Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective



Abstract
1. The Physiology of Vision
2. Retinogenesis
3. The Genetics of Retinal Development
4. Morphological and Molecular Recapitulation of the Human Fetal Retina
5. Light Responsiveness and Synaptic Transmission of the Fetal Retina and Stem Cell-Derived Retinal Organoids
6. Improved Retinal Organoid Modelling
7. The Apical CRB and PAR Complexes
8. The Localization of the Mammalian Retinal CRB Complex
9. CRB1 and Leber Congenital Amaurosis
10. Gene Augmentation for Hereditary Retinopathies
11. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vu, H.T.V.; Keeffe, J.E.; McCarty, C.A.; Taylor, H.R. Impact of unilateral and bilateral vision loss on quality of life. Br. J. Ophthalmol. 2005, 89, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.N.; Sheibani, N.; Albert, D.M. Retinal light toxicity. Eye 2011, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, T.; Gruss, P. Generating neuronal diversity in the retina: One for nearly all. Trends Neurosci. 2002, 25, 32–38. [Google Scholar] [CrossRef]
- Xiang, M. Intrinsic control of mammalian retinogenesis. Cell. Mol. Life Sci. 2013, 70, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.G.; Hankins, M.W. Non-rod, non-cone photoreception in the vertebrates. Prog. Retin. Eye Res. 2002, 21, 507–527. [Google Scholar] [CrossRef]
- Ebrey, T.; Koutalos, Y. Vertebrate photoreceptors. Prog. Retin. Eye Res. 2001, 20, 49–94. [Google Scholar] [CrossRef]
- Xiao, M.; Hendrickson, A. Spatial and Temporal Expression of Short, Long/Medium, or Both Opsins in Human Fetal Cones. J. Comp. Neurol. 2000, 559, 545–559. [Google Scholar] [CrossRef]
- Roorda, A.; Williams, D.R. The arrangement of the three cone classes in the living human eye. Nature 1999, 397, 520–522. [Google Scholar] [CrossRef]
- Grimes, W.N.; Songco-Aguas, A.; Rieke, F. Parallel Processing of Rod and Cone Signals: Retinal Function and Human Perception. Annu. Rev. Vis. Sci 2018, 4, 123–141. [Google Scholar] [CrossRef]
- Franze, K.; Grosche, J.; Skatchkov, S.N.; Schinkinger, S.; Foja, C.; Schild, D.; Uckermann, O.; Travis, K.; Reichenbach, A.; Guck, J. Muller cells are living optical fibers in the vertebrate retina. Proc. Natl. Acad. Sci. USA 2007, 104, 8287–8292. [Google Scholar] [CrossRef] [PubMed]
- Labin, A.M.; Ribak, E.N. Retinal glial cells enhance human vision acuity. Phys. Rev. Lett. 2010, 104, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Labin, A.M.; Safuri, S.K.; Ribak, E.N.; Perlman, I. Muller cells separate between wavelengths to improve day vision with minimal effect upon night vision. Nat. Commun. 2014, 5, 4319. [Google Scholar] [CrossRef] [PubMed]
- Kefalov, V.J. Rod and cone visual pigments and phototransduction through pharmacological, genetic, and physiological approaches. J. Biol. Chem. 2012, 287, 1635–1641. [Google Scholar] [CrossRef]
- Lee, B.B.; Martin, P.R.; Grünert, U. Retinal connectivity and primate vision. Prog. Retin. Eye Res. 2010, 29, 622–639. [Google Scholar] [CrossRef]
- Rivlin-Etzion, M.; Grimes, W.N.; Rieke, F. Flexible Neural Hardware Supports Dynamic Computations in Retina. Trends Neurosci. 2018, 41, 224–237. [Google Scholar] [CrossRef]
- Kolb, H.; Marshak, D. The midget pathways of the primate retina. Doc. Ophthalmol. 2003, 106, 67–81. [Google Scholar] [CrossRef]
- Dacey, D.M. The mosaic of midget ganglion cells in the human retina. J. Neurosci. 1993, 13, 5334–5355. [Google Scholar] [CrossRef]
- Provis, J.M.; Penfold, P.L.; Cornish, E.E.; Sandercoe, T.M.; Madigan, M.C. Anatomy and development of the macula: Specialisation and the vulnerability to macular degeneration. Clin. Exp. Optom. 2005, 88, 269–281. [Google Scholar] [CrossRef]
- Ellis, E.M.; Gauvain, G.; Sivyer, B.; Murphy, G.J. Shared and distinct retinal input to the mouse superior colliculus and dorsal lateral geniculate nucleus. J. Neurophysiol. 2016, 116, 602–610. [Google Scholar] [CrossRef]
- Erskine, L.; Herreral, E. Connecting the retina to the brain. ASN Neuro 2015, 6. [Google Scholar] [CrossRef]
- Ahmadlou, M.; Zweifel, L.S.; Heimel, J.A. Functional modulation of primary visual cortex by the superior colliculus in the mouse. Nat. Commun. 2018, 9, 3895. [Google Scholar] [CrossRef] [PubMed]
- Warner, C.E.; Kwan, W.C.; Wright, D.; Johnston, L.A.; Egan, G.F.; Bourne, J.A. Preservation of vision by the pulvinar following early-life primary visual cortex lesions. Curr. Biol. 2015, 25, 424–434. [Google Scholar] [CrossRef]
- Ashtari, M.; Zhang, H.; Cook, P.A.; Cyckowski, L.L.; Shindler, K.S.; Marshall, K.A.; Aravand, P.; Vossough, A.; Gee, J.C.; Maguire, A.M.; et al. Plasticity of the human visual system after retinal gene therapy in patients with Leber ’ s congenital amaurosis. Sci. Transl. Med. 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Benhar, I.; London, A.; Schwartz, M. The privileged immunity of immune privileged organs: The case of the eye. Front. Immunol. 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Kenyon, K.L.; Zaghloul, N.; Moody, S.A. Transcription factors of the anterior neural plate alter cell movements of epidermal progenitors to specify a retinal fate. Dev. Biol. 2001, 240, 77–91. [Google Scholar] [CrossRef]
- Li, H.; Tierney, C.; Wen, L.; Wu, J.Y.; Rao, Y. A single morphogenetic field gives rise to two retina primordia under the influence of the prechordal plate. Development 1997, 124, 603–615. [Google Scholar]
- Heavner, W.; Pevny, L. Eye development and retinogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Zaghloul, N.A.; Yan, B.; Moody, S.A. Step-wise specification of retinal stem cells during normal embryogenesis. Biol. Cell 2005, 97, 321–337. [Google Scholar] [CrossRef]
- Graw, J. Eye development. Curr. Top. Dev. Biol. 2010, 90, 343–386. [Google Scholar]
- O’Rahilly, R. The prenatal development of the human eye. Exp. Eye Res. 1975, 21, 93–112. [Google Scholar] [CrossRef]
- Randlett, O.; Norden, C.; Harris, W.A. The vertebrate retina: A model for neuronal polarization in vivo. Dev. Neurobiol. 2011, 71, 567–583. [Google Scholar] [CrossRef]
- Baye, L.M.; Link, B.A. Nuclear migration during retinal development. Brain Res. 2008, 1192, 29–36. [Google Scholar] [CrossRef]
- Baye, L.M.; Link, B.A. Interkinetic Nuclear Migration and the Selection of Neurogenic Cell Divisions during Vertebrate Retinogenesis. J. Neurosci. 2007, 27, 10143–10152. [Google Scholar] [CrossRef]
- Norden, C.; Young, S.; Link, B.A.; Harris, W.A. Actomyosin Is the Main Driver of Interkinetic Nuclear Migration in the Retina. Cell 2009, 138, 1195–1208. [Google Scholar] [CrossRef]
- Cayouette, M. The orientation of cell division influences cell-fate choice in the developing mammalian retina. Development 2003, 130, 2329–2339. [Google Scholar] [CrossRef][Green Version]
- Yang, X.J. Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Semin. Cell Dev. Biol. 2004, 15, 91–103. [Google Scholar] [CrossRef]
- Jin, K. Transitional Progenitors during Vertebrate Retinogenesis. Mol. Neurobiol. 2017, 54, 3565–3576. [Google Scholar] [CrossRef]
- Cepko, C.L.; Austin, C.P.; Yang, X.; Alexiades, M.; Ezzeddine, D. Cell fate determination in the vertebrate retina. Proc. Natl. Acad. Sci. USA 1996, 93, 589–595. [Google Scholar] [CrossRef]
- Aldiri, I.; Xu, B.; Wang, L.; Chen, X.; Hiler, D.; Griffiths, L.; Valentine, M.; Shirinifard, A.; Thiagarajan, S.; Sablauer, A.; et al. The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and Tumorigenesis. Neuron 2017, 94, 550–568.e10. [Google Scholar] [CrossRef]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.R.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779.e4. [Google Scholar] [CrossRef]
- Reese, B.E.; Harvey, A.R.; Tan, S.S. Radial and tangential dispersion patterns in the mouse retina are cell-class specific. Proc. Natl. Acad. Sci. USA 1995, 92, 2494–2498. [Google Scholar] [CrossRef]
- Reese, B.E.; Necessary, B.D.; Tam, P.P.; Faulkner-Jones, B.; Tan, S.S. Clonal expansion and cell dispersion in the developing mouse retina. Eur. J. Neurosci. 1999, 11, 2965–2978. [Google Scholar] [CrossRef]
- Pearson, R.A.; Lüneborg, N.L.; Becker, D.L.; Mobbs, P. Gap junctions modulate interkinetic nuclear movement in retinal progenitor cells. J. Neurosci. 2005, 25, 10803–10814. [Google Scholar] [CrossRef][Green Version]
- Amini, R.; Rocha-martins, M.; Norden, C. Neuronal Migration and Lamination in the Vertebrate Retina. 2018, 11, 1–16. [Google Scholar] [CrossRef]
- Kay, J.N. Radial migration: Retinal neurons hold on for the ride. J. Cell Biol. 2016, 215, 147–149. [Google Scholar] [CrossRef]
- Icha, J.; Kunath, C.; Martins, M.R.; Norden, C. Independent modes of ganglion cell translocation ensure correct lamination of the zebrafish retina. J. Cell Biol. 2016, 215, 259–275. [Google Scholar] [CrossRef]
- Reese, B.E.; Galli-Resta, L. The role of tangential dispersion in retinal mosaic formation. Prog. Retin. Eye Res. 2002, 21, 153–168. [Google Scholar] [CrossRef]
- Reese, B.E.; Keeley, P.W. Design principles and developmental mechanisms underlying retinal mosaics. Biol. Rev. Camb. Philos. Soc. 2015, 90, 854–876. [Google Scholar] [CrossRef]
- Davidson, E.H. cis -Regulatory Modules, and the Structure/Function Basis. In The Regulatory Genome Gene Regulatory Networks In Development And Evolution; Academic Press: Cambridge, MA, USA, 2006; pp. 31–86. [Google Scholar]
