The Molecular Genetics of Gordon Syndrome



Abstract
1. Introduction
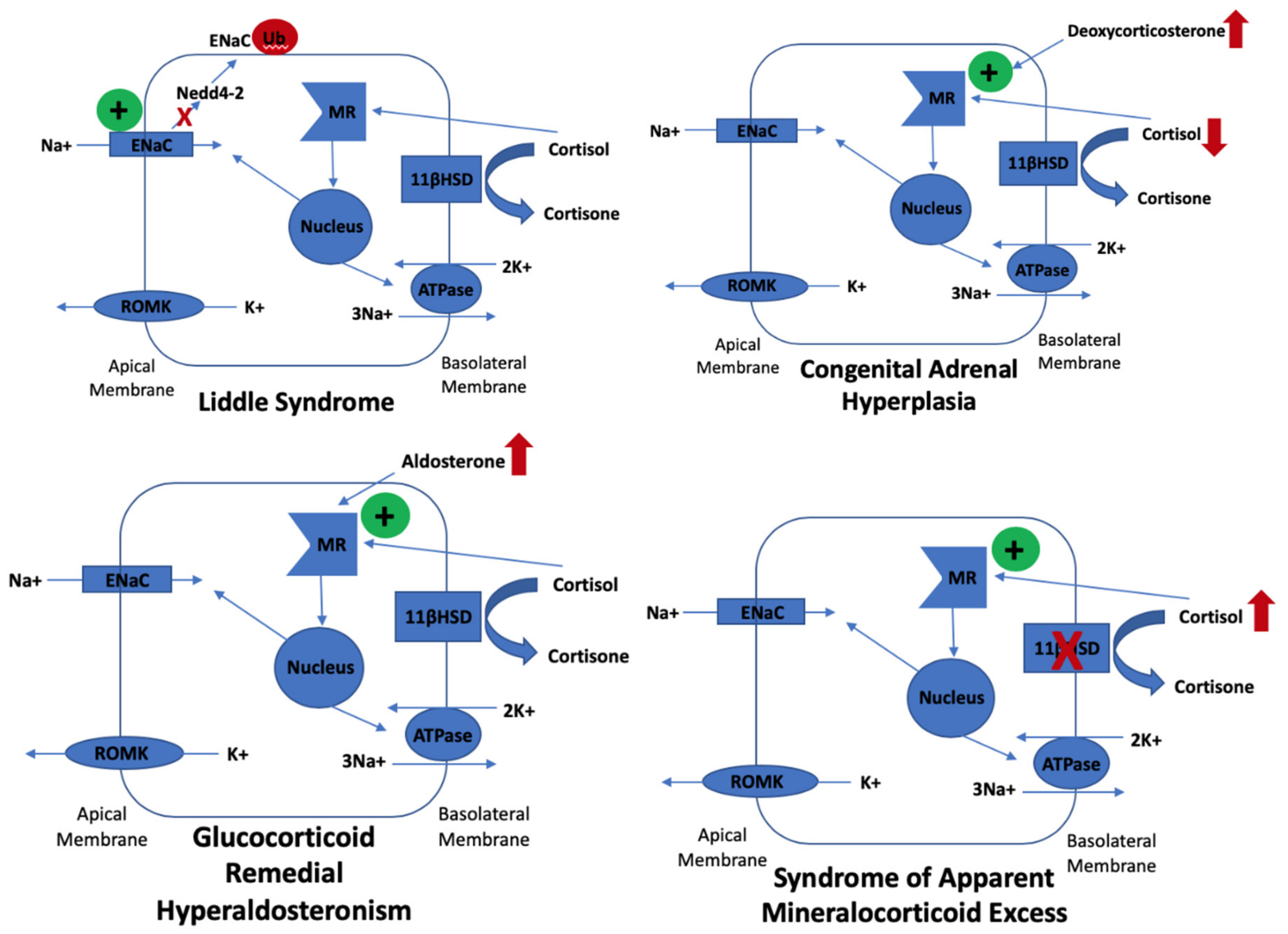
2. Clinical Presentations
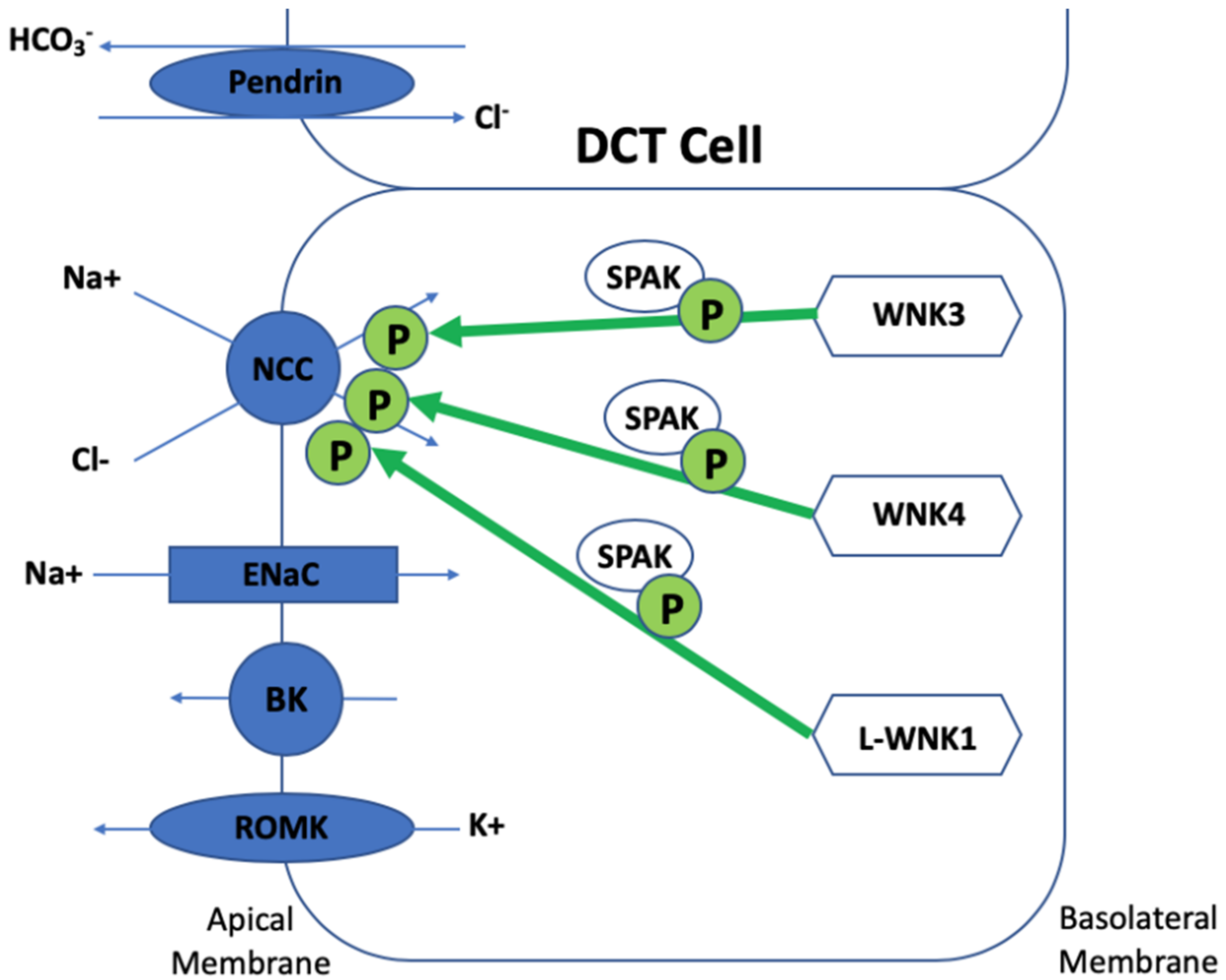
3. The Regulation of NCC
4. Discovery of SPAK and OSR1
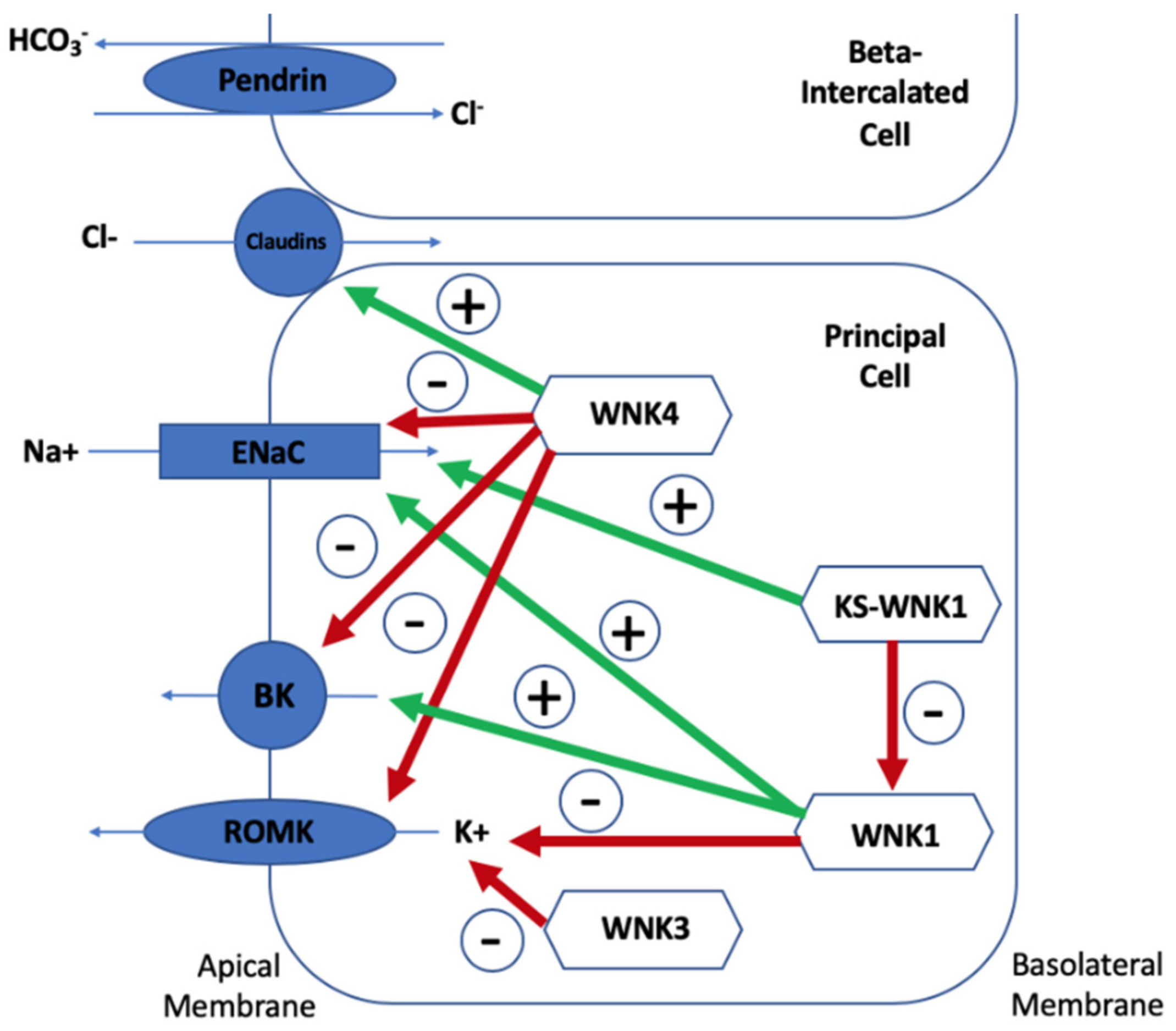
5. Mechanisms of Hyperkalaemia in Gordon Syndrome
5.1. Lack of Na+ Reaching ENaC for Adequate Exchange with K+
5.2. Direct Reduction on K+ Secretion
5.3. A Secondary Deficiency in K+ Secretion Due to Lack of Chloride Reaching ENaC
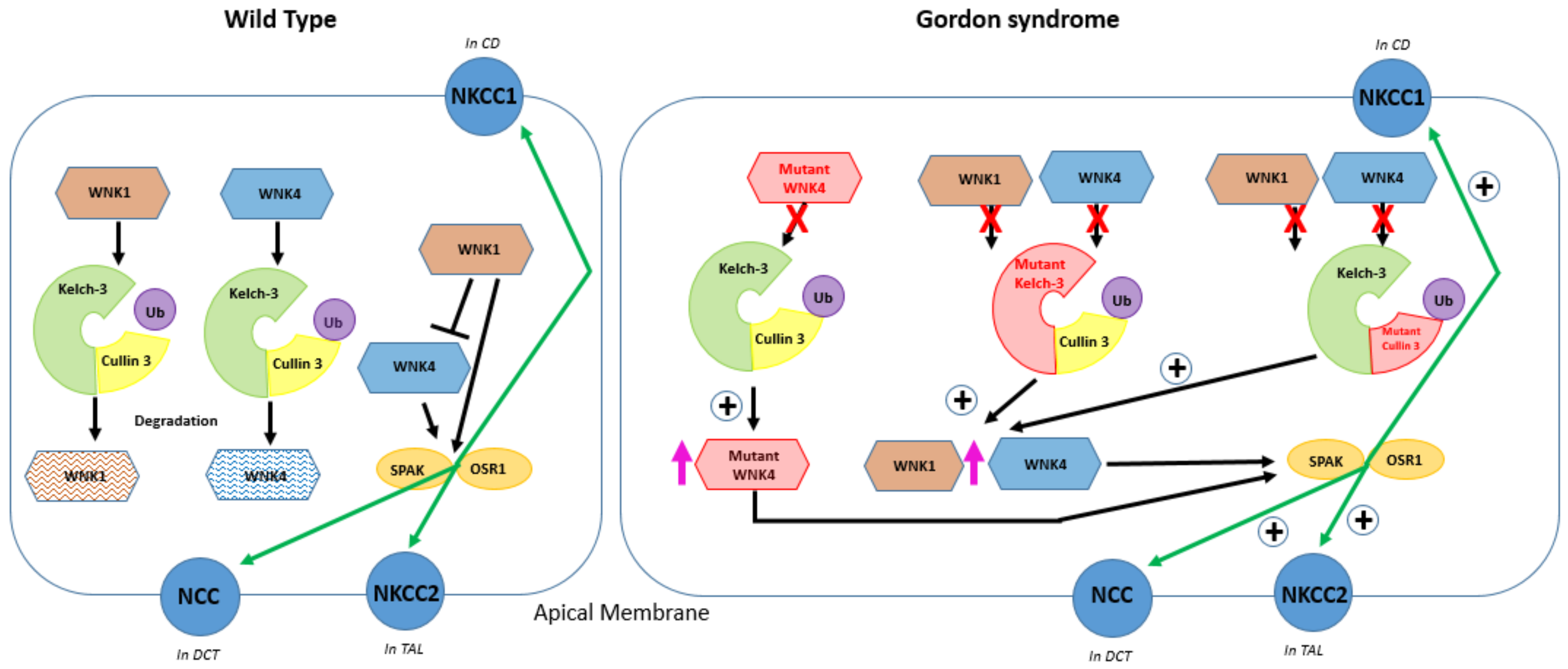
6. The Role of Kelch-like 3 and Cullin 3
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paver, W.; Pauline, G. Hypertension and hyperpotassaemia without renal disease in a young male. Med. J. Aust. 1964, 2, 305–306. [Google Scholar] [CrossRef]
- Arnold, J.E.; Healy, J.K. Hyperkalemia, hypertension and systemic acidosis without renal failure associated with a tubular defect in potassium excretion. Am. J. Med. 1969, 47, 461–472. [Google Scholar] [CrossRef]
- Gordon, R.D. The syndrome of hypertension and hyperkalemia with normal glomerular filtration rate: Gordon’s syndrome. Aust. N. Z. J. Med. 1986, 16, 183–184. [Google Scholar] [CrossRef]
- Schambelan, M.; Sebastian, A.; Rector, F.J. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): Role of increased renal chloride reabsorption. Kidney Int. 1981, 19, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Toka, H.R.; Koshy, J.M.; Hariri, A. The molecular basis of blood pressure variation. Pediatr. Nephrol. 2013, 28, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.D.; Geddes, R.A.; Pawsey, C.G.; O’Halloran, M.W. Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction. Australas. Ann. Med. 1970, 19, 287–294. [Google Scholar] [CrossRef]
- Achard, J.M.; Disse-Nicodeme, S.; Fiquet-Kempf, B.; Jeunemaitre, X. Phenotypic and genetic heterogeneity of familial hyperkalaemic hypertension (Gordon syndrome). Clin. Exp. Pharmacol. Physiol. 2001, 28, 1048–1052. [Google Scholar] [CrossRef]
- Mayan, H.; Vered, I.; Mouallem, M.; Tzadok-Witkon, M.; Pauzner, R.; Farfel, Z. Pseudohypoaldosteronism type II: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J. Clin. Endocrinol. Metab. 2002, 87, 3248–3254. [Google Scholar] [CrossRef]
- Achard, J.M.; Warnock, D.G.; Disse-Nicodeme, S.; Fiquet-Kempf, B.; Corvol, P.; Fournier, A.; Jeunemaitre, X. Familial hyperkalemic hypertension: Phenotypic analysis in a large family with the WNK1 deletion mutation. Am. J. Med. 2003, 114, 495–498. [Google Scholar] [CrossRef]
- Hadchouel, J.; Delaloy, C.; Fauré, S.; Achard, J.; Jeunemaitre, X. Familial Hyperkalaemic Hypertension. JASN 2006, 17, 208–217. [Google Scholar] [CrossRef]
- Mansfield, T.A.; Simon, D.B.; Farfel, Z.; Bia, M.; Tucci, J.R.; Lebel, M.; Gutkin, M.; Vialettes, B.; Christofilis, M.A.; Kauppinen-Makelin, R.; et al. Multilocus linkage of familial hyperkalaemia and hypertension, pseudohypoaldosteronism type II, to chromosomes 1q31-42 and 17p11-q21. Nat. Genet. 1997, 16, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Disse-Nicodeme, S.; Achard, J.M.; Desitter, I.; Houot, A.M.; Fournier, A.; Corvol, P.; Jeunemaitre, X. A new locus on chromosome 12p13.3 for pseudohypoaldosteronism type II, an autosomal dominant form of hypertension. Am. J. Hum. Genet. 2000, 67, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Sohara, E.; Rai, T.; Sasaki, S. Regulation of with-nolysine kinase signaling by Kelch-like proteins. Biol. Cell 2014, 106, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Glover, M.; Ware, J.S.; Henry, A.; Wolley, M.; Walsh, R.; Wain, L.V.; Xu, S.; Van’t Hoff, W.G.; Tobin, M.D.; Hall, I.P.; et al. Detection of mutations in KLHL3 and CUL3 in families with FHHt (familial hyperkalaemic hypertension or Gordon’s syndrome). Clin. Sci. 2014, 126, 721–726. [Google Scholar] [CrossRef]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of world- wide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Rettig, R.; Grisk, O. The kidney as a determinant of genetic hypertension: Evidence from renal transplantation studies. Hypertension 2005, 46, 463–468. [Google Scholar] [CrossRef]
- Curtis, J.J.; Luke, R.G.; Dustan, H.P.; Kashgarian, M.; Whelchel, J.D.; Jones, P.; Diethelm, A.G. Remission of essential hypertension after renal transplantation. N. Engl. J. Med. 1983, 309, 1009–1015. [Google Scholar] [CrossRef]
- Viera, A.J.; Neutze, D.M. Diagnosis of secondary hypertension: An age-based approach. Am. Fam. Physician 2010, 82, 1471–1478. [Google Scholar]
- Rimoldi, S.; Scherrer, U.; Messerli, F.H. Secondary arterial hypertension: When, who, and how to screen? Eur. Heart J. 2014, 35, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; Ferreira, T.; Chasman, D.I.; Jackson, A.U.; Schmidt, E.M.; Johnson, T.; Thorleifsson, G.; Luan, J.A.; Donnelly, L.A.; Kanoni, S.; et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat. Genet. 2016, 48, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, G.D.; Mohaupt, M.G.; Bianchetti, M.G. Monogenic forms of hypertension. Eur. J. Pediatr. 2012, 171, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Mayan, H.; Munter, G.; Shaharabany, M.; Mouallem, M.; Pauzner, R.; Holtzman, E.J.; Farfel, Z. Hypercalciuria in Familial Hyperkalemia and Hypertension Accompanies Hyperkalemia and Precedes Hypertension: Description of a Large Family with the Q565E WNK4 Mutation. J. Clin. Endocrinol. 2004, 89, 4025–4030. [Google Scholar] [CrossRef]
- Peng, J.B.; Liang, Y.; Warnock, D.G. WNK4 kinase enhances ECaC-mediated calcium transport. J. Am. Soc. Nephrol. 2004, 15, 62A. [Google Scholar]
- Kunchaparty, S.; Palcso, M.; Berkman, J.; Vela’zquez, H.; Bernstein, P.; Reilly, R.F.; Ellison, D.H. Defective processing and expression of the thiazide sensitive Na-Cl cotransporter as a cause Gitelman’s Syndrome. Am. J. Physiol. 1999, 277, 643–649. [Google Scholar] [CrossRef]
- Golbang, A.P.; Cope, G.; Hamad, A.; Murthy, M.; Liu, C.H.; Cuthbert, A.W.; O’Shaughnessy, K.M. Regulation of the expression of the Na/Cl cotransporter (NCCT) by WNK4 and WNK1: Evidence that accelerated dynamin-dependent endocytosis is not involved. Am. J. Physiol. Renal Physiol. 2006, 291, 1369–1376. [Google Scholar] [CrossRef]
- Kahle, K.T.; Wilson, F.H.; Leng, Q.; Lalioti, M.D.; O’Connell, A.D.; Dong, K.; Rapson, A.K.; MacGregor, G.G.; Giebisch, G.; Hebert, S.C.; et al. WNK4 regulates the balance between renal NaCl reabsorption and K secretion. Nat. Genet. 2003, 35, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Angell, J.; Mitchell, R.; Ellison, D.H. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Invest. 2003, 111, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Naray-Fejes-Toth, A.; Snyder, P.M.; Fejes-Toth, G. The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na transport. Proc. Natl. Acad. Sci. USA 2004, 101, 17434–17439. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Macgregor, G.G.; Wilson, F.H.; Van Hoek, A.N.; Brown, D.; Ardito, T.; Kashgarian, M.; Giebisch, G.; Hebert, S.C.; Boulpaep, E.L.; et al. Paracellular Cl—Permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 14877–14882. [Google Scholar] [CrossRef]
- Melo, Z.; Cruz-Rangel, S.; Bautista, R.; Vazquez, N.; Castaneda-Bueno, M.; Mount, D.B.; Pasantes-Morales, H.; Mercado, A.; Gamba, G. Molecular evidence for a role for K(+)–Cl(−) cotransporters in the kidney. Am. J. Physiol. Renal Physiol. 2013, 305, 1402–1411. [Google Scholar] [CrossRef]
- Leng, Q.; Kahle, K.T.; Rinehart, J.; MacGregor, G.G.; Wilson, F.H.; Canessa, C.M.; Lifton, R.P.; Hebert, S.C. WNK3, a kinase related to genes mutated in hereditary hypertension with hyperkalaemia, regulates the K+ channel ROMK1. J. Physiol. 2006, 571, 275–286. [Google Scholar] [CrossRef]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The slc12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef]
- Lo, Y.F.; Lin, Y.W.; Lin, S.H.; Huang, C.L.; Cheng, C.J. WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 4502–4507. [Google Scholar]
- Murthy, M.; Kurz, T.; O’Shaughnessy, K.M. WNK signalling pathways in blood pressure regulation. Cell. Mol. Life Sci. 2017, 74, 1261–1280. [Google Scholar] [CrossRef]
- Moriguchi, T.; Urushiyama, S.; Hisamoto, N.; Iemura, S.I.; Uchida, S.; Natsume, T.; Matsumoto, K.; Shibuya, H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 2006, 280, 42685–42693. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Zhu, X.; Wang, Z.; Subramanya, A.R.; Ellison, D.H. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive nacl cotransport. J. Clin. Invest. 2005, 115, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H. The thiazide-sensitive Na–Cl cotransporter and human disease: Reemergence of an old player. J. Am. Soc. Nephrol. 2003, 14, 538–540. [Google Scholar] [PubMed]
- Richardson, C.; Rafiqi, F.H.; Karlsson, H.K.; Moleleki, N.; Vandewalle, A.; Campbell, D.G.; Morrice, N.A.; Alessi, D.R. Activation of the thiazide-sensitive Na+–Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 2008, 121, 675–684. [Google Scholar] [CrossRef]
- Vidal-Petiot, E.; Elvira-Matelot, E.; Mutig, K.; Soukaseum, C.; Baudrie, V.; Wu, S.; Cheval, L.; Huc, E.; Cambillau, M.; Bachmann, S.; et al. WNK1-related familial hyperkalemic hypertension results from an increased expression of l-WNK1 specifically in the distal nephron. Proc. Natl. Acad. Sci. USA 2013, 110, 14366–14371. [Google Scholar] [CrossRef]
- