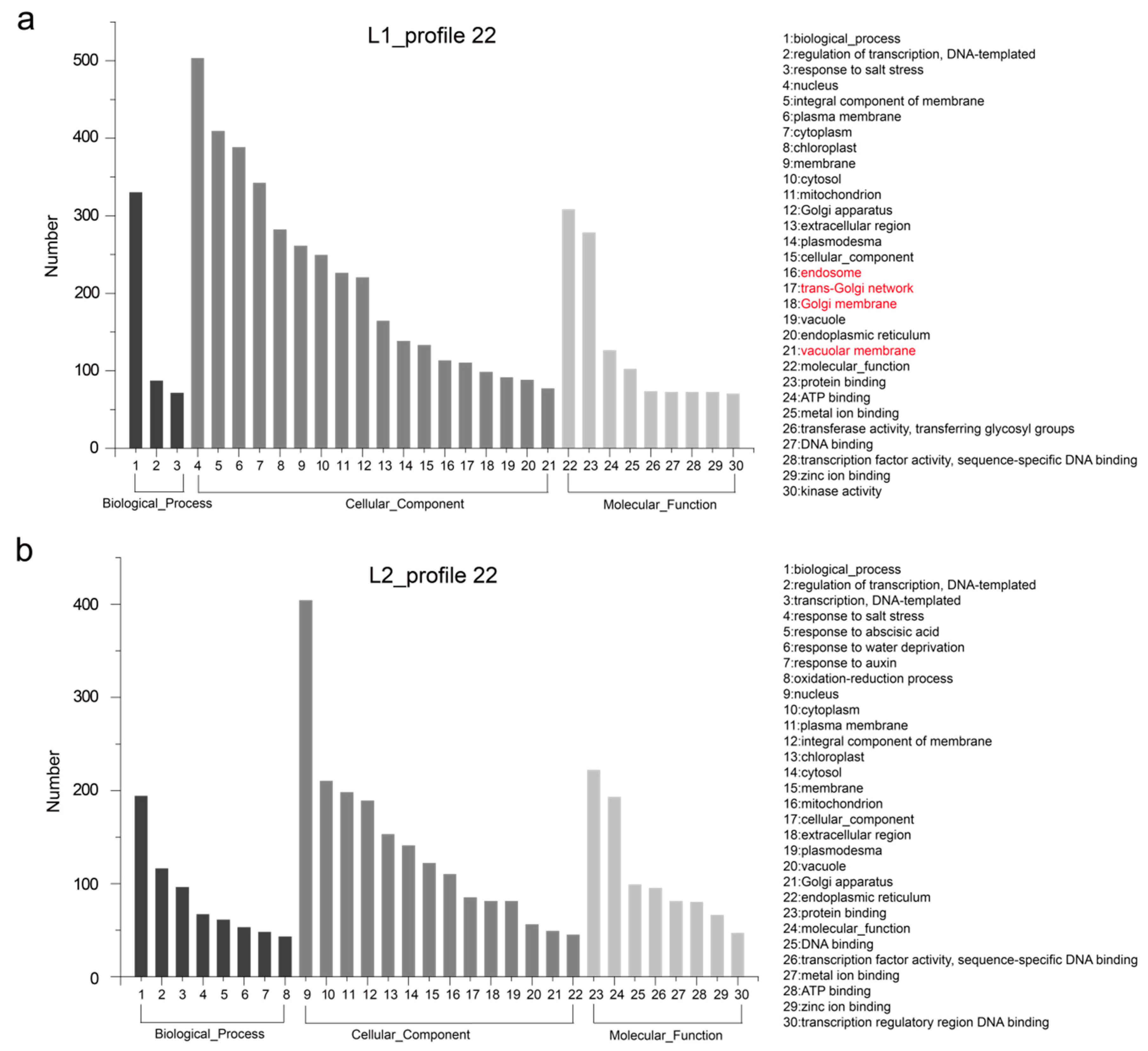
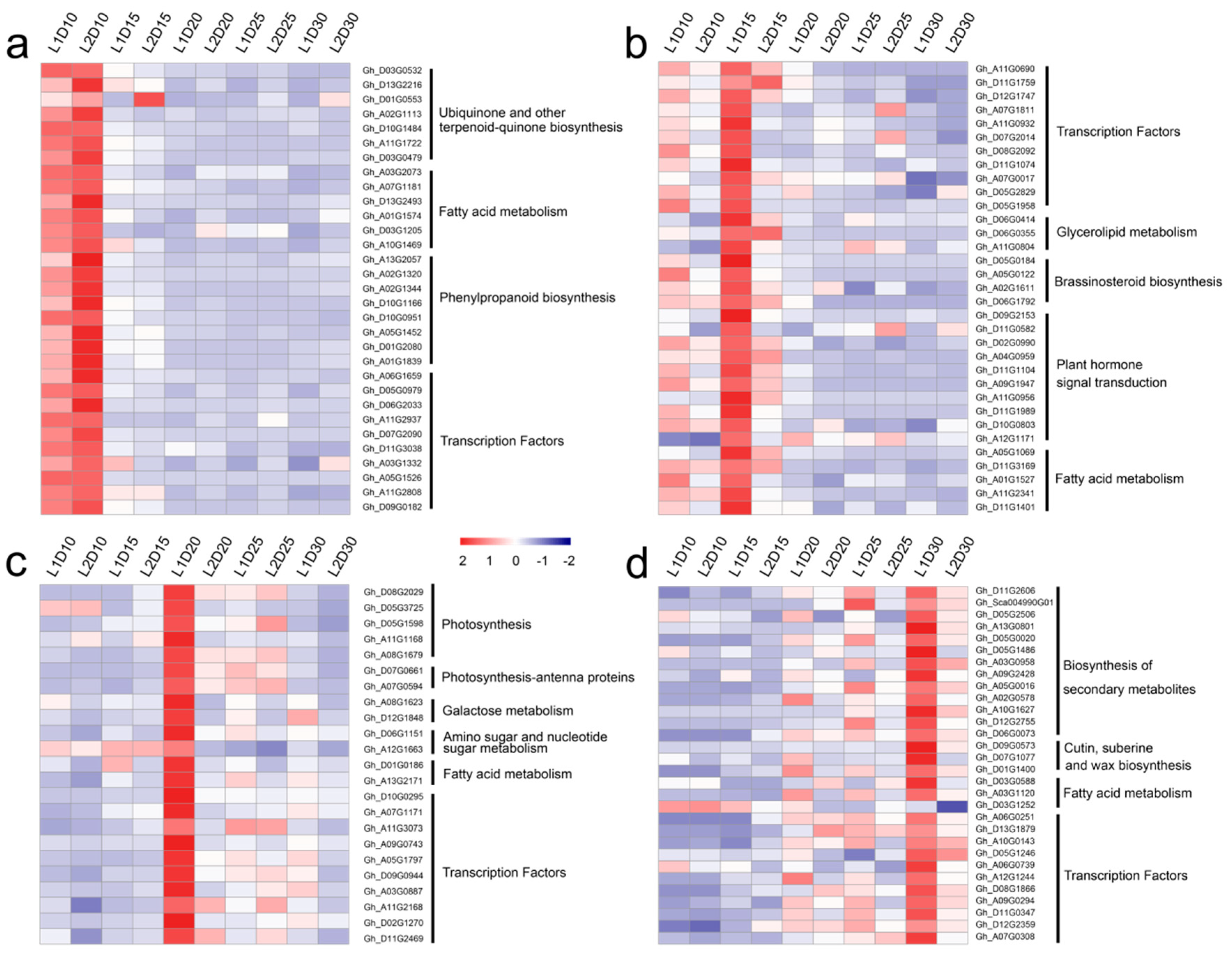
3.4. Temporal Gene Expression Patterns
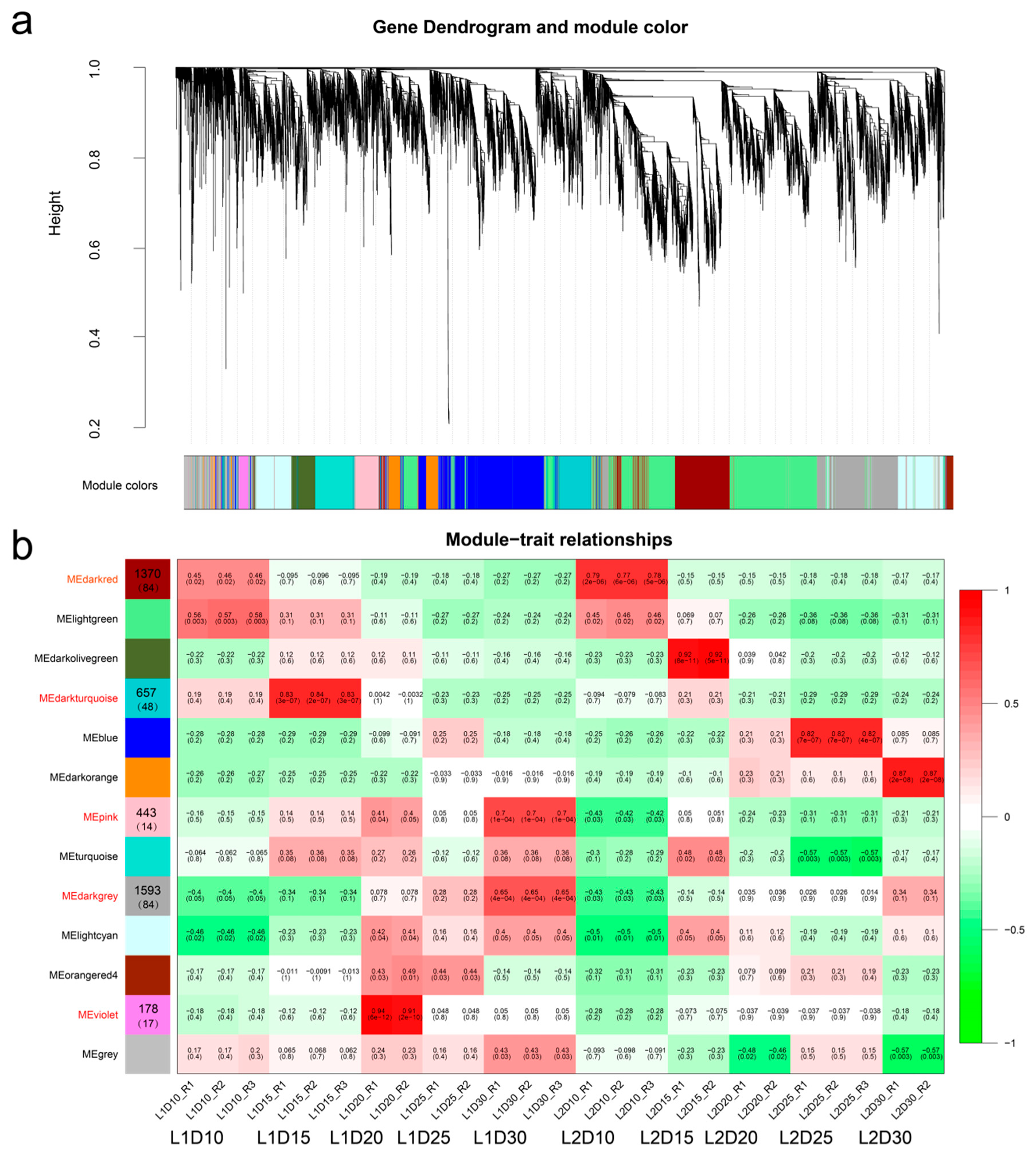
3.5. Gene Co-Expression Network Analysis
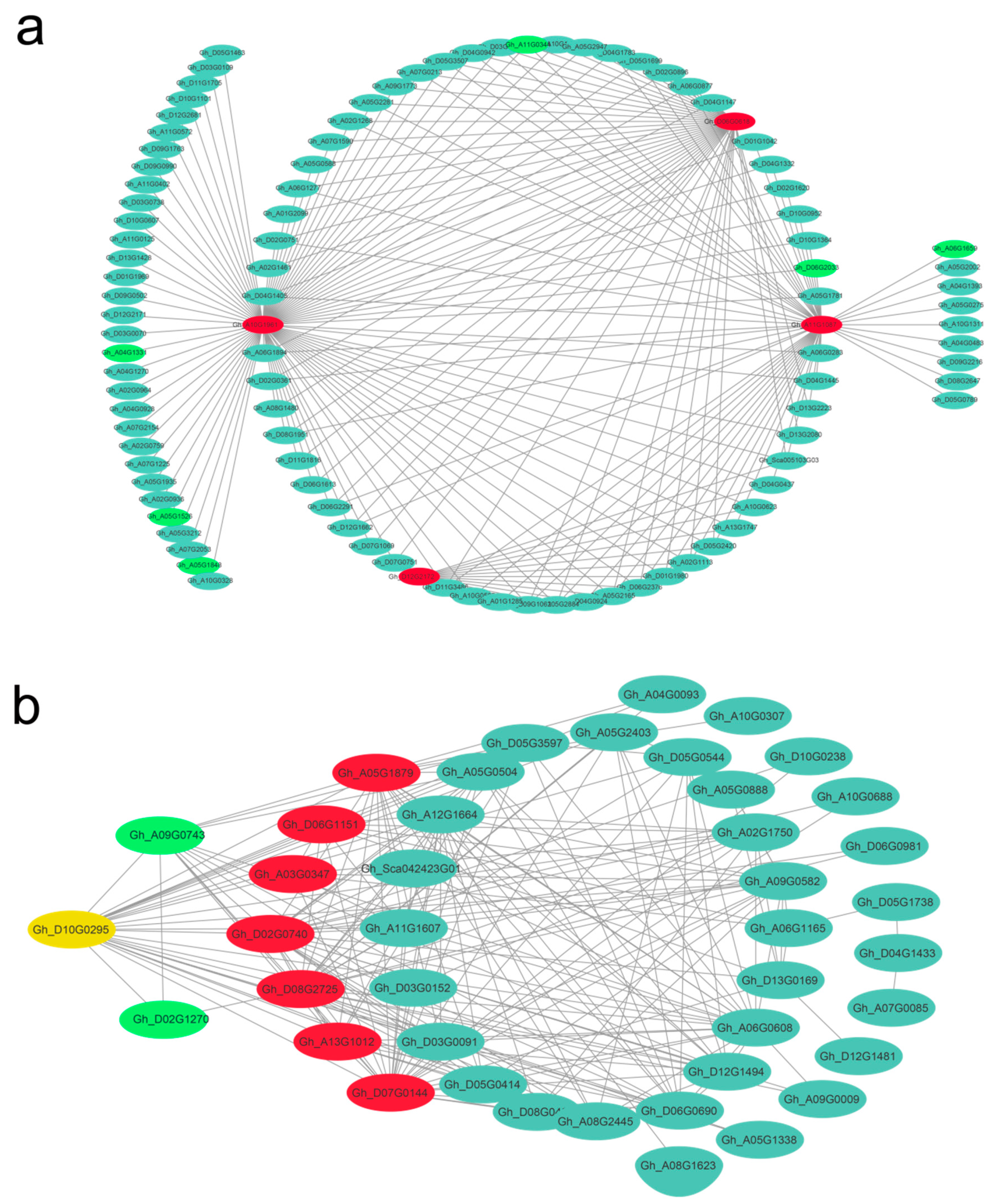
3.6. Identification of Hub Genes for Elite Fiber Quality and Visualization of Correlation Networks
4. Discussion
4.1. Transcriptome Sequencing of Elite and Poor-Quality Fiber Lines Mapped to the Reference Genome of Upland Cotton
4.2. RNA-Seq Provides Potentia information by Comparison of Expression Profiles between the Two Recombinant Inbred Lines
4.3. Hub Genes Identified by WGCNA May Play Critical Roles in Improvement of Fiber Quality
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoo, M.-J.; Wendel, J.F. Comparative evolutionary and developmental dynamics of the cotton (Gossypium hirsutum) fiber transcriptome. PLoS Genet. 2014, 10, e1004073. [Google Scholar] [CrossRef] [PubMed]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, M.; Liu, G.; Pei, W.; Zhai, H.; Yu, J.; Zhang, J.; Yu, S. Identification of candidate genes for fiber length quantitative trait loci through RNA-Seq and linkage and physical mapping in cotton. BMC Genom. 2017, 18, 427. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-T.; Wang, M.; Lu, Q.-W.; Ge, Q.; Rashid, M.H.O.; Liu, A.-Y.; Gong, J.-W.; Shang, H.-H.; Gong, W.-K.; Li, J.-W.; et al. Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and Chromosome Segment Substitution Lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Shi, Y.; Xiao, X.; Li, P.; Gong, J.; Gong, W.; Liu, A.; Shang, H.; Li, J.; Ge, Q.; et al. Transcriptome analysis suggests that chromosome introgression fragments from sea island cotton (Gossypium barbadense) increase fiber strength in upland cotton (Gossypium hirsutum). G3 Genes|Genomes|Genet. 2017, 7, 3469–3479. [Google Scholar] [CrossRef] [PubMed]
- Applequist, W.L.; Cronn, R.; Wendel, J.F. Comparative development of fiber in wild and cultivated cotton. Evol. Dev. 2001, 3, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Fang, D.D.; Thyssen, G.N.; Delhom, C.D.; Liu, Y.; Kim, H.J. Comparative fiber property and transcriptome analyses reveal key genes potentially related to high fiber strength in cotton (Gossypium hirsutum L.) line MD52ne. BMC Plant Biol. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.K.; Kim, H.J.; Tang, Y.; Naoumkina, M.; Fang, D.D. Comparative Transcriptome Analysis of Short Fiber Mutants Ligon-Lintless 1 And 2 Reveals Common Mechanisms Pertinent to Fiber Elongation in Cotton (Gossypium hirsutum L.). PLoS ONE 2014, 9, e95554. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, Q.; Liu, A.; Li, J.; Gong, J.; Shang, H.; Shi, Y.; Chen, T.; Wang, Y.; Palanga, K.K.; et al. Construction of a High-Density Genetic Map and Its Application to QTL Identification for Fiber Strength in Upland Cotton. Crop Sci. 2017, 57, 774. [Google Scholar] [CrossRef]
- Sun, F.D.; Zhang, J.H.; Wang, S.F.; Gong, W.K.; Shi, Y.Z.; Liu, A.Y.; Li, J.W.; Gong, J.W.; Shang, H.H.; Yuan, Y.L. QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 2012, 30, 569–582. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Muhammad, J.; Cai, J.; Jia, F.; Shi, Y.; Gong, J.; Shang, H.; Liu, A.; Chen, T.; et al. High Resolution Consensus Mapping of Quantitative Trait Loci for Fiber Strength, Length and Micronaire on Chromosome 25 of the Upland Cotton (Gossypium hirsutum L.). PLoS ONE 2015, 10, e0135430. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jung, S.; Cheng, C.H.; Ficklin, S.P.; Lee, T.; Zheng, P.; Jones, D.; Percy, R.G.; Main, D. CottonGen: A genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 2014, 42, 1229–1236. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Nau, G.J.; Bar-Joseph, Z. Clustering Short Time Series Gene Expression Data; Oxford University Press: Oxford, UK, 2005; pp. 159–168. [Google Scholar]
- Ernst, J.; Barjoseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Tong, H.; Chu, C. Functional Specificities of Brassinosteroid and Potential Utilization for Crop Improvement. Trends Plant Sci. 2018. [Google Scholar] [CrossRef]
- Feng, S.; Yue, R.; Tao, S.; Yang, Y.; Zhang, L.; Xu, M.; Wang, H.; Shen, C. Genome-wide identification, expression analysis of auxin-responsive GH3 family genes in maize (Zea mays L.) under abiotic stresses. J. Integr. Plant Biol. 2015, 57, 783–795. [Google Scholar] [CrossRef]
- Liu, H.; Yang, W.; Liu, D.; Han, Y.; Zhang, A.; Li, S. Ectopic expression of a grapevine transcription factor VvWRKY11 contributes to osmotic stress tolerance in Arabidopsis. Mol. Biol. Rep. 2011, 38, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Journot-Catalino, N.; Somssich, I.E.; Roby, D.; Kroj, T. The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell 2006, 18, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.M.; Hu, C.Y.; Pang, Y.; Kastaniotis, A.J.; Hiltunen, J.K.; Zhu, Y.X. Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 2007, 19, 3692–3704. [Google Scholar] [CrossRef] [PubMed]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmulling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef]
- Shi, Y.L.; Guo, S.D.; Zhang, R.; Meng, Z.G.; Ren, M.Z. The role of Somatic embryogenesis receptor-like kinase 1 in controlling pollen production of the Gossypium anther. Mol. Biol. Rep. 2014, 41, 411–422. [Google Scholar] [CrossRef]
- Brill, E.; van Thournout, M.; White, R.G.; Llewellyn, D.; Campbell, P.M.; Engelen, S.; Ruan, Y.L.; Arioli, T.; Furbank, R.T. A novel isoform of sucrose synthase is targeted to the cell wall during secondary cell wall synthesis in cotton fiber. Plant Physiol. 2011, 157, 40–54. [Google Scholar] [CrossRef]
- Molhoj, M.; Verma, R.; Reiter, W.D. The biosynthesis of D-Galacturonate in plants. functional cloning and characterization of a membrane-anchored UDP-D-Glucuronate 4-epimerase from Arabidopsis. Plant Physiol. 2004, 135, 1221–1230. [Google Scholar] [CrossRef]
- Tuttle, J.R.; Nah, G.; Duke, M.V.; Alexander, D.C.; Guan, X.; Song, Q.; Chen, Z.J.; Scheffler, B.E.; Haigler, C.H. Metabolomic and transcriptomic insights into how cotton fiber transitions to secondary wall synthesis, represses lignification, and prolongs elongation. BMC Genom. 2015, 16, 477. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Veerabomma, S.; Abdel-Mageed, H.A.; Fokar, M.; Asami, T.; Yoshida, S.; Allen, R.D. Brassinosteroid regulates fiber development on cultured cotton ovules. Plant Cell Physiol. 2005, 46, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.C.; Martin, C.; Toledo-Ortiz, G.; Quail, P.H.; Huq, E.; Heim, M.A.; Jakoby, M.; Werber, M.; Weisshaar, B. Update on the basic helix-loop-helix transcription factor gene family in Arabidopsis thaliana. Plant Cell 2003, 15, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Hohner, K.; Berken, A.; Wittinghofer, A. The unique plant RhoGAPs are dimeric and contain a CRIB motif required for affinity and specificity towards cognate small G proteins. Biopolymers 2011, 95, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qin, T.; Ma, Q.; Sun, J.; Liu, Z.; Yuan, M.; Mao, T. Light-Regulated Hypocotyl Elongation Involves Proteasome-Dependent Degradation of the Microtubule Regulatory Protein WDL3 in Arabidopsis. Plant Cell 2013, 25, 1740–1755. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ma, Q.; Mao, T. Ethylene Regulates the Arabidopsis Microtubule-Associated Protein WAVE-DAMPENED2-LIKE5 in Etiolated Hypocotyl Elongation. Plant Physiol. 2015, 169, 325–337. [Google Scholar] [CrossRef]
- An, R.; Liu, X.; Wang, R.; Wu, H.; Liang, S.; Shao, J.; Qi, Y.; An, L.; Yu, F. The over-expression of two transcription factors, ABS5/bHLH30 and ABS7/MYB101, leads to upwardly curly leaves. PLoS ONE 2014, 9, e107637. [Google Scholar] [CrossRef]
- Louvet, R.; Cavel, E.; Gutierrez, L.; Guénin, S.; Roger, D.; Gillet, F.; Guerineau, F.; Pelloux, J. Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana. Planta 2006, 224, 782–791. [Google Scholar] [CrossRef]
- Gonzalez, A.; Mendenhall, J.; Huo, Y.; Lloyd, A. TTG1 complex MYBs, MYB5 and TT2, control outer seed coat differentiation. Dev. Biol. 2009, 325, 412–421. [Google Scholar] [CrossRef]
- Li, S.F.; Milliken, O.N.; Pham, H.; Seyit, R.; Napoli, R.; Preston, J.; Koltunow, A.M.; Parish, R.W. The Arabidopsis MYB5 transcription factor regulates mucilage synthesis, seed coat development, and trichome morphogenesis. Plant Cell 2009, 21, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Saez-Aguayo, S.; Rautengarten, C.; Temple, H.; Sanhueza, D.; Ejsmentewicz, T.; Sandoval-Ibañez, O.; Doñas-Cofré, D.A.; Parra-Rojas, J.P.; Ebert, B.; Lehner, A. UUAT1 Is a Golgi-Localized UDP-Uronic Acid Transporter that Modulates the Polysaccharide Composition of Arabidopsis Seed Mucilage. Plant Cell 2017, 29, tpc.00465.02016. [Google Scholar] [CrossRef] [PubMed]
- Matija, D.; Anne-Ruxandra, C.; Benoit, C.; Mary, G.; Samuel, J.P.; Murat, T. Evidence for Network Evolution in an Arabidopsis Interactome Map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Huffaker, A.; Bryan, A.C.; Tax, F.E.; Ryan, C.A. PEPR2 is a second receptor for the Pep1 and Pep2 peptides and contributes to defense responses in Arabidopsis. Plant Cell 2010, 22, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Krupkova, E.; Immerzeel, P.; Pauly, M.; Schmulling, T. The TUMOROUS SHOOT DEVELOPMENT2 gene of Arabidopsis encoding a putative methyltransferase is required for cell adhesion and co-ordinated plant development. Plant J. Cell Mol. Biol. 2007, 50, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. RNA-seq analysis of short fiber mutants Ligon-lintless-1 (Li 1) and -2 (Li 2) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, H.; Liu, K.; Jian, G.; Qi, F.; Si, N. Large-scale identification of Gossypium hirsutum genes associated with Verticillium dahliae by comparative transcriptomic and reverse genetics analysis. PLoS ONE 2017, 12, e0181609. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhu, L.; Tu, L.; Liu, L.; Yuan, D.; Jin, L.; Long, L.; Zhang, X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J. Exp. Bot. 2011, 62, 5607–5621. [Google Scholar] [CrossRef]
- Artico, S.; Ribeiro-Alves, M.; Oliveira-Neto, O.B.; de Macedo, L.L.; Silveira, S.; Grossi-de-Sa, M.F.; Martinelli, A.P.; Alves-Ferreira, M. Transcriptome analysis of Gossypium hirsutum flower buds infested by cotton boll weevil (Anthonomus grandis) larvae. BMC Genom. 2014, 15, 854. [Google Scholar] [CrossRef]
- Bowman, M.J.; Park, W.; Bauer, P.J.; Udall, J.A.; Page, J.T.; Raney, J.; Scheffler, B.E.; Jones, D.C.; Campbell, B.T. RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS ONE 2013, 8, e82634. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 20582. [Google Scholar] [CrossRef] [PubMed]
- Stiff, M.R.; Haigler, C.H. Recent Advances in Cotton Fiber Development; Flowering and Fruiting in Cotton Tennessee; The Cotton Foundation: Cordova, TN, USA, 2012; pp. 163–192. [Google Scholar]
- Yang, Z.; Zhang, C.; Yang, X.; Liu, K.; Wu, Z.; Zhang, X.; Zheng, W.; Xun, Q.; Liu, C.; Lu, L. PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef]
- He, X.C.; Qin, Y.M.; Xu, Y.; Hu, C.Y.; Zhu, Y.X. Molecular cloning, expression profiling, and yeast complementation of 19 beta-tubulin cDNAs from developing cotton ovules. J. Exp. Bot. 2008, 59, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Javelle, M.; Vernoud, V.; Rogowsky, P.M.; Ingram, G.C. Epidermis: The formation and functions of a fundamental plant tissue. New Phytol. 2011, 189, 17–39. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, J.; Liu, D.; Wei, Y.L.; Wang, Y.; Li, X.B. Characterization of bHLH/HLH genes that are involved in brassinosteroid (BR) signaling in fiber development of cotton (Gossypium hirsutum). BMC Plant Biol. 2018, 18, 304. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hu, L.; Fan, J.; Amombo, E.; Khaldun, A.B.M.; Zheng, Y.; Chen, L. Cotton GhERF38 gene is involved in plant response to salt/drought and ABA. Ecotoxicology 2017, 26, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Petrovska, B.; Jerabkova, H.; Kohoutova, L.; Cenklova, V.; Pochylova, Z.; Gelova, Z.; Kocarova, G.; Vachova, L.; Kurejova, M.; Tomastikova, E.; et al. Overexpressed TPX2 causes ectopic formation of microtubular arrays in the nuclei of acentrosomal plant cells. J. Exp. Bot. 2013, 64, 4575–4587. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | L1D10 | L2D10 | Description in Arabidopsis thaliana |
|---|---|---|---|
| Transcription factors | |||
| Gh_A11G0692 | 6.49 | 0.9 | homeobox-leucine zipper protein 4 |
| Gh_D07G0362 | 8.39 | 1.6 | Integrase-type DNA-binding superfamily protein |
| Gh_D10G0270 | 49.31 | 11.65 | homeobox-3 |
| Arginine biosynthesis | |||
| Gh_A05G2143 | 11.59 | 2 | arginase |
| Gh_A11G2354 | 6.86 | 1.47 | glutamate dehydrogenase 1 |
| Brassinosteroid biosynthesis | |||
| Gh_D05G0184 | 19.25 | 2.36 | Cytochrome P450 superfamily protein |
| Gh_A05G0122 | 41.06 | 11.02 | Cytochrome P450 superfamily protein |
| Metabolic pathways | |||
| Gh_A03G0507 | 19.78 | 1.02 | Glycosyl hydrolases family 32 protein |
| Gh_A05G3473 | 66.67 | 5.21 | 10-formyltetrahydrofolate synthetase |
| Gh_A05G3997 | 54.47 | 5.12 | 4-coumarate:CoA ligase 2 |
| Gh_A03G1938 | 10.11 | 1.62 | N-acetylglucosamine-1-phosphate uridylyltransferase 2 |
| Gh_A05G0479 | 5.74 | 1.09 | aldehyde dehydrogenase 11A3 |
| Gh_D07G0692 | 13.39 | 2.57 | UDP-glucose 6-dehydrogenase family protein |
| Gh_D02G0761 | 9.2 | 1.95 | Thiamin diphosphate-binding fold (THDP-binding) protein |
| Gh_A10G2327 | 7.37 | 1.75 | ACT domain-containing small subunit of acetolactate synthase protein |
| Gh_A09G2449 | 20.78 | 5.87 | NAD(P)-binding Rossmann-fold superfamily protein |
| mRNA surveillance pathway | |||
| Gh_A05G0238 | 6.55 | 0.21 | homolog of CFIM-25 |
| Gh_A03G1654 | 133.17 | 39.45 | poly(A) binding protein 2 |
| Gh_D02G2070 | 108.02 | 33.65 | poly(A) binding protein 2 |
| Plant hormone signal transduction | |||
| Gh_D11G1989 | 15.5 | 5.38 | Auxin-responsive GH3 family protein |
| Gh_D09G1585 | 14.32 | 1.17 | PYR1-like 4 |
| Gh_A09G2421 | 11.06 | 1.06 | PYR1-like 4 |
| Gene Name | L1D10 | L2D10 | Description in A. thaliana |
|---|---|---|---|
| Phenylalanine, tyrosine and tryptophan biosynthesis | |||
| Gh_A13G0258 | 1.59 | 12.58 | aspartate aminotransferase |
| Gh_A13G0603 | 1.36 | 7.68 | 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase |
| Biosynthesis of amino acids | |||
| Gh_A07G0619 | 0.42 | 9.8 | 3-phosphoserine phosphatase |
| Nicotinate and nicotinamide metabolism | |||
| Gh_D01G1478 | 1.61 | 6.54 | quinolinate phoshoribosyltransferase |
| Gh_A06G0128 | 5.31 | 48.44 | GA requiring 3 |
| Phagosome | |||
| Gh_D13G1883 | 11.97 | 96.74 | calreticulin 1a |
| Transcription factors | |||
| Gh_A05G0528 | 2.73 | 11.38 | WRKY DNA-binding protein 11 |
| Gh_A13G1728 | 9.91 | 46.36 | Zinc finger C-x8-C-x5-C-x3-H type family protein |
| Genes id | L1D20 | L2D20 | Description |
|---|---|---|---|
| Biosynthesis of unsaturated fatty acids | |||
| Gh_Sca007938G01 | 10.65 | 1.5 | beta-ketoacyl reductase 1 |
| Gh_D01G0186 | 139.55 | 29.59 | acyl-CoA oxidase 3 |
| Gh_A13G2171 | 112.41 | 36.72 | acyl-CoA oxidase 4 |
| Butanoate metabolism | |||
| Gh_D12G0436 | 13.79 | 1.9 | short-chain dehydrogenase-reductase B |
| Gh_A12G1414 | 56.97 | 16.99 | glutamate decarboxylase 4 |
| Gh_A07G0810 | 110.4 | 46.02 | glyoxylate reductase 1 |
| Cutin, suberine and wax biosynthesis | |||
| Gh_A07G0991 | 5.36 | 0.72 | Jojoba acyl CoA reductase-related male sterility protein |
| Gh_D05G1294 | 67.39 | 11.9 | Glucose-methanol-choline (GMC) oxidoreductase family protein |
| Gh_D01G1400 | 172.1 | 49 | Caleosin-related family protein |
| Fatty acid elongation | |||
| Gh_A01G1563 | 150.26 | 29.07 | 3-ketoacyl-CoA synthase 6 |
| Gh_D01G1810 | 213.82 | 54.5 | 3-ketoacyl-CoA synthase 6 |
| Gh_A09G0749 | 19.87 | 7.06 | 3-ketoacyl-acyl carrier protein synthase I |
| Gh_Sca006141G01 | 23.73 | 8.81 | acetyl-CoA carboxylase carboxyl transferase subunit β |
| Phagosome | |||
| Gh_A08G2381 | 635.59 | 71.34 | β-6 tubulin |
| Gh_A11G2095 | 1198.32 | 173.46 | tubulin α-3 |
| Gh_Sca012883G01 | 2274.54 | 359.05 | tubulin β 8 |
| Gh_D08G1960 | 460.89 | 75.32 | β-6 tubulin |
| Gh_D05G1052 | 572.74 | 176.55 | tubulin β 8 |
| Gh_A02G0819 | 357.99 | 136.45 | tubulin α-2 chain |
| Gh_D13G1047 | 259.25 | 106.17 | tubulin α-3 |
| Transcription factors | |||
| Gh_D12G0607 | 16.71 | 4.38 | basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| Gh_A07G1171 | 82.82 | 17.27 | basic region/leucine zipper motif 53 |
| Gh_D05G0463 | 37.9 | 13.35 | CCCH-type zinc finger family protein |
| Gh_A12G0561 | 40.16 | 15.17 | B-box zinc finger family protein |
| Gh_D05G2596 | 267.05 | 106.76 | K-box region and MADS-box transcription factor family protein |
| Gh_A12G1244 | 21.19 | 4.2 | MYB-like 102 |
| Gh_D08G0157 | 10.98 | 2.96 | AP2/B3 transcription factor family protein |
| Gh_D02G0043 | 40.69 | 11.26 | WRKY family transcription factor family protein |
| Genes Name | L1D20 | L2D20 | Description in A. thaliana |
|---|---|---|---|
| Biosynthesis of secondary metabolites | |||
| Gh_D06G0096 | 0.12 | 12.6 | cytochrome P450, family 82, subfamily C, polypeptide 4 |
| Gh_D04G0605 | 0.38 | 9.89 | glycerol-3-phosphate acyltransferase 5 |
| Gh_A13G0603 | 1.47 | 10.65 | 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase |
| Gh_D05G2554 | 4.26 | 22.71 | acetyl-CoA carboxylase 1 |
| Nitrogen metabolism | |||
| Gh_Sca069862G01 | 18.38 | 189.16 | glutamine synthase clone R1 |
| Gh_D07G1773 | 54.01 | 333.88 | glutamine synthase clone R1 |
| Gh_A01G1586 | 1.4 | 7.56 | nitrate reductase 2 |
| Gh_D01G1872 | 7.83 | 26.87 | nitrate reductase 2 |
| Phenylpropanoid biosynthesis | |||
| Gh_D10G0473 | 11.3 | 63.26 | 4-coumarate:CoA ligase 1 |
| Gh_D08G1135 | 15.62 | 62.86 | O-methyltransferase 1 |
| Gh_A08G0711 | 32.62 | 105.85 | Peroxidase superfamily protein |
| Gh_A04G1032 | 29.29 | 80.06 | S-adenosyl-L-methionine-dependent methyltransferases |
| Zeatin biosynthesis | |||
| Gh_D05G1813 | 9.23 | 43.01 | cytokinin oxidase 5 |
| Gh_A05G0290 | 6.64 | 25.84 | cytokinin oxidase 7 |
| Gh_A05G1631 | 7.6 | 26.27 | cytokinin oxidase 5 |
| Gh_D05G0391 | 12.27 | 39.85 | cytokinin oxidase 7 |
| Transcription factors | |||
| Gh_A02G1575 | 9.12 | 28.68 | GRAS family transcription factor |
| Gh_D01G0974 | 102.38 | 371.7 | basic leucine-zipper 44 |
| Gh_A11G2875 | 3.42 | 15.35 | myb-like transcription factor family protein |
| Gh_A11G2522 | 3.52 | 19.11 | myb domain protein 120 |
| Gh_A05G3778 | 2.5 | 24.64 | LOB domain-containing protein 13 |
| Gh_D07G1330 | 0.53 | 5.69 | NAC domain transcriptional regulator protein |
| Gh_D06G0254 | 0.52 | 6.37 | GATA-type zinc finger protein with TIFY domain |
| Gh_A10G0516 | 2.06 | 26.94 | myb domain protein 26 |
| Gh_D08G1424 | 0.11 | 6.98 | WRKY DNA-binding protein 3 |
| Gene Name | KME | Arabidopsis ID | Function Description in Arabidopsis thaliana Gene |
|---|---|---|---|
| 10 DPA specific darkred module | |||
| Gh_D12G2172 | 0.991 | AT1G12240 | Glycosyl hydrolases family 32 protein |
| Gh_A10G1961 | 0.991 | AT2G37620 | actin 1 |
| Gh_A11G1087 | 0.997 | AT4G03100 | Rho GTPase activating protein with PAK-box [46] |
| Gh_D06G0618 | 0.991 | AT4G28950 | RHO-related protein from plants 9 |
| Gh_D05G0591 | 0.992 | AT4G31890 | ARM repeat superfamily protein |
| 15 DPA of L1 specific darkturquoise module | |||
| Gh_A02G1692 | 0.968 | AT3G51430 | Calcium-dependent phosphotriesterase superfamily protein |
| Gh_A07G0225 | 0.983 | AT4G32330 | TPX2 (targeting protein for Xklp2) protein family [47,48] |
| Gh_A12G0226 | 0.971 | AT2G15780 | Cupredoxin superfamily protein |
| Gh_D03G1054 | 0.973 | AT1G01630 | Sec14p-like phosphatidylinositol transfer family protein |
| Gh_D12G0026 | 0.964 | AT3G51895 | sulfate transporter 3;1 |
| 20 DPA of L1 specific violet module | |||
| Gh_A03G0347 | 0.967 | AT2G28790 | Pathogenesis-related thaumatin superfamily protein |
| Gh_A05G1879 | 0.981 | AT4G33580 | beta carbonic anhydrase 5 |
| Gh_A13G1012 | 0.965 | AT4G09510 | cytosolic invertase 2 |
| Gh_D06G1151 | 0.968 | AT4G16130 | arabinose kinase |
| Gh_D10G0295 | 0.974 | AT1G68810 | basic helix-loop-helix (bHLH) DNA-binding superfamily protein 30 [49] |
| Gh_D02G0740 | 0.97 | AT1G21460 | Nodulin MtN3 family protein |
| Gh_D07G0144 | 0.969 | AT5G20860 | Plant invertase/pectin methylesterase inhibitor superfamily [50] |
| Gh_D08G2725 | 0.973 | AT3G62690 | AtL5 |
| 20&30 DPA of L1 specific pink module | |||
| Gh_A08G1486 | 0.95 | AT5G47120 | BAX inhibitor 1 |
| Gh_A08G2508 | 0.944 | AT4G27000 | RNA-binding (RRM/RBD/RNP motifs) family protein |
| Gh_A11G2726 | 0.961 | AT3G13540 | myb domain protein 5 [51,52] |
| Gh_D03G1843 | 0.958 | AT1G32050 | SCAMP family protein |
| Gh_D09G0347 | 0.962 | AT5G14240 | Thioredoxin superfamily protein |
| 30 DPA of L1 specific darkgrey module | |||
| Gh_A09G1137 | 0.981 | AT5G04160 | UDP-URONIC acid transporter 1 [53,54]; |
| Gh_A10G1754 | 0.989 | AT5G06390 | FASCICLIN-like arabinogalactan protein 17 precursor |
| Gh_D01G1697 | 0.982 | AT1G20550 | O-fucosyltransferase family protein |
| Gh_D08G2152 | 0.985 | AT2G33170 | Leucine-rich repeat receptor-like protein kinase family protein [55] |
| Gh_D10G1056 | 0.99 | AT5G64030 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein [56] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B.; et al. Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis. Genes 2019, 10, 119. https://doi.org/10.3390/genes10020119
Zou X, Liu A, Zhang Z, Ge Q, Fan S, Gong W, Li J, Gong J, Shi Y, Tian B, et al. Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis. Genes. 2019; 10(2):119. https://doi.org/10.3390/genes10020119
Chicago/Turabian StyleZou, Xianyan, Aiying Liu, Zhen Zhang, Qun Ge, Senmiao Fan, Wankui Gong, Junwen Li, Juwu Gong, Yuzhen Shi, Baoming Tian, and et al. 2019. "Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis" Genes 10, no. 2: 119. https://doi.org/10.3390/genes10020119
APA StyleZou, X., Liu, A., Zhang, Z., Ge, Q., Fan, S., Gong, W., Li, J., Gong, J., Shi, Y., Tian, B., Wang, Y., Liu, R., Lei, K., Zhang, Q., Jiang, X., Feng, Y., Zhang, S., Jia, T., Zhang, L., ... Shang, H. (2019). Co-Expression Network Analysis and Hub Gene Selection for High-Quality Fiber in Upland Cotton (Gossypium hirsutum) Using RNA Sequencing Analysis. Genes, 10(2), 119. https://doi.org/10.3390/genes10020119