- Peter, I.S.; Davidson, E.H. Evolution of gene regulatory networks controlling body plan development. Cell 2011, 144, 970–985. [Google Scholar] [CrossRef]
- Allan, D.W.; Thor, S. Transcriptional selectors, masters, and combinatorial codes: Regulatory principles of neural subtype specification. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 505–528. [Google Scholar] [CrossRef] [PubMed]
- Suryamohan, K.; Halfon, M.S. Identifying transcriptional cis-regulatory modules in animal genomes. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 59–84. [Google Scholar] [CrossRef]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.L.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef]
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Hu, B.; Mao, Y.; Chen, Y.; Yan, L.; Yong, J.; Dong, J.; Wei, Y.; Wang, W.; et al. Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis. PLOS Biol. 2019, 17, e3000365. [Google Scholar] [CrossRef]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 116, 10824–10833. [Google Scholar] [CrossRef]
- Phillips, M.J.; Jiang, P.; Howden, S.; Barney, P.; Min, J.; York, N.W.; Chu, L.-F.; Capowski, E.E.; Cash, A.; Jain, S.; et al. A Novel Approach to Single Cell RNA-Sequence Analysis Facilitates In Silico Gene Reporting of Human Pluripotent Stem Cell-Derived Retinal Cell Types. Stem Cells 2018, 36, 313–324. [Google Scholar] [CrossRef]
- Collin, J.; Queen, R.; Zerti, D.; Dorgau, B.; Hussain, R.; Coxhead, J.; Cockell, S.; Lako, M. Deconstructing Retinal Organoids: Single Cell RNA-Seq Reveals the Cellular Components of Human Pluripotent Stem Cell-Derived Retina. Stem Cells 2019, 37, 593–598. [Google Scholar] [CrossRef]
- Langer, K.B.; Ohlemacher, S.K.; Phillips, M.J.; Fligor, C.M.; Jiang, P.; Gamm, D.M.; Meyer, J.S. Retinal Ganglion Cell Diversity and Subtype Specification from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1282–1293. [Google Scholar] [CrossRef]
- Mao, X.; An, Q.; Xi, H.; Yang, X.-J.; Zhang, X.; Yuan, S.; Wang, J.; Hu, Y.; Liu, Q.; Fan, G. Single-Cell RNA Sequencing of hESC-Derived 3D Retinal Organoids Reveals Novel Genes Regulating RPC Commitment in Early Human Retinogenesis. Stem Cell Rep. 2019, 13, 747–760. [Google Scholar] [CrossRef]
- Kim, J.W.; Yang, H.J.; Brooks, M.J.; Zelinger, L.; Karakülah, G.; Gotoh, N.; Boleda, A.; Gieser, L.; Giuste, F.; Whitaker, D.T.; et al. NRL-Regulated Transcriptome Dynamics of Developing Rod Photoreceptors. Cell Rep. 2016, 17, 2460–2473. [Google Scholar] [CrossRef]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126.e5. [Google Scholar] [CrossRef]
- Kim, J.W.; Yang, H.J.; Oel, A.P.; Brooks, M.J.; Jia, L.; Plachetzki, D.C.; Li, W.; Allison, W.T.; Swaroop, A. Recruitment of Rod Photoreceptors from Short-Wavelength-Sensitive Cones during the Evolution of Nocturnal Vision in Mammals. Dev. Cell 2016, 37, 520–532. [Google Scholar] [CrossRef]
- Mo, A.; Luo, C.; Davis, F.P.; Mukamel, E.A.; Henry, G.L.; Nery, J.R.; Urich, M.A.; Picard, S.; Lister, R.; Eddy, S.R.; et al. Epigenomic landscapes of retinal rods and cones. Elife 2016, 5, 1–29. [Google Scholar] [CrossRef]
- Hughes, A.E.O.; Enright, J.M.; Myers, C.A.; Shen, S.Q.; Corbo, J.C. Cell Type-Specific Epigenomic Analysis Reveals a Uniquely Closed Chromatin Architecture in Mouse Rod Photoreceptors. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Norrie, J.L.; Lupo, M.S.; Xu, B.; Al Diri, I.; Valentine, M.; Putnam, D.; Griffiths, L.; Zhang, J.; Johnson, D.; Easton, J.; et al. Nucleome Dynamics during Retinal Development. Neuron 2019, 104, 521–528.e11. [Google Scholar] [CrossRef]
- Blackshaw, S.; Harpavat, S.; Trimarchi, J.; Cai, L.; Huang, H.; Kuo, W.P.; Weber, G.; Lee, K.; Fraioli, R.E.; Cho, S.-H.; et al. Genomic Analysis of Mouse Retinal Development. PLoS Biol. 2004, 2, e247. [Google Scholar] [CrossRef]
- Lo Giudice, Q.; Leleu, M.; La Manno, G.; Fabre, P.J. Single-cell transcriptional logic of cell-fate specification and axon guidance in early-born retinal neurons. Development 2019, 146, dev178103. [Google Scholar] [CrossRef]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Diacou, R.; Zhao, Y.; Zheng, D.; Cvekl, A.; Liu, W. Six3 and Six6 Are Jointly Required for the Maintenance of Multipotent Retinal Progenitors through Both Positive and Negative Regulation. Cell Rep. 2018, 25, 2510–2523.e4. [Google Scholar] [CrossRef]
- Liu, W.; Cvekl, A. Six3 in a small population of progenitors at E8.5 is required for neuroretinal specification via regulating cell signaling and survival in mice. Dev. Biol. 2017, 428, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lagutin, O.; Swindell, E.; Jamrich, M.; Oliver, G. Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J. Clin. Investig. 2010, 120, 3568–3577. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.; Novak, J.; Liang, M.Y.; Basu, S.; Ploder, L.; Hawes, N.L.; Vidgen, D.; Hoover, F.; Goldman, D.; Kalnins, V.I.; et al. Ocular retardation mouse caused by Chx10 homeobox null allele: Impaired retinal progenitor proliferation and bipolar cell differentiation. Nat. Genet. 1996, 12, 376–384. [Google Scholar] [CrossRef]
- Li, X.; Perissi, V.; Liu, F.; Rose, D.W.; Rosenfeld, M.G. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science 2002, 297, 1180–1183. [Google Scholar] [CrossRef]
- Marquardt, T.; Ashery-Padan, R.; Andrejewski, N.; Scardigli, R.; Guillemot, F.; Gruss, P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 2001, 105, 43–55. [Google Scholar] [CrossRef]
- Gordon, P.J.; Yun, S.; Clark, A.M.; Monuki, E.S.; Murtaugh, L.C.; Levine, E.M. Lhx2 balances progenitor maintenance with neurogenic output and promotes competence state progression in the developing retina. J. Neurosci. 2013, 33, 12197–12207. [Google Scholar] [CrossRef]
- Zibetti, C.; Liu, S.; Wan, J.; Qian, J.; Blackshaw, S. Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun. Biol. 2019, 2, 1–13. [Google Scholar] [CrossRef]
- Tétreault, N.; Champagne, M.-P.; Bernier, G. The LIM homeobox transcription factor Lhx2 is required to specify the retina field and synergistically cooperates with Pax6 for Six6 trans-activation. Dev. Biol. 2009, 327, 541–550. [Google Scholar] [CrossRef]
- Azuma, N.; Nishina, S.; Yanagisawa, H.; Okuyama, T.; Yamada, M. PAX6 missense mutation in isolated foveal hypoplasia. Nat. Genet. 1996, 13, 141–142. [Google Scholar] [CrossRef]
- Voronina, V.A.; Kozhemyakina, E.A.; O’Kernick, C.M.; Kahn, N.D.; Wenger, S.L.; Linberg, J.V.; Schneider, A.S.; Mathers, P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2004, 13, 315–322. [Google Scholar] [CrossRef]
- Mattar, P.; Ericson, J.; Blackshaw, S.; Cayouette, M. A conserved regulatory logic controls temporal identity in mouse neural progenitors. Neuron 2015, 85, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Jolicoeur, C.; Ramamurthy, V.; Cayouette, M. Ikaros Confers Early Temporal Competence to Mouse Retinal Progenitor Cells. Neuron 2008, 60, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Hamon, A.; Masson, C.; Bitard, J.; Gieser, L.; Roger, J.E.; Perron, M. Retinal Degeneration Triggers the Activation of YAP/TEAD in Reactive Müller Cells. Investig. Opthalmol. Vis. Sci. 2017, 58, 1941. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, R.; Lee, J.H.J.; Shin, J.; Nickas, J.; Kim, S.; Cho, S.H. Yap is essential for retinal progenitor cell cycle progression and RPE cell fate acquisition in the developing mouse eye. Dev. Biol. 2016, 419, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Rueda, E.M.; Hall, B.M.; Hill, M.C.; Swinton, P.G.; Tong, X.; Martin, J.F.; Poché, R.A. The Hippo Pathway Blocks Mammalian Retinal Müller Glial Cell Reprogramming. Cell Rep. 2019, 27, 1637–1649.e6. [Google Scholar] [CrossRef]
- Hamon, A.; García-García, D.; Ail, D.; Bitard, J.; Chesneau, A.; Dalkara, D.; Locker, M.; Roger, J.E.; Perron, M. Linking YAP to Müller Glia Quiescence Exit in the Degenerative Retina. Cell Rep. 2019, 27, 1712–1725.e6. [Google Scholar] [CrossRef]
- Furukawa, T.; Mukherjee, S.; Bao, Z.Z.; Morrow, E.M.; Cepko, C.L. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 2000, 26, 383–394. [Google Scholar] [CrossRef]
- Hojo, M.; Ohtsuka, T.; Hashimoto, N.; Gradwohl, G.; Guillemot, F.; Kageyama, R. Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development 2000, 127, 2515–2522. [Google Scholar]
- Poché, R.A.; Furuta, Y.; Chaboissier, M.-C.; Schedl, A.; Behringer, R.R. Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Müller glial cell development. J. Comp. Neurol. 2008, 510, 237–250. [Google Scholar] [CrossRef]
- de Melo, J.; Zibetti, C.; Clark, B.S.; Hwang, W.; Miranda-Angulo, A.L.; Qian, J.; Blackshaw, S. Lhx2 Is an Essential Factor for Retinal Gliogenesis and Notch Signaling. J. Neurosci. 2016, 36, 2391–2405. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Sullivan, L.S.; Mintz-Hittner, H.A.; Birch, D.; Heckenlively, J.R.; Freund, C.L.; Mclnnes, R.R.; Daiger, S.P. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am. J. Hum. Genet. 1998, 63, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Swain, P.K.; Chen, S.; Wang, Q.L.; Affatigato, L.M.; Coats, C.L.; Brady, K.D.; Fishman, G.A.; Jacobson, S.G.; Swaroop, A.; Stone, E.; et al. Mutations in the cone-rod homeobox gene are associated with the cone- rod dystrophy photoreceptor degeneration. Neuron 1997, 19, 1329–1336. [Google Scholar] [CrossRef]
- Newman, H.; Blumen, S.C.; Braverman, I.; Hanna, R.; Tiosano, B.; Perlman, I.; Ben-Yosef, T. Homozygosity for a recessive loss-of-function mutation of the NRL gene is associated with a variant of enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5361–5371. [Google Scholar] [CrossRef]
- DeAngelis, M.M.; Grimsby, J.L.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Novel mutations in the NRL gene and associated clinical findings in patients with dominant retinitis pigmentosa. Arch. Ophthalmol. 2002, 120, 369–375. [Google Scholar] [CrossRef]
- Gire, A.I.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1-2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar]
- Nishida, A.; Furukawa, A.; Koike, C.; Tano, Y.; Aizawa, S.; Matsuo, I.; Furukawa, T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci. 2003, 6, 1255–1263. [Google Scholar] [CrossRef]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Mears, A.J.; Kondo, M.; Swain, P.K.; Takada, Y.; Bush, R.A.; Saunders, T.L.; Sieving, P.A.; Swaroop, A. Nrl is required for rod photoreceptor development. Nat. Genet. 2001, 29, 447–452. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, H.; Ng, L.; Kim, J.W.; Hao, H.; Swaroop, A.; Forrest, D. Feedback induction of a photoreceptor-specific isoform of retinoid-related orphan nuclear receptor β by the rod transcription factor NRL. J. Biol. Chem. 2014, 289, 32469–32480. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rattner, A.; Nathans, J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef]
- Oh, E.C.T.; Khan, N.; Novelli, E.; Khanna, H.; Strettoi, E.; Swaroop, A. Transformation of cone precursors to functional rod photoreceptors by bZIP transcription factor NRL. Proc. Natl. Acad. Sci. USA 2007, 104, 1679–1684. [Google Scholar] [CrossRef]
- Wang, S.; Sengel, C.; Emerson, M.M.; Cepko, C.L. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev. Cell 2014, 30, 513–527. [Google Scholar] [CrossRef]
- Brzezinski, J.A.; Lamba, D.A.; Reh, T.A. Blimp1 controls photoreceptor versus bipolar cell fate choice during retinal development. Development 2010, 137, 619–629. [Google Scholar] [CrossRef]
- Ng, L.; Hurley, J.B.; Dierks, B.; Srinivas, M.; Saltó, C.; Vennström, B.; Reh, T.A.; Forrest, D. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 2001, 27, 94–98. [Google Scholar] [CrossRef]
- Roberts, M.R.; Hendrickson, A.; McGuire, C.R.; Reh, T.A. Retinoid X receptor γ is necessary to establish the S-opsin gradient in cone photoreceptors of the developing mouse retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2897–2904. [Google Scholar] [CrossRef]
- Roberts, M.R.; Srinivas, M.; Forrest, D.; De Escobar, G.M.; Reh, T.A. Making the gradient: Thyroid hormone regulates cone opsin expression in the develoninn mouse retina. Proc. Natl. Acad. Sci. USA 2006, 103, 6218–6223. [Google Scholar] [CrossRef]
- Emerson, M.M.; Surzenko, N.; Goetz, J.J.; Trimarchi, J.; Cepko, C.L. Otx2 and Onecut1 promote the fates of cone photoreceptors and horizontal cells and repress rod photoreceptors. Dev. Cell 2013, 26, 59–72. [Google Scholar] [CrossRef]
- Schick, E.; McCaffery, S.D.; Keblish, E.E.; Thakurdin, C.; Emerson, M.M. Lineage tracing analysis of cone photoreceptor associated cis-regulatory elements in the developing chicken retina. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Chow, R.L.; Volgyi, B.; Szilard, R.K.; Ng, D.; McKerlie, C.; Bloomfield, S.A.; Birch, D.G.; McInnes, R.R. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc. Natl. Acad. Sci. USA 2004, 101, 1754–1759. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, F.; Feng, L.; Luo, X.-J.; Liang, G.; Zeng, X.-Y.; Yi, J.-L.; Gan, L. Bhlhb5 is required for the subtype development of retinal amacrine and bipolar cells in mice. Dev. Dyn. 2014, 243, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y.; Fujitani, S.; Luo, H.; Qiu, F.; Burlison, J.; Long, Q.; Kawaguchi, Y.; Edlund, H.; MacDonald, R.J.; Furukawa, T.; et al. Ptf1a determines horizontal and amacrine cell fates during mouse retinal development. Development 2006, 133, 4439–4450. [Google Scholar] [CrossRef]
- Li, S.; Mo, Z.; Yang, X.; Price, S.M.; Shen, M.M.; Xiang, M. Foxn4 controls the genesis of amacrine and horizontal cells by retinal progenitors. Neuron 2004, 43, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Livesey, F.J.; Cepko, C.L.; Oliver, G. Prox1 function controls progenitor cell proliferation and horizontal cell genesis in the mammalian retina. Nat. Genet. 2003, 34, 53–58. [Google Scholar] [CrossRef]
- Wu, F.; Li, R.; Umino, Y.; Kaczynski, T.J.; Sapkota, D.; Li, S.; Xiang, M.; Fliesler, S.J.; Sherry, D.M.; Gannon, M.; et al. Onecut1 is essential for horizontal cell genesis and retinal integrity. J. Neurosci. 2013, 33, 13053–13065. [Google Scholar] [CrossRef]
- Liu, W.; Wang, J.H.; Xiang, M. Specific expression of the LIM/homeodomain protein Lim-1 in horizontal cells during retinogenesis. Dev. Dyn. 2000, 217, 320–325. [Google Scholar] [CrossRef]
- Edqvist, P.H.; Myers, S.M.; Hallböök, F. Early identification of retinal subtypes in the developing, pre-laminated chick retina using the transcription factors Prox1, Lim1, Ap2α, Pax6, Isl1, Isl2, Lim3 and Chx10. Eur. J. Histochem. 2006, 50, 147–154. [Google Scholar]
- Poché, R.A.; Kin, M.K.; Raven, M.A.; Furuta, Y.; Reese, B.E.; Behringer, R.R. Lim1 is essential for the correct laminar positioning of retinal horizontal cells. J. Neurosci. 2007, 27, 14099–14107. [Google Scholar] [CrossRef]
- Jin, K.; Jiang, H.; Xiao, D.; Zou, M.; Zhu, J.; Xiang, M. Tfap2a and 2b act downstream of Ptf1a to promote amacrine cell differentiation during retinogenesis. Mol. Brain 2015, 8, 1–14. [Google Scholar] [CrossRef]
- Elshatory, Y.; Everhart, D.; Deng, M.; Xie, X.; Barlow, R.B.; Gan, L. Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J. Neurosci. 2007, 27, 12707–12720. [Google Scholar] [CrossRef]
- Cherry, T.J.; Wang, S.; Bormuth, I.; Schwab, M.; Olson, J.; Cepko, C.L. NeuroD factors regulate cell fate and neurite stratification in the developing retina. J. Neurosci. 2011, 31, 7365–7379. [Google Scholar] [CrossRef]
- Ding, Q.; Chen, H.; Xie, X.; Libby, R.T.; Tian, N.; Gan, L. BARHL2 differentially regulates the development of retinal amacrine and ganglion neurons. J. Neurosci. 2009, 29, 3992–4003. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Kaczynski, T.J.; Sethuramanujam, S.; Li, R.; Jain, V.; Slaughter, M.; Mu, X. Two transcription factors, Pou4f2 and Isl1, are sufficient to specify the retinal ganglion cell fate. Proc. Natl. Acad. Sci. USA 2015, 112, E1559–E1568. [Google Scholar] [CrossRef]
- Mu, X.; Fu, X.; Beremand, P.D.; Thomas, T.L.; Klein, W.H. Gene-regulation logic in retinal ganglion cell development: Isl1 defines a critical branch distinct from but overlapping with Pou4f2. Proc. Natl. Acad. Sci. USA 2008, 105, 6942–6947. [Google Scholar] [CrossRef]
- Mao, C.A.; Wang, S.W.; Pan, P.; Klein, W.H. Rewiring the retinal ganglion cell gene regulatory network: Neurod1 promotes retinal ganglion cell fate in the absence of Math5. Development 2008, 135, 3379–3388. [Google Scholar] [CrossRef]
- Jiang, Y.; Ding, Q.; Xie, X.; Libby, R.T.; Lefebvre, V.; Gan, L. Transcription factors SOX4 and SOX11 function redundantly to regulate the development of mouse retinal ganglion cells. J. Biol. Chem. 2013, 288, 18429–18438. [Google Scholar] [CrossRef]
- Quinn, P.M.; Buck, T.M.; Mulder, A.A.; Ohonin, C.; Alves, C.H.; Vos, R.M.; Bialecka, M.; van Herwaarden, T.; van Dijk, E.H.C.; Talib, M.; et al. Human iPSC-Derived Retinas Recapitulate the Fetal CRB1 CRB2 Complex Formation and Demonstrate that Photoreceptors and Müller Glia Are Targets of AAV5. Stem Cell Rep. 2019, 12, 906–919. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Kantardzhieva, A.; Malysheva, A.; Meuleman, J.; Versteeg, I.; Levelt, C.; Klooster, J.; Geiger, S.; Seeliger, M.W.; Rashbass, P.; et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J. Cell Sci. 2004, 117, 4169–4177. [Google Scholar] [CrossRef]
- Alves, C.H.; Pellissier, L.P.; Vos, R.M.; Garcia Garrido, M.; Sothilingam, V.; Seide, C.; Beck, S.C.; Klooster, J.; Furukawa, T.; Flannery, J.G.; et al. Targeted ablation of Crb2 in photoreceptor cells induces retinitis pigmentosa. Hum. Mol. Genet. 2014, 23, 3384–3401. [Google Scholar] [CrossRef]
- Quinn, P.M.; Mulder, A.A.; Henrique Alves, C.; Desrosiers, M.; de Vries, S.I.; Klooster, J.; Dalkara, D.; Koster, A.J.; Jost, C.R.; Wijnholds, J. Loss of CRB2 in Müller glial cells modifies a CRB1-associated retinitis pigmentosa phenotype into a Leber congenital amaurosis phenotype. Hum. Mol. Genet. 2019, 28, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Patrakka, J.; Nukui, M.; Chi, L.; Niu, D.; Betsholtz, C.; Pikkarainen, T.; Pikkarainan, T.; Vainio, S.; Tryggvason, K. Deficiency in Crumbs homolog 2 (Crb2) affects gastrulation and results in embryonic lethality in mice. Dev. Dyn. 2011, 240, 2646–2656. [Google Scholar] [CrossRef]
- Lamont, R.E.; Tan, W.-H.; Innes, A.M.; Parboosingh, J.S.; Schneidman-Duhovny, D.; Rajkovic, A.; Pappas, J.; Altschwager, P.; DeWard, S.; Fulton, A.; et al. Expansion of phenotype and genotypic data in CRB2-related syndrome. Eur. J. Hum. Genet. 2016, 24, 1436–1444. [Google Scholar] [CrossRef]
- Charrier, L.E.; Loie, E.; Laprise, P. Mouse Crumbs3 sustains epithelial tissue morphogenesis in vivo. Sci. Rep. 2015, 5, 1–16. [Google Scholar] [CrossRef]
- Whiteman, E.L.; Fan, S.; Harder, J.L.; Walton, K.D.; Liu, C.-J.; Soofi, A.; Fogg, V.C.; Hershenson, M.B.; Dressler, G.R.; Deutsch, G.H.; et al. Crumbs3 Is Essential for Proper Epithelial Development and Viability. Mol. Cell. Biol. 2014, 34, 43–56. [Google Scholar] [CrossRef]
- Haverkamp, S.; Haeseleer, F.; Hendrickson, A. A comparison of immunocytochemical markers to identify bipolar cell types in human and monkey retina. Vis. Neurosci. 2003, 20, 589–600. [Google Scholar] [CrossRef]
- Abramov, I.; Gordon, J.; Hendrickson, A.; Hainline, L.; Dobson, V.; Labossiere, E. The retina of the newborn human infant. Science 1982, 217, 265–267. [Google Scholar] [CrossRef]
- Bibb, L.C.; Holt, J.K.; Tarttelin, E.E.; Hodges, M.D.; Gregory-Evans, K.; Rutherford, A.; Lucas, R.J.; Sowden, J.C.; Gregory-Evans, C.Y. Temporal and spatial expression patterns of the CRX transcription factor and its downstream targets. Critical differences during human and mouse eye development. Hum. Mol. Genet. 2001, 10, 1571–1579. [Google Scholar] [CrossRef]
- Hendrickson, A. A morphological comparison of foveal development in man and monkey. Eye 1992, 6, 136–144. [Google Scholar] [CrossRef]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.-A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.; Buck, T.M.; Ohonin, C.; Mikkers, H.M.M.; Wijnholds, J. Production of iPS-Derived Human Retinal Organoids for Use in Transgene Expression Assays. Methods Mol. Biol. 2018, 1715, 261–273. [Google Scholar]
- Quinn, P.M.; Pellissier, L.P.; Wijnholds, J. The CRB1 complex: Following the trail of Crumbs to a feasible gene therapy strategy. Front. Neurosci. 2017, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Eldred, K.C.; Hadyniak, S.E.; Hussey, K.A.; Brenerman, B.; Zhang, P.; Chamling, X.; Sluch, V.M.; Welsbie, D.S.; Hattar, S.; Taylor, J.; et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 2018, 362, 359950. [Google Scholar] [CrossRef]
- Lakowski, J.; Welby, E.; Budinger, D.; Di Marco, F.; Di Foggia, V.; Bainbridge, J.W.B.; Wallace, K.; Gamm, D.M.; Ali, R.R.; Sowden, J.C. Isolation of Human Photoreceptor Precursors via a Cell Surface Marker Panel from Stem Cell-Derived Retinal Organoids and Fetal Retinae. Stem Cells 2018, 36, 709–722. [Google Scholar] [CrossRef]
- Dorgau, B.; Felemban, M.; Sharpe, A.; Bauer, R.; Hallam, D.; Steel, D.H.; Lindsay, S.; Mellough, C.; Lako, M. Laminin γ3 plays an important role in retinal lamination, photoreceptor organisation and ganglion cell differentiation. Cell Death Dis. 2018, 9, 615. [Google Scholar] [CrossRef]
- Felemban, M.; Dorgau, B.; Hunt, N.C.; Hallam, D.; Zerti, D.; Bauer, R.; Ding, Y.; Collin, J.; Steel, D.; Krasnogor, N.; et al. Extracellular matrix components expression in human pluripotent stem cell-derived retinal organoids recapitulates retinogenesis in vivo and reveals an important role for IMPG1 and CD44 in the development of photoreceptors and interphotoreceptor matrix. Acta Biomater. 2018, 74, 207–221. [Google Scholar] [CrossRef]
- Hendrickson, A.; Drucker, D. The development of parafoveal and mid-peripheral human retina. Behav. Brain Res. 1992, 49, 21–31. [Google Scholar] [CrossRef]
- Hendrickson, A.; Bumsted-O’Brien, K.; Natoli, R.; Ramamurthy, V.; Possin, D.; Provis, J. Rod photoreceptor differentiation in fetal and infant human retina. Exp. Eye Res. 2008, 87, 415–426. [Google Scholar] [CrossRef]
- Hendrickson, A.; Possin, D.; Vajzovic, L.; Toth, C.A. Histologic Development of the Human Fovea From Midgestation to Maturity. Am. J. Ophthalmol. 2012, 154, 767–778.e2. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, A. Development of Retinal Layers in Prenatal Human Retina. Am. J. Ophthalmol. 2016, 161, 29–35.e1. [Google Scholar] [CrossRef] [PubMed]
- Kozulin, P.; Natoli, R.; O’Brien, K.M.B.; Madigan, M.C.; Provis, J.M. Differential expression of anti-angiogenic factors and guidance genes in the developing macula. Mol. Vis. 2009, 15, 45–59. [Google Scholar] [PubMed]
- O’Brien, K.M.B.; Schulte, D.; Hendrickson, A.E. Expression of photoreceptor-associated molecules during human fetal eye development. Mol. Vis. 2003, 9, 401–409. [Google Scholar]
- Curcio, C.A.; Sloan, K.R.; Kalina, R.E.; Hendrickson, A.E. Human photoreceptor topography. J. Comp. Neurol. 1990, 292, 497–523. [Google Scholar] [CrossRef]
- Narayanan, K.; Wadhwa, S. Photoreceptor morphogenesis in the human retina: A scanning electron microscopic study. Anat. Rec. 1998, 252, 133–139. [Google Scholar] [CrossRef]
- Cornish, E.E.; Xiao, M.; Yang, Z.; Provis, J.M.; Hendrickson, A.E. The role of opsin expression and apoptosis in determination of cone types in human retina. Exp. Eye Res. 2004, 78, 1143–1154. [Google Scholar] [CrossRef]
- Cornish, E.E.; Hendrickson, A.E.; Provis, J.M. Distribution of short-wavelength-sensitive cones in human fetal and postnatal retina: Early development of spatial order and density profiles. Vision Res. 2004, 44, 2019–2026. [Google Scholar] [CrossRef]
- Hendrickson, A.E.; Yuodelis, C. The Morphological Development of the Human Fovea. Ophthalmology 1984, 91, 603–612. [Google Scholar] [CrossRef]
- Fligor, C.M.; Langer, K.B.; Sridhar, A.; Ren, Y.; Shields, P.K.; Edler, M.C.; Ohlemacher, S.K.; Sluch, V.M.; Zack, D.J.; Zhang, C.; et al. Three-Dimensional Retinal Organoids Facilitate the Investigation of Retinal Ganglion Cell Development, Organization and Neurite Outgrowth from Human Pluripotent Stem Cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human induced pluripotent stem cells generate light responsive retinal organoids with variable and nutrient dependent efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhong, X.; Li, K.; Xie, B.; Liu, Y.; Ye, M.; Li, K.; Xu, C.; Ge, J. An Optimized System for Effective Derivation of Three-dimensional Retinal Tissue via Wnt Signaling Regulation. Stem Cells 2018, 36, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, A.; Ozone, C.; Nakano, T.; Saito, K.; Eiraku, M.; Sasai, Y. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015, 6, 6286. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nogales, M.; Murcia-Belmonte, V.; Chen, H.Y.; Herrera, E. The peripheral eye: A neurogenic area with potential to treat retinal pathologies? Prog. Retin. Eye Res. 2018, 68, 110–123. [Google Scholar] [CrossRef]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Rep. 2018, 10, 300–313. [Google Scholar] [CrossRef]
- Ovando-Roche, P.; West, E.L.; Branch, M.J.; Sampson, R.D.; Fernando, M.; Munro, P.; Georgiadis, A.; Rizzi, M.; Kloc, M.; Naeem, A.; et al. Use of bioreactors for culturing human retinal organoids improves photoreceptor yields. Stem Cell Res. Ther. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Kaewkhaw, R.; Kaya, K.D.; Brooks, M.; Homma, K.; Zou, J.; Chaitankar, V.; Rao, M.; Swaroop, A. Transcriptome Dynamics of Developing Photoreceptors in Three-Dimensional Retina Cultures Recapitulates Temporal Sequence of Human Cone and Rod Differentiation Revealing Cell Surface Markers and Gene Networks. Stem Cells 2015, 33, 3504–3518. [Google Scholar] [CrossRef]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor Outer Segment-like Structures in Long-Term 3D Retinas from Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef]
- Li, M.; Jia, C.; Kazmierkiewicz, K.L.; Bowman, A.S.; Tian, L.; Liu, Y.; Gupta, N.A.; Gudiseva, H.V.; Yee, S.S.; Kim, M.; et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum. Mol. Genet. 2014, 23, 4001–4014. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Murcia-Belmonte, V.; Wang, Q.; Coca, Y.; Ferreiro-Galve, S.; Kuwajima, T.; Khalid, S.; Ross, M.E.; Mason, C.; Herrera, E. The Ciliary Margin Zone of the Mammalian Retina Generates Retinal Ganglion Cells. Cell Rep. 2016, 17, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.-C.; Robert, B.; Cayouette, M. Msx1-Positive Progenitors in the Retinal Ciliary Margin Give Rise to Both Neural and Non-neural Progenies in Mammals. Dev. Cell 2017, 40, 137–150. [Google Scholar] [CrossRef]
- Provis, J.M.; Hendrickson, A.E. The foveal avascular region of developing human retina. Arch. Ophthalmol. 2008, 126, 507–511. [Google Scholar] [CrossRef]
- Yuodelis, C.; Hendrickson, A. A qualitative and quantitative analysis of the human fovea during development. Vision Res. 1986, 26, 847–855. [Google Scholar] [CrossRef]
- Vajzovic, L.; Hendrickson, A.E.; O’Connell, R.V.; Clark, L.A.; Tran-Viet, D.; Possin, D.; Chiu, S.J.; Farsiu, S.; Toth, C.A. Maturation of the human fovea: Correlation of spectral-domain optical coherence tomography findings with histology. Am. J. Ophthalmol. 2012, 154, 779–789.e2. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.J.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef]
- Simic, N.; Westall, C.; Astzalos, E.V.; Rovet, J. Visual abilities at 6 months in preterm infants: Impact of thyroid hormone deficiency and neonatal medical morbidity. Thyroid 2010, 20, 309–315. [Google Scholar] [CrossRef]
- Duerksen, K.; Barlow, W.E.; Stasior, O.G. Fused eyelids in premature infants. Ophthal. Plast. Reconstr. Surg. 1994, 10, 234–240. [Google Scholar] [CrossRef]
- Smyth, C.N. Exploratory methods for testing the integrity of the foetus and neonate. BJOG Int. J. Obstet. Gynaecol. 1965, 72, 920–925. [Google Scholar] [CrossRef]
- Polishuk, W.Z.; Laufer, N.; Sadovsky, E. Fetal reaction to external light. Harefuah 1975, 89, 395–396. [Google Scholar] [PubMed]
- Peleg, D.; Goldman, J.A. Fetal heart rate acceleration in response to light stimulation as a clinical measure of fetal well-being. A preliminary report. J. Perinat. Med. 1980, 8, 38–41. [Google Scholar] [PubMed]
- Tatsumura, M. Studies on features of fetal movement and development of human fetus with use of fetal actogram. Nihon Sanka Fujinka Gakkai Zasshi 1991, 43, 864–873. [Google Scholar] [PubMed]
- Kiuchi, M.; Nagata, N.; Ikeno, S.; Terakawa, N. The relationship between the response to external light stimulation and behavioral states in the human fetus: How it differs from vibroacoustic stimulation. Early Hum. Dev. 2000, 58, 153–165. [Google Scholar] [CrossRef]
- Thanaboonyawat, I.; Wataganara, T.; Boriboonhiransarn, D.; Viboonchart, S.; Tontisirin, P. Effect of halogen light in fetal stimulation for fetal well-being assessment. J. Med. Assoc. Thailand 2006, 89, 1376–1380. [Google Scholar]
- Eswaran, H.; Wilson, J.; Preissl, H.; Robinson, S.; Vrba, J.; Murphy, P.; Rose, D.; Lowery, C. Magnetoencephalographic recordings of visual evoked brain activity in the human fetus. Lancet 2002, 360, 779–780. [Google Scholar] [CrossRef]
- Eswaran, H.; Lowery, C.L.; Wilson, J.D.; Murphy, P.; Preissl, H. Functional development of the visual system in human fetus using magnetoencephalography. Exp. Neurol. 2004, 190, S52–S58. [Google Scholar] [CrossRef]
- McCubbin, J.; Murphy, P.; Eswaran, H.; Preissl, H.; Yee, T.; Robinson, S.E.; Vrba, J. Validation of the flash-evoked response from fetal MEG. Phys. Med. Biol. 2007, 52, 5803–5813. [Google Scholar] [CrossRef]
- Matuz, T.; Govindan, R.B.; Preissl, H.; Siegel, E.R.; Muenssinger, J.; Murphy, P.; Ware, M.; Lowery, C.L.; Eswaran, H. Habituation of visual evoked responses in neonates and fetuses: A MEG study. Dev. Cogn. Neurosci. 2012, 2, 303–316. [Google Scholar] [CrossRef]
- Fulford, J.; Vadeyar, S.H.; Dodampahala, S.H.; Moore, R.J.; Young, P.; Baker, P.N.; James, D.K.; Gowland, P.A. Fetal brain activity in response to a visual stimulus. Hum. Brain Mapp. 2003, 20, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Schwindt, E.; Giordano, V.; Rona, Z.; Czaba-Hnizdo, C.; Olischar, M.; Waldhoer, T.; Werther, T.; Fuiko, R.; Berger, A.; Klebermass-Schrehof, K. The impact of extrauterine life on visual maturation in extremely preterm born infants. Pediatr. Res. 2018, 84, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Menzies, R.; MacMillan, L.J.; Whyte, H.E. VEPs in normal full-term and premature neonates: Longitudinal versus cross-sectional data. Electroencephalogr. Clin. Neurophysiol. 1987, 68, 20–27. [Google Scholar] [CrossRef]
- Tsuneishi, S.; Casaer, P. Effects of preterm extrauterine visual experience on the development of the human visual system: A flash VEP study. Dev. Med. Child Neurol. 2000, 42, 663–668. [Google Scholar] [CrossRef]
- Leaf, A.A.; Green, C.R.; Escwk, A.; Costeloe, K.L.; Prior, P.F. Maturation of Electroretinograms and Visual Evoked Potentials in Preterm Infants. Dev. Med. Child Neurol. 1995, 37, 814–826. [Google Scholar] [CrossRef]
- Mactier, H.; Dexter, J.D.; Hewett, J.E.; Latham, C.B.; Woodruff, C.W. The electroretinogram in preterm infants. J. Pediatr. 1988, 113, 607–612. [Google Scholar] [CrossRef]
- Mets, M.B.; Smith, V.C.; Pokorny, J.; Pass, A. Postnatal retinal development as measured by the electroretinogram in premature infants. Doc. Ophthalmol. 1995, 90, 111–127. [Google Scholar] [CrossRef]
- Mactier, H.; Hamilton, R.; Bradnam, M.S.; Turner, T.L.; Dudgeon, J. Contact lens electroretinography in preterm infants from 32 weeks after conception: A development in current methodology. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 82, F233–F236. [Google Scholar] [CrossRef]
- Fulton, A.B. The development of scotopic retinal function in human infants. Doc. Ophthalmol. 1988, 69, 101–109. [Google Scholar] [CrossRef]
- Fulton, A.B.; Hansen, R.M. The development of scotopic sensitivity. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1588–1596. [Google Scholar]
- Altschwager, P.; Moskowitz, A.; Fulton, A.B.; Hansen, R.M. Multifocal erg responses in subjects with a history of preterm birth. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2603–2608. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Åkerblom, H.; Andreasson, S.; Larsson, E.; Holmström, G. Photoreceptor Function in School-Aged Children is Affected by Preterm Birth. Transl. Vis. Sci. Technol. 2014, 3, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hansen, R.M.; Moskowitz, A.; Akula, J.D.; Fulton, A.B. The neural retina in retinopathy of prematurity. Prog. Retin. Eye Res. 2017, 56, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.; Harris, R.; Bhansali, P.; Cvekl, A.; Liu, W. Intercellular Adhesion-Dependent Cell Survival and ROCK-Regulated Actomyosin-Driven Forces Mediate Self-Formation of a Retinal Organoid. Stem Cell Rep. 2016, 6, 743–756. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146, dev171686. [Google Scholar] [CrossRef]
- Cowan, C.S.; Renner, M.; Gross-Scherf, B.; Goldblum, D.; Munz, M.; Krol, J.; Szikra, T.; Papasaikas, P.; Cuttat, R.; Waldt, A.; et al. Cell types of the human retina and its organoids at single-cell resolution: Developmental convergence, transcriptomic identity, and disease map. bioRxiv 2019, 703348. [Google Scholar] [CrossRef]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic Comparison of Retinal Organoid Differentiation from Human Pluripotent Stem Cells Reveals Stage Specific, Cell Line, and Methodological Differences. Stem Cells Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef]
- Chichagova, V.; Hilgen, G.; Ghareeb, A.; Georgiou, M.; Carter, M.; Sernagor, E.; Lako, M.; Armstrong, L. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cells 2019. [Google Scholar] [CrossRef]
- Zerti, D.; Dorgau, B.; Felemban, M.; Ghareeb, A.E.; Yu, M.; Ding, Y.; Krasnogor, N.; Lako, M. Developing a simple method to enhance the generation of cone and rod photoreceptors in pluripotent stem cell-derived retinal organoids. Stem Cells 2019, 50, e95. [Google Scholar] [CrossRef]
- Brooks, M.J.; Chen, H.Y.; Kelley, R.A.; Mondal, A.K.; Nagashima, K.; De Val, N.; Li, T.; Chaitankar, V.; Swaroop, A. Improved Retinal Organoid Differentiation by Modulating Signaling Pathways Revealed by Comparative Transcriptome Analyses with Development In Vivo. Stem Cell Rep. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kaya, K.D.; Chen, H.Y.; Brooks, M.J.; Kelley, R.A.; Shimada, H.; Nagashima, K.; de Val, N.; Drinnan, C.T.; Gieser, L.; Kruczek, K.; et al. Transcriptome-based molecular staging of human stem cell-derived retinal organoids uncovers accelerated photoreceptor differentiation by 9-cis retinal. bioRxiv 2019, 733071. [Google Scholar] [CrossRef]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2018, 140, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human Retina-on-a-Chip platform. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Iraha, S.; Tu, H.Y.; Yamasaki, S.; Kagawa, T.; Goto, M.; Takahashi, R.; Watanabe, T.; Sugita, S.; Yonemura, S.; Sunagawa, G.A.; et al. Establishment of Immunodeficient Retinal Degeneration Model Mice and Functional Maturation of Human ESC-Derived Retinal Sheets after Transplantation. Stem Cell Rep. 2018, 10, 1059–1074. [Google Scholar] [CrossRef]
- Gagliardi, G.; Ben M’Barek, K.; Chaffiol, A.; Slembrouck-Brec, A.; Conart, J.B.; Nanteau, C.; Rabesandratana, O.; Sahel, J.A.; Duebel, J.; Orieux, G.; et al. Characterization and Transplantation of CD73-Positive Photoreceptors Isolated from Human iPSC-Derived Retinal Organoids. Stem Cell Rep. 2018, 11, 665–680. [Google Scholar] [CrossRef]
- McLelland, B.T.; Lin, B.; Mathur, A.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Transplanted hESC-derived retina organoid sheets differentiate, integrate, and improve visual function in retinal degenerate rats. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2586–2603. [Google Scholar] [CrossRef]
- Pearson, R.A.; Gonzalez-Cordero, A.; West, E.L.; Ribeiro, J.R.; Aghaizu, N.; Goh, D.; Sampson, R.D.; Georgiadis, A.; Waldron, P.V.; Duran, Y.; et al. Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors. Nat. Commun. 2016, 7, 13029. [Google Scholar] [CrossRef]
- Singh, M.S.; Balmer, J.; Barnard, A.R.; Aslam, S.A.; Moralli, D.; Green, C.M.; Barnea-Cramer, A.; Duncan, I.; MacLaren, R.E. Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat. Commun. 2016, 7, 13537. [Google Scholar] [CrossRef]
- Santos-Ferreira, T.; Llonch, S.; Borsch, O.; Postel, K.; Haas, J.; Ader, M. Retinal transplantation of photoreceptors results in donor-host cytoplasmic exchange. Nat. Commun. 2016, 7, 13028. [Google Scholar] [CrossRef]
- Garita-Hernandez, M.; Lampic, M.; Chaffiol, A.; Guibbal, L.; Routet, F.; Santos-Ferreira, T.; Gagliardi, G.; Reichman, S.; Picaud, S.; Sahel, J.-A.; et al. Restoration of visual function by transplantation of optogenetically engineered photoreceptors. bioRxiv 2018, 399725. [Google Scholar] [CrossRef] [PubMed]
- Hoek, R.M.; Quinn, P.M.; Hooibrink, B.; Wijnholds, J. Transplantation of NTPDase2-positive Sorted Müller Glial Cells into the Mouse Retina. J. Neurosci. Neurosurg. 2018, 1. [Google Scholar] [CrossRef]
- Hoek, R.M.; Quinn, P.M.; Alves, C.H.; Hooibrink, B.; Wijnholds, J. NTPDase2 as a Surface Marker to Isolate Flow Cytometrically a Müller Glial Cell Enriched Population from Dissociated Neural Retinae. J. Neurosci. Neurosurg. 2018, 1. [Google Scholar] [CrossRef]
- Santos-Ferreira, T.F.; Borsch, O.; Ader, M. Rebuilding the Missing Part—A Review on Photoreceptor Transplantation. Front. Syst. Neurosci. 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sayadian, A.-C.; Lowe, N.; Lovegrove, H.E.; St Johnston, D. An alternative mode of epithelial polarity in the Drosophila midgut. PLoS Biol. 2018, 16, e3000041. [Google Scholar] [CrossRef] [PubMed]
- Tree, D.R.P.; Ma, D.; Axelrod, J.D. A three-tiered mechanism for regulation of planar cell polarity. Semin. Cell Dev. Biol. 2002, 13, 217–224. [Google Scholar] [CrossRef]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Tepass, U.; Theres, C.; Knust, E. crumbs Encodes an EGFlike Protein Expressed on Apical Membranes of Drosophila Epithelial Cells and Required for Organization of Epithelia. Cell 1990, 61, 787–799. [Google Scholar] [CrossRef]
- Alves, C.H.; Bossers, K.; Vos, R.M.; Essing, A.H.W.; Swagemakers, S.; van der Spek, P.J.; Verhaagen, J.; Wijnholds, J. Microarray and Morphological Analysis of Early Postnatal CRB2 Mutant Retinas on a Pure C57BL/6J Genetic Background. PLoS ONE 2013, 8, e82532. [Google Scholar] [CrossRef]
- Park, B.; Alves, C.H.; Lundvig, D.M.; Tanimoto, N.; Beck, S.C.; Huber, G.; Richard, F.; Klooster, J.; Andlauer, T.F.M.; Swindell, E.C.; et al. PALS1 is essential for retinal pigment epithelium structure and neural retina stratification. J. Neurosci. 2011, 31, 17230–17241. [Google Scholar] [CrossRef]
- Alves, C.H.; Sanz, A.S.; Park, B.; Pellissier, L.P.; Tanimoto, N.; Beck, S.C.; Huber, G.; Murtaza, M.; Richard, F.; Sridevi Gurubaran, I.; et al. Loss of CRB2 in the mouse retina mimics human retinitis pigmentosa due to mutations in the CRB1 gene. Hum. Mol. Genet. 2013, 22, 35–50. [Google Scholar] [CrossRef]
- Dudok, J.J.; Sanz, A.S.; Lundvig, D.M.S.; Sothilingam, V.; Garrido, M.G.; Klooster, J.; Seeliger, M.W.; Wijnholds, J. MPP3 regulates levels of PALS1 and adhesion between photoreceptors and Müller cells. Glia 2013, 61, 1629–1644. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.C.N.; Pichaud, F. Crumbs is required to achieve proper organ size control during Drosophila head development. Development 2010, 137, 641–650. [Google Scholar] [CrossRef]
- Alves, C.H.; Pellissier, L.P.; Wijnholds, J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog. Retin. Eye Res. 2014, 40, 35–52. [Google Scholar] [CrossRef]
- Bulgakova, N.A.; Knust, E. The Crumbs complex: From epithelial-cell polarity to retinal degeneration. J. Cell Sci. 2009, 122, 2587–2596. [Google Scholar] [CrossRef]
- Fan, S.; Fogg, V.; Wang, Q.; Chen, X.-W.; Liu, C.-J.; Margolis, B. A novel Crumbs3 isoform regulates cell division and ciliogenesis via importin beta interactions. J. Cell Biol. 2007, 178, 387–398. [Google Scholar] [CrossRef]
- Bazellières, E.; Aksenova, V.; Barthélémy-Requin, M.; Massey-Harroche, D.; Le Bivic, A. Role of the Crumbs proteins in ciliogenesis, cell migration and actin organization. Semin. Cell Dev. Biol. 2018, 81, 13–20. [Google Scholar] [CrossRef]
- Lee, J.D.; Silva-Gagliardi, N.F.; Tepass, U.; McGlade, C.J.; Anderson, K.V. The FERM protein Epb4.1l5 is required for organization of the neural plate and for the epithelial-mesenchymal transition at the primitive streak of the mouse embryo. Development 2007, 134, 2007–2016. [Google Scholar] [CrossRef]
- Hirano, M.; Hashimoto, S.; Yonemura, S.; Sabe, H.; Aizawa, S. EPB41L5 functions to post-transcriptionally regulate cadherin and integrin during epithelial-mesenchymal transition. J. Cell Biol. 2008, 182, 1217–1230. [Google Scholar] [CrossRef]
- Christensen, A.K.; Jensen, A.M. Tissue-specific requirements for specific domains in the FERM protein Moe/Epb4.1l5 during early zebrafish development. BMC Dev. Biol. 2008, 8, 3. [Google Scholar] [CrossRef]
- Gosens, I.; Sessa, A.; den Hollander, A.I.; Letteboer, S.J.F.; Belloni, V.; Arends, M.L.; Le Bivic, A.; Cremers, F.P.M.; Broccoli, V.; Roepman, R. FERM protein EPB41L5 is a novel member of the mammalian CRB-MPP5 polarity complex. Exp. Cell Res. 2007, 313, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Laprise, P.; Beronja, S.; Silva-Gagliardi, N.F.; Pellikka, M.; Jensen, A.M.; McGlade, C.J.; Tepass, U. The FERM protein Yurt is a negative regulatory component of the Crumbs complex that controls epithelial polarity and apical membrane size. Dev. Cell 2006, 11, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, C.L.; Parent-Prévost, F.; Jacquet, K.; Biehler, C.; Jetté, A.; Laprise, P. Oligomerization of the FERM-FA protein Yurt controls epithelial cell polarity. J. Cell Biol. 2018, 217, 3853. [Google Scholar] [CrossRef] [PubMed]
- Schell, C.; Rogg, M.; Suhm, M.; Helmstädter, M.; Sellung, D.; Yasuda-Yamahara, M.; Kretz, O.; Küttner, V.; Suleiman, H.; Kollipara, L.; et al. The FERM protein EPB41L5 regulates actomyosin contractility and focal adhesion formation to maintain the kidney filtration barrier. Proc. Natl. Acad. Sci. USA 2017, 114, E4621–E4630. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, C.L.; Hardy, É.J.-L.; Chartier, F.J.-M.; Bisson, N.; Laprise, P. A bidirectional antagonism between aPKC and Yurt regulates epithelial cell polarity. J. Cell Biol. 2014, 204, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.; Schneider, M.; Theilenberg, E.; Grawe, F.; Knust, E. Drosophila Stardust is a partner of Crumbs in the control of epithelial cell polarity. Nature 2001, 414, 638–643. [Google Scholar] [CrossRef]
- Lemmers, C.; Michel, D.; Lane-Guermonprez, L.; Delgrossi, M.-H.; Médina, E.; Arsanto, J.-P.; Le Bivic, A. CRB3 binds directly to Par6 and regulates the morphogenesis of the tight junctions in mammalian epithelial cells. Mol. Biol. Cell 2004, 15, 1324–1333. [Google Scholar] [CrossRef]
- Roh, M.H.; Fan, S.; Liu, C.-J.; Margolis, B. The Crumbs3-Pals1 complex participates in the establishment of polarity in mammalian epithelial cells. J. Cell Sci. 2003, 116, 2895–2906. [Google Scholar] [CrossRef]
- Roh, M.H.; Makarova, O.; Liu, C.-J.; Shin, K.; Lee, S.; Laurinec, S.; Goyal, M.; Wiggins, R.; Margolis, B. The Maguk protein, Pals1, functions as an adapter, linking mammalian homologues of Crumbs and Discs Lost. J. Cell Biol. 2002, 157, 161–172. [Google Scholar] [CrossRef]
- Kantardzhieva, A.; Alexeeva, S.; Versteeg, I.; Wijnholds, J. MPP3 is recruited to the MPP5 protein scaffold at the retinal outer limiting membrane. FEBS J. 2006, 273, 1152–1165. [Google Scholar] [CrossRef]
- Kantardzhieva, A.; Gosens, I.; Alexeeva, S.; Punte, I.M.; Versteeg, I.; Krieger, E.; Neefjes-Mol, C.A.; den Hollander, A.I.; Letteboer, S.J.F.; Klooster, J.; et al. MPP5 recruits MPP4 to the CRB1 complex in photoreceptors. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yamanaka, T.; Hirose, T.; Manabe, N.; Mizuno, K.; Shimizu, M.; Akimoto, K.; Izumi, Y.; Ohnishi, T.; Ohno, S. Atypical protein kinase C is involved in the evolutionarily conserved PAR protein complex and plays a critical role in establishing epithelia-specific junctional structures. J. Cell Biol. 2001, 152, 1183–1196. [Google Scholar] [CrossRef]
- Suzuki, A.; Akimoto, K.; Ohno, S. Protein kinase C λ/ι (PKCλ/ι: A PKC isotype essential for the development of multicellular organisms. J. Biochem. 2003, 133, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Joberty, G.; Petersen, C.; Gao, L.; Macara, I.G. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2000, 2, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Edwards, A.S.; Fawcett, J.P.; Mbamalu, G.; Scott, J.D.; Pawson, T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat. Cell Biol. 2000, 2, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Horikoshi, Y.; Suzuki, A.; Sugiyama, Y.; Kitamura, K.; Maniwa, R.; Nagai, Y.; Yamashita, A.; Hirose, T.; Ishikawa, H.; et al. PAR-6 regulates aPKC activity in a novel way and mediates cell-cell contact-induces formation of the epithelial junctional complex. Genes Cells 2001, 6, 721–731. [Google Scholar] [CrossRef]
- Atwood, S.X.; Chabu, C.; Penkert, R.R.; Doe, C.Q.; Prehoda, K.E. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J. Cell Sci. 2007, 120, 3200–3206. [Google Scholar] [CrossRef]
- Graybill, C.; Wee, B.; Atwood, S.X.; Prehoda, K.E. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J. Biol. Chem. 2012, 287, 21003–21011. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Umeda, M. FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc. Natl. Acad. Sci. USA 2010, 107, 748–753. [Google Scholar] [CrossRef]
- Klarlund, J.K.; Holik, J.; Chawla, A.; Park, J.G.; Buxton, J.; Czech, M.P. Signaling complexes of the FERM domain-containing protein GRSP1 bound to ARF exchange factor GRP1. J. Biol. Chem. 2001, 276, 40065–40070. [Google Scholar] [CrossRef]
- Kong, Y.; Zhao, L.; Charette, J.R.; Hicks, W.L.; Stone, L.; Nishina, P.M.; Naggert, J.K. An FRMD4B variant suppresses dysplastic photoreceptor lesions in models of enhanced S-cone syndrome and of Nrl deficiency. Hum. Mol. Genet. 2018, 27, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Aragon, M.; Elbediwy, A.; Foglizzo, V.; Fletcher, G.C.; Li, V.S.W.; Thompson, B.J. Pak1 Kinase Maintains Apical Membrane Identity in Epithelia. Cell Rep. 2018, 22, 1639–1646. [Google Scholar] [CrossRef]
- Belmonte, M.A.; Santos, M.F.; Kihara, A.H.; Yan, C.Y.I.; Hamassaki, D.E. Light-Induced photoreceptor degeneration in the mouse involves activation of the small GTPase Rac1. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Gao, L.; Roh, M.H.; Macara, I.G.; Margolis, B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat. Cell Biol. 2003, 5, 137–142. [Google Scholar] [CrossRef]
- Koike, C.; Nishida, A.; Akimoto, K.; Nakaya, M.; Noda, T.; Ohno, S.; Furukawa, T. Function of atypical protein kinase C lambda in differentiating photoreceptors is required for proper lamination of mouse retina. J. Neurosci. 2005, 25, 10290–10298. [Google Scholar] [CrossRef]
- Heynen, S.R.; Tanimoto, N.; Joly, S.; Seeliger, M.W.; Samardzija, M.; Grimm, C. Retinal degeneration modulates intracellular localization of CDC42 in photoreceptors. Mol. Vis. 2011, 17, 2934–2946. [Google Scholar]
- Santos-Bredariol, A.S.; Santos, M.F.; Hamassaki-Britto, D.E. Distribution of the small molecular weight GTP-binding proteins Rac1, CdC42, RhoA and RhoB in the developing chick retina. J. Neurocytol. 2002, 31, 149–159. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Ghiani, M.; de Kok, Y.J.M.; Wijnholds, J.; Ballabio, A.; Cremers, F.P.M.; Broccoli, V. Isolation of Crb1, a mouse homologue of Drosophila crumbs, and analysis of its expression pattern in eye and brain. Mech. Dev. 2002, 110, 203–207. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Lundvig, D.M.S.; Tanimoto, N.; Klooster, J.; Vos, R.M.; Richard, F.; Sothilingam, V.; Garcia Garrido, M.; Le Bivic, A.; Seeliger, M.W.; et al. CRB2 acts as a modifying factor of CRB1-related retinal dystrophies in mice. Hum. Mol. Genet. 2014, 23, 3759–3771. [Google Scholar] [CrossRef]
- van Rossum, A.G.S.H.; Aartsen, W.M.; Meuleman, J.; Klooster, J.; Malysheva, A.; Versteeg, I.; Arsanto, J.-P.; Le Bivic, A.; Wijnholds, J. Pals1/Mpp5 is required for correct localization of Crb1 at the subapical region in polarized Muller glia cells. Hum. Mol. Genet. 2006, 15, 2659–2672. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Quinn, P.M.; Alves, C.H.; Vos, R.M.; Klooster, J.; Flannery, J.G.; Heimel, J.A.; Wijnholds, J. Gene therapy into photoreceptors and Müller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum. Mol. Genet. 2015, 24, 3104–3118. [Google Scholar] [CrossRef]
- Herranz-Martín, S.; Jimeno, D.; Paniagua, A.E.; Velasco, A.; Lara, J.M.; Aijón, J.; Lillo, C. Immunocytochemical evidence of the localization of the Crumbs homologue 3 protein (CRB3) in the developing and mature mouse retina. PLoS ONE 2012, 7, e50511. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Groot, A.J.; Van Der Groep, P.; Sersansie, R.; Vooijs, M.; Van Diest, P.J.; Van Noorden, C.J.F.; Schlingemann, R.O.; Klaassen, I. Active HIF-1 in the normal human retina. J. Histochem. Cytochem. 2010, 58, 247–254. [Google Scholar] [CrossRef]
- Cabral, T.; Toral, M.A.; Velez, G.; DiCarlo, J.E.; Gore, A.M.; Mahajan, M.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Dissection of Human Retina and RPE-Choroid for Proteomic Analysis. J. Vis. Exp. 2017, 1–5. [Google Scholar] [CrossRef]
- Paniagua, A.E.; Herranz-Martín, S.; Jimeno, D.; Jimeno, Á.M.; López-Benito, S.; Carlos Arévalo, J.; Velasco, A.; Aijón, J.; Lillo, C. CRB2 completes a fully expressed Crumbs complex in the Retinal Pigment Epithelium. Sci. Rep. 2015, 5, 14504. [Google Scholar] [CrossRef]
- Vallespin, E.; Cantalapiedra, D.; Riveiro-Alvarez, R.; Wilke, R.; Aguirre-Lamban, J.; Avila-Fernandez, A.; Lopez-Martinez, M.A.; Gimenez, A.; Trujillo-Tiebas, M.J.; Ramos, C.; et al. Mutation screening of 299 Spanish families with retinal dystrophies by Leber congenital amaurosis genotyping microarray. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5653–5661. [Google Scholar] [CrossRef]
- Corton, M.; Tatu, S.D.; Avila-Fernandez, A.; Vallespín, E.; Tapias, I.; Cantalapiedra, D.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Bernal, S.; García-Sandoval, B.; et al. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet J. Rare Dis. 2013, 8, 20. [Google Scholar] [CrossRef]
- Henderson, R.H.; Mackay, D.S.; Li, Z.; Moradi, P.; Sergouniotis, P.; Russell-Eggitt, I.; Thompson, D.A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br. J. Ophthalmol. 2011, 95, 811–817. [Google Scholar] [CrossRef]
- Mathijssen, I.B.; Florijn, R.J.; van den Born, L.I.; Zekveld-Vroon, R.C.; Ten Brink, J.B.; Plomp, A.S.; Baas, F.; Meijers-Heijboer, H.; Bergen, A.A.; van Schooneveld, M.J. Long-term follow-up of patients with retinitis pigmentosa type 12 caused by CRB1 mutations: A severe phenotype with considerable interindividual variability. Retina 2017, 37, 161–172. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Hasan, S.M.; Azmeh, A.; Mostafa, O.; Megarbane, A. Coat’s like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: A case report and discussion of the management options. BMC Res. Notes 2016, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Leber, T. Ueber Retinitis pigmentosa und angeborene Amaurose. Albr. Von Graefes Arch. Klin Exp. Ophthalmol. 1869, 15, 1–25. [Google Scholar] [CrossRef]
- Franceschetti, A.; Dieterle, P. Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia. Confin. Neurol. 1954, 14, 184–186. (In French) [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef]
- Richard, M.; Roepman, R.; Aartsen, W.M.; van Rossum, A.G.S.H.; den Hollander, A.I.; Knust, E.; Wijnholds, J.; Cremers, F.P.M. Towards understanding CRUMBS function in retinal dystrophies. Hum. Mol. Genet. 2006, 15, 235–243. [Google Scholar] [CrossRef]
- Aleman, T.S.; Cideciyan, A.V.; Aguirre, G.K.; Huang, W.C.; Mullins, C.L.; Roman, A.J.; Sumaroka, A.; Olivares, M.B.; Tsai, F.F.; Schwartz, S.B.; et al. Human CRB1-associated retinal degeneration: Comparison with the rd8 Crb1-mutant mouse model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6898–6910. [Google Scholar] [CrossRef]
- McKay, G.J.; Clarke, S.; Davis, J.A.; Simpson, D.A.C.; Silvestri, G. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 322–328. [Google Scholar] [CrossRef]
- Simonelli, F.; Ziviello, C.; Testa, F.; Rossi, S.; Fazzi, E.; Bianchi, P.E.; Fossarello, M.; Signorini, S.; Bertone, C.; Galantuomo, S.; et al. Clinical and Molecular Genetics of Leber’s Congenital Amaurosis: A Multicenter Study of Italian Patients. Investig. Opthalmology Vis. Sci. 2007, 48, 4284–4290. [Google Scholar] [CrossRef]
- den Hollander, A.I.; ten Brink, J.B.; de Kok, Y.J.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [CrossRef]
- Heckenlively, J.R. Preserved para-arteriole retinal pigment epithelium (PPRPE) in retinitis pigmentosa. Br. J. Ophthalmol. 1982, 66, 26–30. [Google Scholar] [CrossRef]
- Talib, M.; Schooneveld, V.M.J.; Genderen, V.M.M.; Wijnholds, J.; Florijn, R.J.; ten Brink, J.B.; Al, E. Genotypic and phenotypic characteristics of CRB1-associated retinal dystrophies: A long-term follow-up study. Ophthalmology 2017, 124, 884–895. [Google Scholar] [CrossRef]
- Tsang, S.H.; Burke, T.; Oll, M.; Yzer, S.; Lee, W.; Xie, Y.A.; Allikmets, R. Whole exome sequencing identifies CRB1 defect in an unusual maculopathy phenotype. Ophthalmology 2014, 121, 1773–1782. [Google Scholar] [CrossRef]
- Tosi, J.; Tsui, I.; Lima, L.H.; Wang, N.-K.; Tsang, S.H. Case report: Autofluorescence imaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function. Curr. Eye Res. 2009, 34, 395–400. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Leber Congenital Amaurosis. Adv. Exp. Med. Biol. 2018, 1085, 131–137. [Google Scholar]
- Vincent, A.; Ng, J.; Gerth-Kahlert, C.; Tavares, E.; Maynes, J.T.; Wright, T.; Tiwari, A.; Tumber, A.; Li, S.; Hanson, J.V.M.; et al. Biallelic Mutations in CRB1 Underlie Autosomal Recessive Familial Foveal Retinoschisis. Investig. Opthalmol. Vis. Sci. 2016, 57, 2637–2646. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Heckenlively, J.R.; van den Born, L.I.; de Kok, Y.J.M.; van der Velde-Visser, S.D.; Kellner, U.; Jurklies, B.; van Schooneveld, M.J.; Blankenagel, A.; Rohrschneider, K.; et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am. J. Hum. Genet. 2001, 69, 198–203. [Google Scholar] [CrossRef]
- Quinn, P.M.; Alves, C.H.; Klooster, J.; Wijnholds, J. CRB2 in immature photoreceptors determines the superior-inferior symmetry of the developing retina to maintain retinal structure and function. Hum. Mol. Genet. 2018, 27, 3137–3153. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Alves, C.H.; Quinn, P.M.; Vos, R.M.; Tanimoto, N.; Lundvig, D.M.S.; Dudok, J.J.; Hooibrink, B.; Richard, F.; Beck, S.C.; et al. Targeted ablation of CRB1 and CRB2 in retinal progenitor cells mimics Leber congenital amaurosis. PLoS Genet. 2013, 9, e1003976. [Google Scholar] [CrossRef]
- Yzer, S.; Fishman, G.A.; Racine, J.; Al-Zuhaibi, S.; Chakor, H.; Dorfman, A.; Szlyk, J.; Lachapelle, P.; Van Den Born, L.I.; Allikmets, R.; et al. CRB1 heterozygotes with regional retinal dysfunction: Implications for genetic testing of leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3736–3744. [Google Scholar] [CrossRef][Green Version]
- Khan, K.N.; Robson, A.; Mahroo, O.A.R.; Arno, G.; Inglehearn, C.F.; Armengol, M.; Waseem, N.; Holder, G.E.; Carss, K.J.; Raymond, L.F.; et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur. J. Hum. Genet. 2018, 26, 687–694. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, C.; Yang, D.; Sun, R.; Wang, M.; Sun, H.; Xu, M.; Zhou, L.; Chen, M.; Xie, P.; et al. CRB2 mutation causes autosomal recessive retinitis pigmentosa. Exp. Eye Res. 2018, 180, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.; Kay, J.N. Single molecule sequencing elucidates retinal mRNA isoform diversity—Implications for CRB1 retinopathies. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2531. [Google Scholar]
- Alves, C.H.; Boon, N.; Mulder, A.A.; Koster, A.J.; Jost, C.R.; Wijnholds, J. CRB2 loss in rod photoreceptors is associated with progressive loss of retinal contrast sensitivity. Int. J. Mol. Sci. 2019, 20, 4069. [Google Scholar] [CrossRef]
- Yuge, K.; Nakajima, M.; Uemura, Y.; Miki, H.; Uyama, M.; Tsubura, A. Immunohistochemical features of the human retina and retinoblastoma. Virchows Arch 1995, 426, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Tulvatana, W.; Adamian, M.; Berson, E.L.; Dryja, T.P. Photoreceptor rosettes in autosomal dominant retinitis pigmentosa with reduced penetrance. Arch. Ophthalmol. 1999, 117, 399–402. [Google Scholar] [CrossRef][Green Version]
- Lahav, M.; Albert, D.M.; Craft, J.L. Light and electron microscopic study of dysplastic rosette-like structures occurring in the disorganized mature retina. Albr. Von Graefes Arch. Klin. Exp. Ophthalmol. 1975, 195, 57–68. [Google Scholar] [CrossRef]
- Rich, K.A.; Figueroa, S.L.; Zhan, Y.; Blanks, J.C. Effects of müller cell disruption on mouse photoreceptor cell development. Exp. Eye Res. 1995, 61, 235–248. [Google Scholar] [CrossRef]
- West, E.L.; Pearson, R.A.; Tschernutter, M.; Sowden, J.C.; MacLaren, R.E.; Ali, R.R. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp. Eye Res. 2008, 86, 601–611. [Google Scholar] [CrossRef]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. Defects in the outer limiting membrane are associated with rosette development in the Nrl−/− retina. PLoS ONE 2012, 7, e32484. [Google Scholar] [CrossRef]
- Damiani, D.; Alexander, J.J.; O’Rourke, J.R.; McManus, M.; Jadhav, A.P.; Cepko, C.L.; Hauswirth, W.W.; Harfe, B.D.; Strettoi, E. Dicer Inactivation Leads to Progressive Functional and Structural Degeneration of the Mouse Retina. J. Neurosci. 2008, 28, 4878–4887. [Google Scholar] [CrossRef]
- Mehalow, A.K.; Kameya, S.; Smith, R.S.; Hawes, N.L.; Denegre, J.M.; Young, J.A.; Bechtold, L.; Haider, N.B.; Tepass, U.; Heckenlively, J.R.; et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 2003, 12, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Krol, I.; Alvarez, C.P.P.; Fiscella, M.; Hierlemann, A.; Roska, B.; Filipowicz, W. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat. Commun. 2015, 6, 7305. [Google Scholar] [CrossRef]
- Serjanov, D.; Bachay, G.; Hunter, D.D.; Brunken, W.J. Laminin β2 Chain Regulates Retinal Progenitor Cell Mitotic Spindle Orientation via Dystroglycan. J. Neurosci. 2018, 38, 5996–6010. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sakaguchi, D.S. Inhibition of integrin-mediated adhesion and signaling disrupts retinal development. Dev. Biol. 2004, 275, 202–214. [Google Scholar] [CrossRef]
- Lunardi, A.; Cremisi, F.; Dente, L. Dystroglycan is required for proper retinal layering. Dev. Biol. 2006, 290, 411–420. [Google Scholar] [CrossRef]
- Ouchi, Y.; Baba, Y.; Koso, H.; Taketo, M.M.; Iwamoto, T.; Aburatani, H.; Watanabe, S. β-Catenin signaling regulates the timing of cell differentiation in mouse retinal progenitor cells. Mol. Cell. Neurosci. 2011, 46, 770–780. [Google Scholar] [CrossRef]
- Thompson, B.J.; Pichaud, F.; Röper, K. Sticking together the Crumbs - an unexpected function for an old friend. Nat. Rev. Mol. Cell Biol. 2013, 14, 307–314. [Google Scholar] [CrossRef]
- Röper, K. Anisotropy of Crumbs and aPKC drives myosin cable assembly during tube formation. Dev. Cell 2012, 23, 939–953. [Google Scholar] [CrossRef]
- Kerman, B.E.; Cheshire, A.M.; Myat, M.M.; Andrew, D.J. Ribbon modulates apical membrane during tube elongation through Crumbs and Moesin. Dev. Biol. 2008, 320, 278–288. [Google Scholar] [CrossRef]
- FDA Approves Novel Gene Therapy to Treat Patients with a Rare form of Inherited Vision Loss. [Internet]. FDA.gov. 19 December 2017. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm589467.htm (accessed on 18 August 2019).
- New Gene Therapy for Rare Inherited Disorder Causing Vision loss Recommended for Approval. [Internet]. EMA.europa.eu. 21 September 2019. Available online: https://www.ema.europa.eu/en/news/new-gene-therapy-rare-inherited-disorder-causing-vision-loss-recommended-approval (accessed on 18 August 2019).
- Henrique Alves, C.; Wijnholds, J. AAV-Mediated Gene Therapy for CRB1-Hereditary Retinopathies; Intechopen: London, UK, 2018. [Google Scholar]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Pellissier, L.P.; Hoek, R.M.; Vos, R.M.; Aartsen, W.M.; Klimczak, R.R.; Hoyng, S.A.; Flannery, J.G.; Wijnholds, J. Specific tools for targeting and expression in Müller glial cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14009. [Google Scholar] [CrossRef] [PubMed]
- Yeste, J.; García-Ramírez, M.; Illa, X.; Guimerà, A.; Hernández, C.; Simó, R.; Villa, R. A compartmentalized microfluidic chip with crisscross microgrooves and electrophysiological electrodes for modeling the blood-retinal barrier. Lab Chip 2017, 18, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.G.; Guibbal, L.; Toualbi, L.; Routet, F.; Chaffiol, A.; Winckler, C.; Harinquet, M.; Robert, C.; Fouquet, S.; Sahel, J.; et al. Optogenetic light sensors in human retinal organoids. Front. Neurosci. 2018, 12, 1–12. [Google Scholar]
- Held, M.; Santeramo, I.; Wilm, B.; Murray, P.; Lévy, R. Ex vivo live cell tracking in kidney organoids using light sheet fluorescence microscopy. PLoS ONE 2018, 13, e0199918. [Google Scholar] [CrossRef]
- Cora, V.; Haderspeck, J.; Antkowiak, L.; Mattheus, U.; Neckel, P.H.; Mack, A.F.; Bolz, S.; Ueffing, M.; Pashkovskaia, N.; Achberger, K.; et al. A Cleared View on Retinal Organoids. Cells 2019, 8, 391. [Google Scholar] [CrossRef]
- Mongera, A.; Rowghanian, P.; Gustafson, H.J.; Shelton, E.; Kealhofer, D.A.; Carn, E.K.; Serwane, F.; Lucio, A.A.; Giammona, J.; Campàs, O. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature 2018, 561, 401–405. [Google Scholar] [CrossRef]
- Serwane, F.; Mongera, A.; Rowghanian, P.; Kealhofer, D.A.; Lucio, A.A.; Hockenbery, Z.M.; Campàs, O. In vivo quantification of spatially varying mechanical properties in developing tissues. Nat. Methods 2017, 14, 181–186. [Google Scholar] [CrossRef]









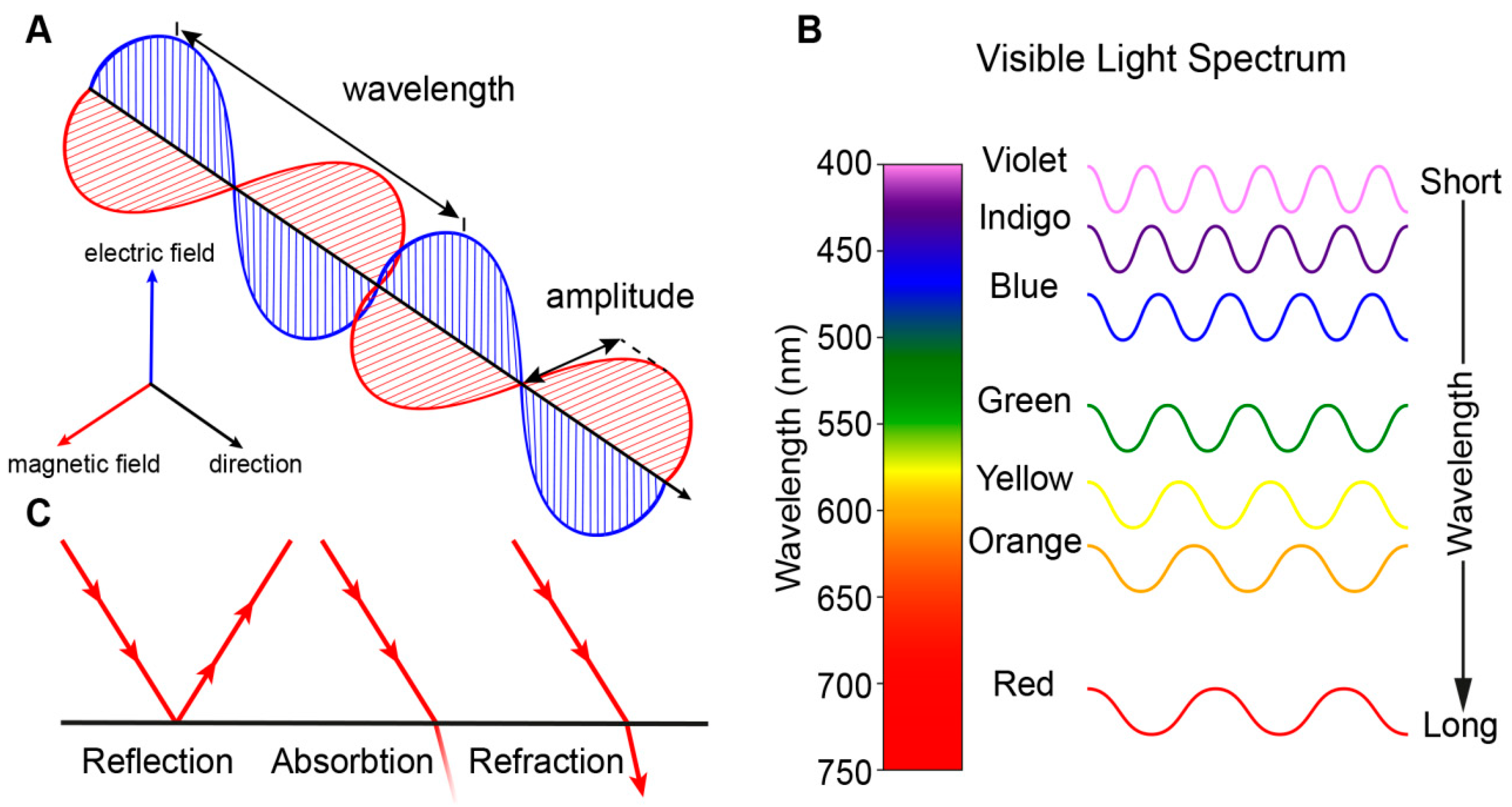{kind=link}
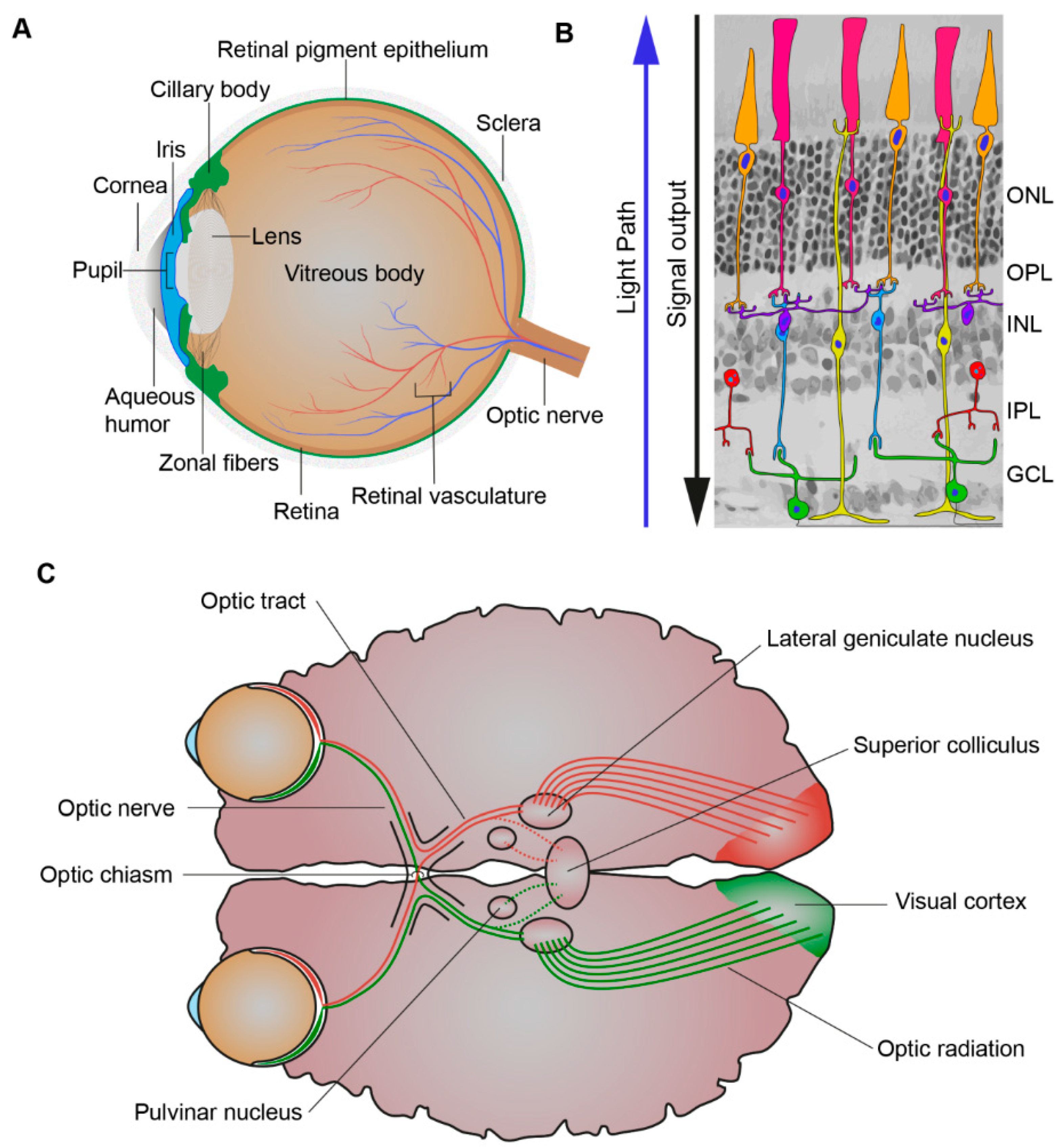{kind=link}
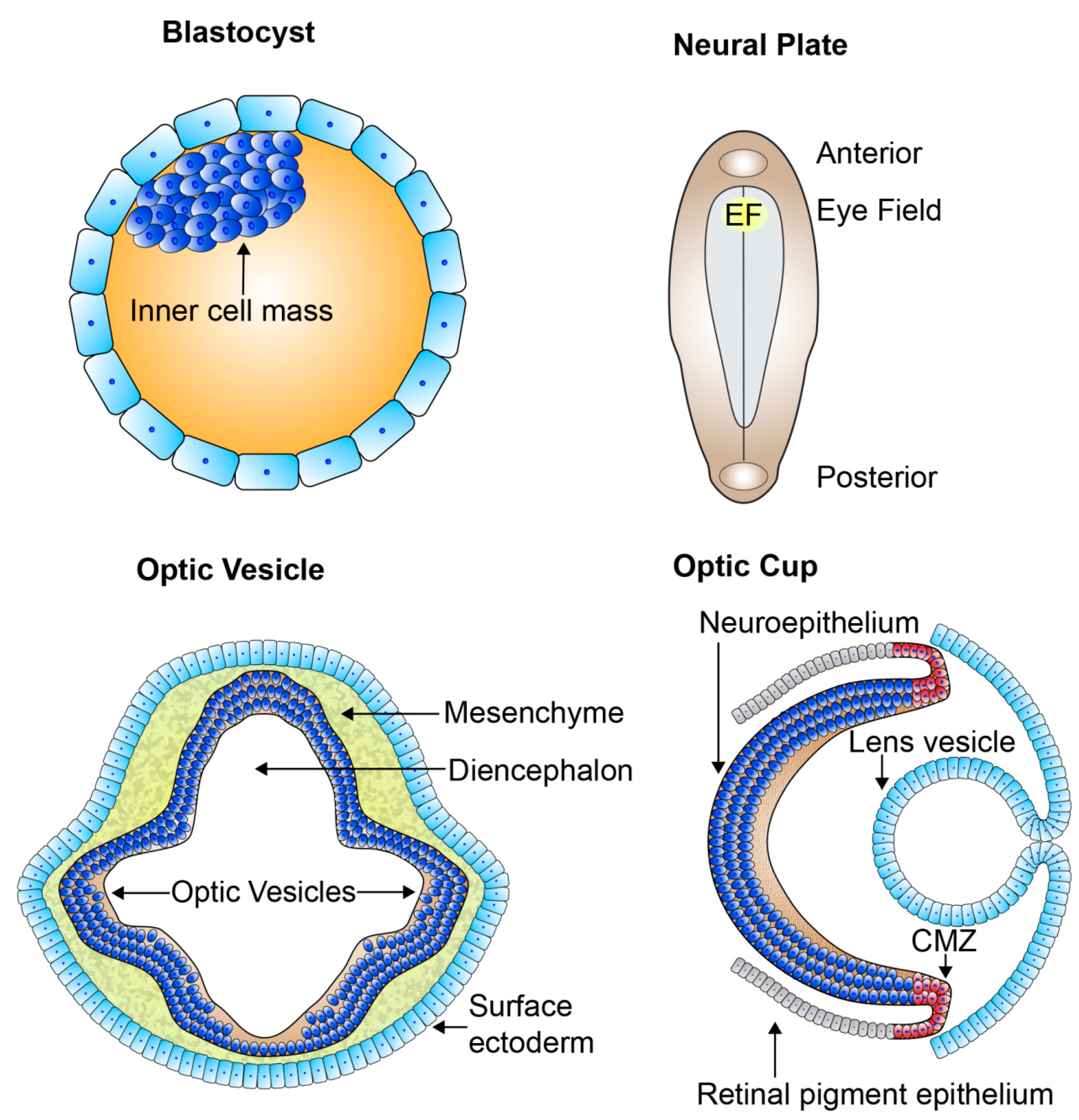{kind=link}
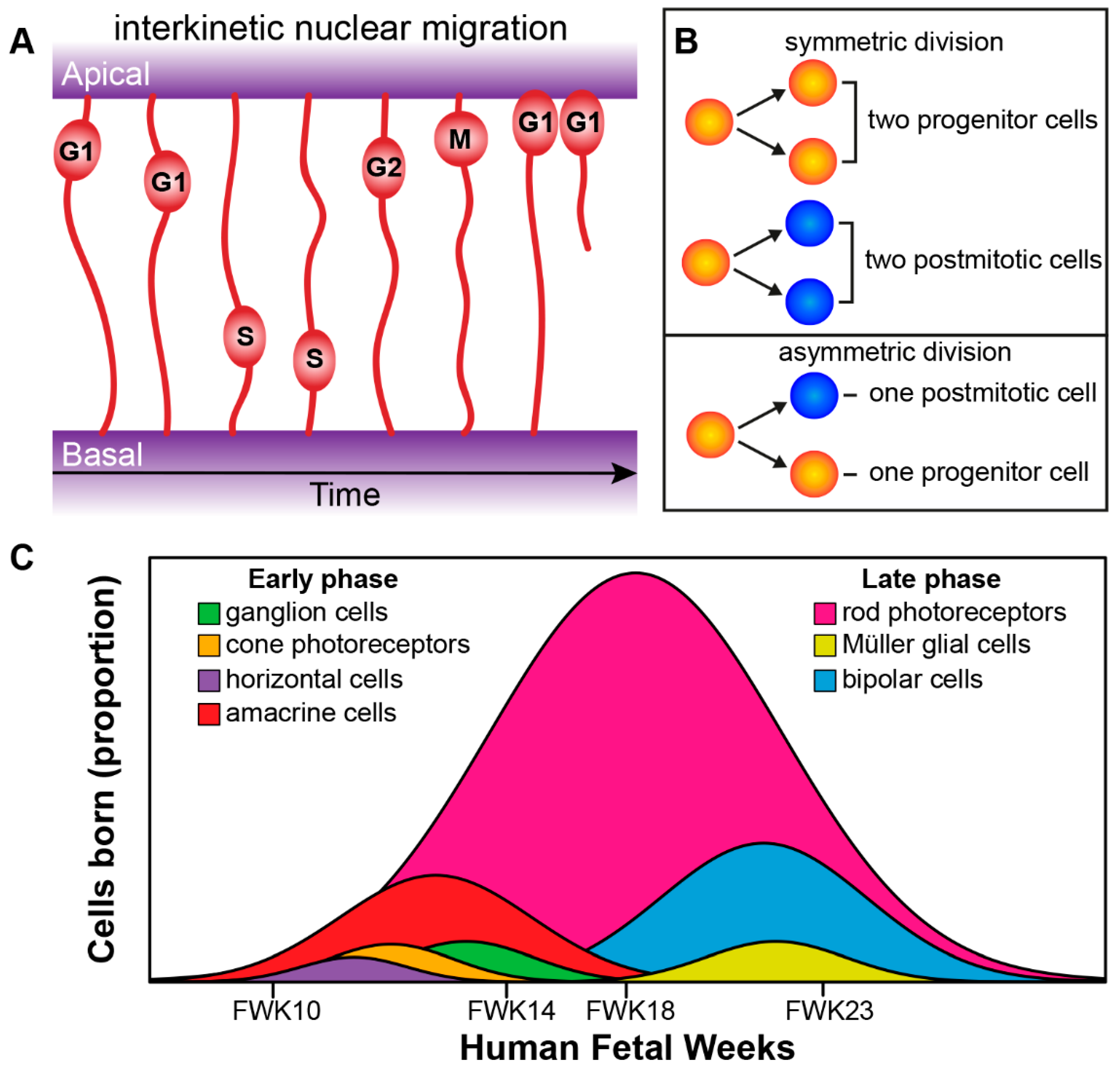{kind=link}
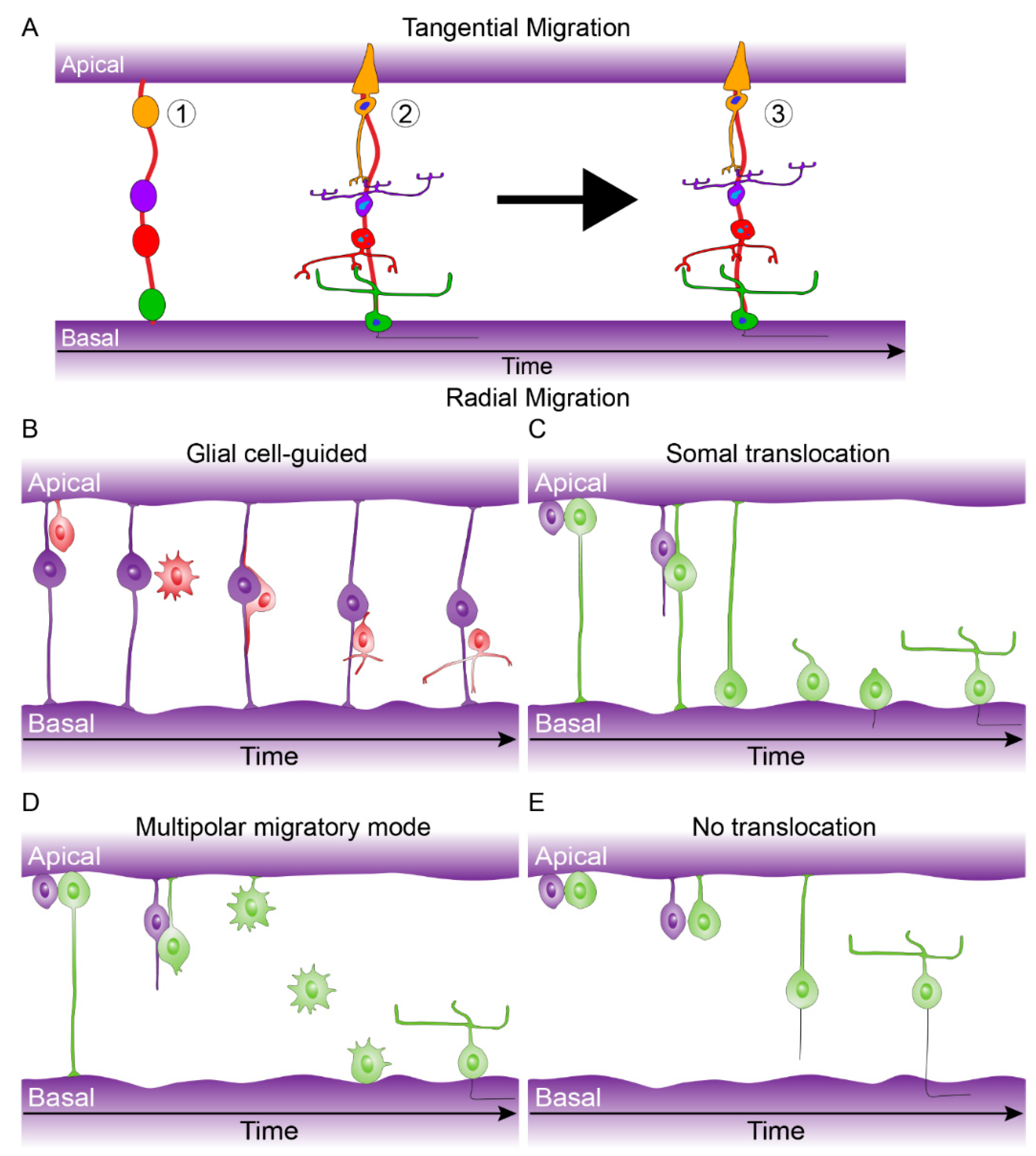{kind=link}
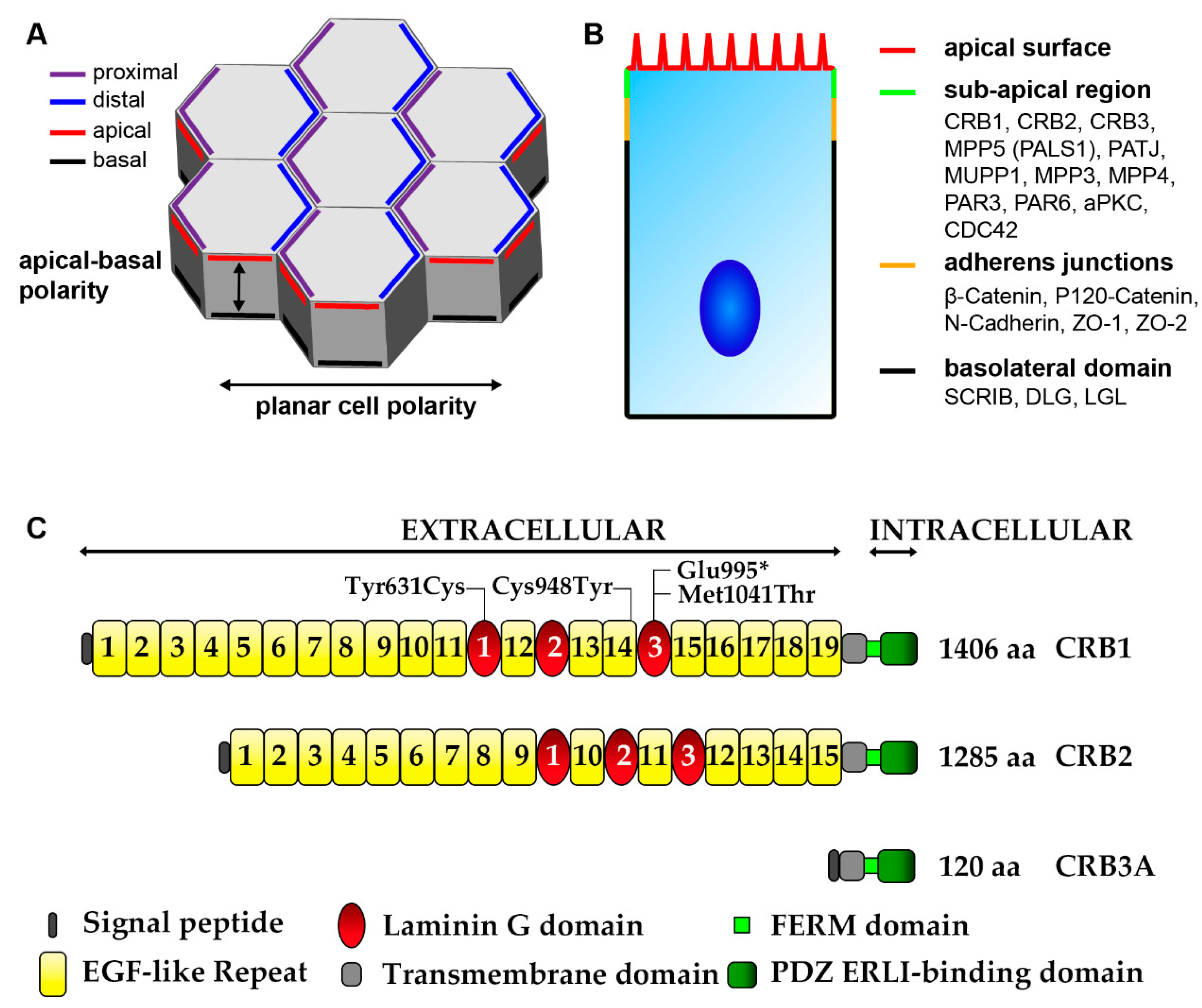{kind=link}
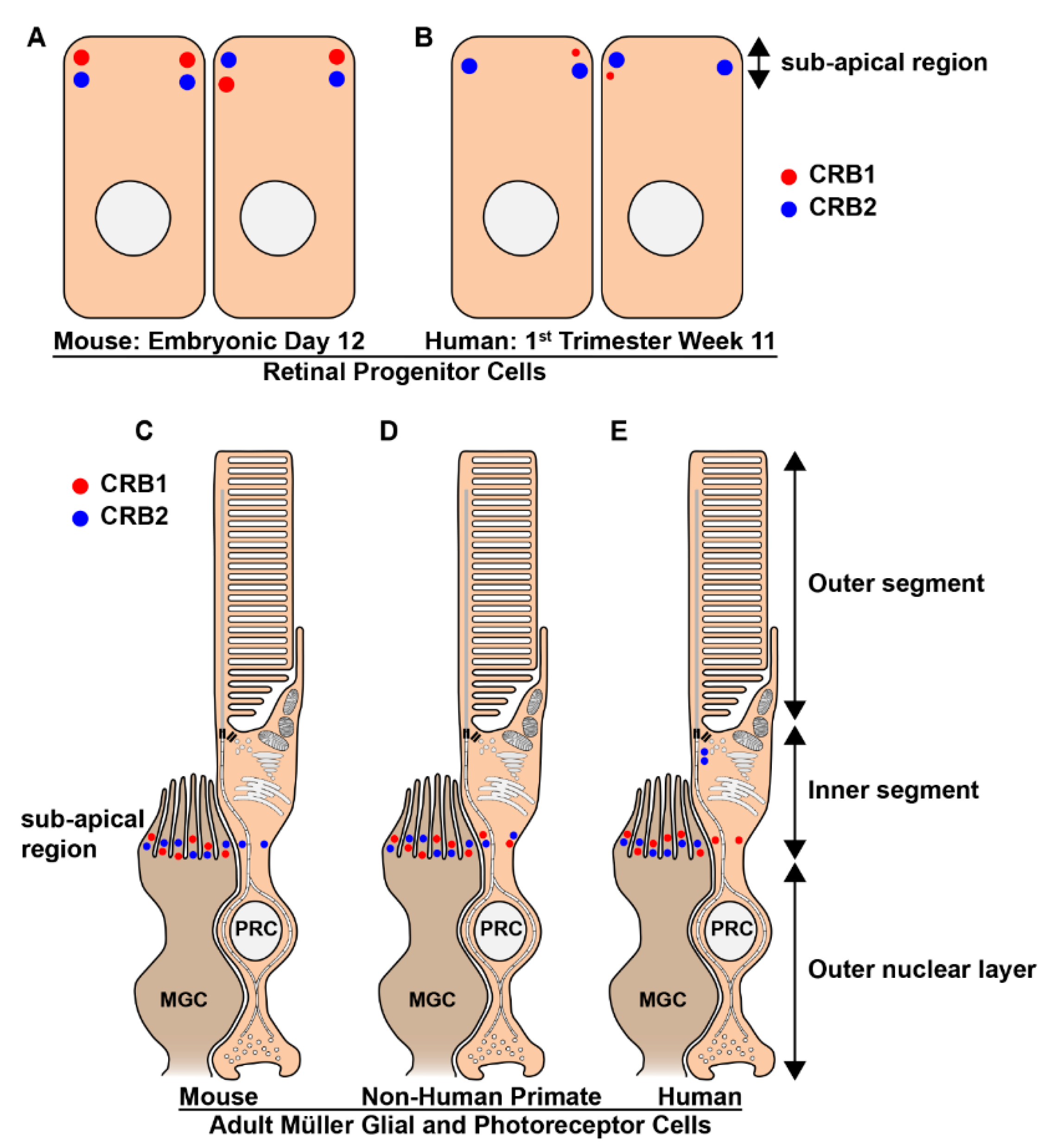{kind=link}
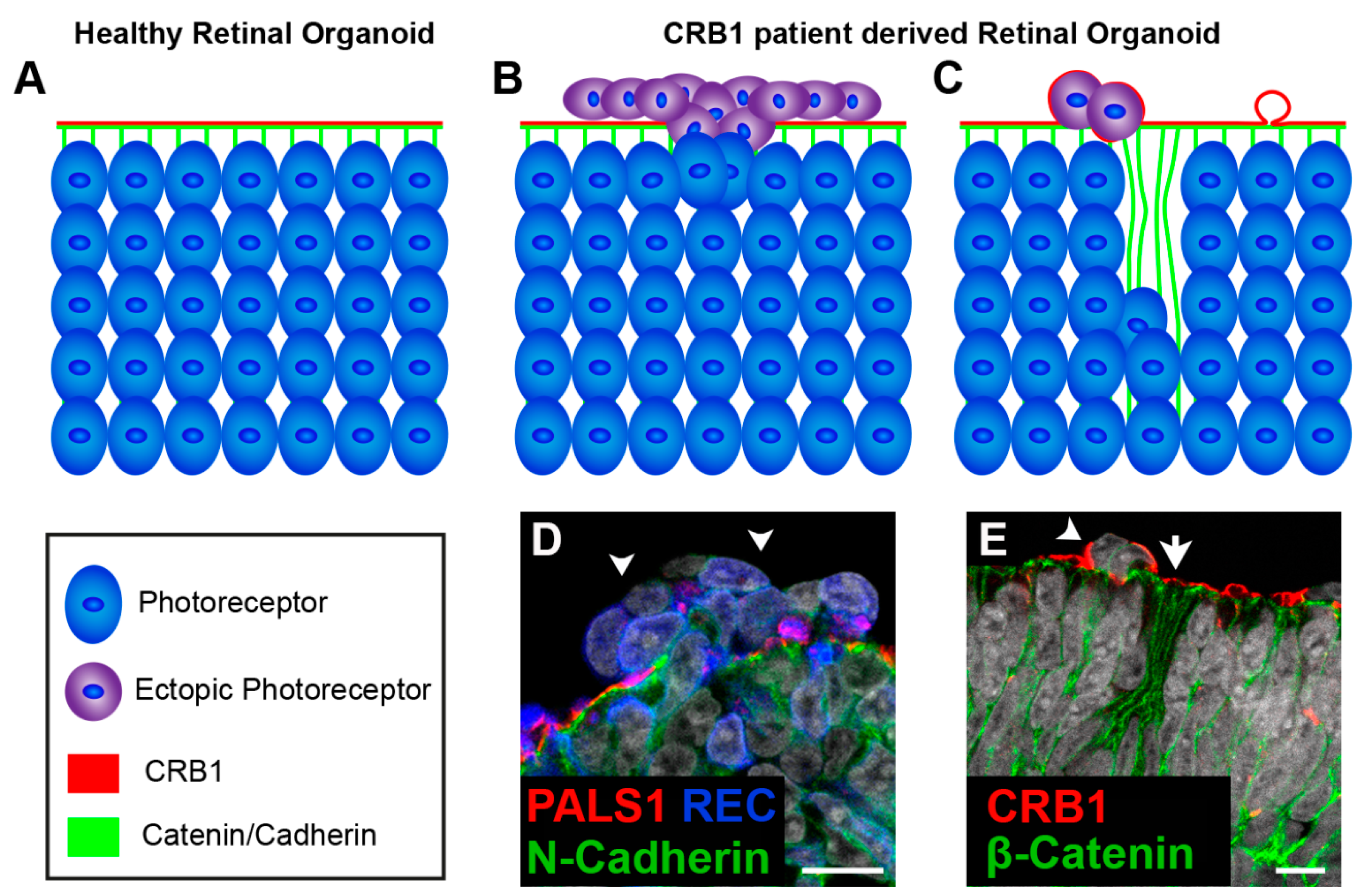{kind=link}
| Crb1KOCrb2ΔRPC | Crb1KOCrb2ΔimPRC | Crb1KO/WTCrb2ΔRPC | Crb1KOCrb2ΔMG | Crb1KOCrb2Δrods | |
|---|---|---|---|---|---|
| Severity | ++++ | +++ | ++ | ++ | + |
| CRB1 + CRB2 in MGC/PRC/RPC | 0/0/0 | 60/0/60 | 20/0/20 | 0/100/60 | 60/03/60 |
| Cre Mosaicism | 50% | 95% | 50% | 95% | 99–100% |
| Phenotype onset | E13 whole retina | E15 whole retina | E15 periphery | E17 periphery | 3M periphery |
| OLM disruption | ++++ | ++++ | ++ | +++ centrally ++++ at periphery | + whole retina |
| Abnormal retinal lamination | YES | YES | YES | YES | NO |
| Ectopic localization retinal cells | YES of all neurons | YES of PRC and RPC | YES of PRC, BC, AC and GC | YES of PRC, MGC, BC, AC and RPC | YES of PRC |
| Transiently thickened retina | YES | YES | YES | YES | NO |
| Intermingling of nuclei of the ONL and INL | YES | YES | YES But moderately | YES at peripheral retina | YES Sporadic |
| Ectopic retinal cells in GCL | ++++ | +++ | (+) sporadic ectopic cells | + periphery | NO |
| ERG max b-waves 1M/3M | 25%/0% | */* | 25%/10% | 50%/0% | 100%/90% |
| IS/OS at P14 | NO | NO | NO | NO | YES |
| ONL at P14 | NO | NO | YES | YES but not at periphery | YES |
| OPL at P14 | NO | NO | At foci | YES but not at periphery | YES |
| INL at P14 | NO | NO | YES | YES | YES |
| IPL at P14 | YES | YES | YES | YES | YES |
| GCL at P14 | Thickened in all of retina | Thickened in majority of retina | Thickened in part of retina | Thickened at peripheral retina | YES |
| Increase number of RPC | YES | YES | YES | NO | NA |
| Severity retinal phenotype | Throughout retina | Superior > inferior | Peripheral > central | Peripheral > central | Peripheral and Central Superior |
| Neovascularization | YES | YES | YES | YES | NA |
| Photoreceptors lost at 3M | YES (some sparse nuclei) | NA | thin ONL | YES (some sparse nuclei) | Minimal |
| Retinal overgrowth by late-born cells | YES | NO | NO | NO | NA |
| Apoptosis increased | YES | YES | YES | NA | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, P.M.J.; Wijnholds, J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes 2019, 10, 987. https://doi.org/10.3390/genes10120987
Quinn PMJ, Wijnholds J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes. 2019; 10(12):987. https://doi.org/10.3390/genes10120987
Chicago/Turabian StyleQuinn, Peter M.J., and Jan Wijnholds. 2019. "Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective" Genes 10, no. 12: 987. https://doi.org/10.3390/genes10120987
APA StyleQuinn, P. M. J., & Wijnholds, J. (2019). Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes, 10(12), 987. https://doi.org/10.3390/genes10120987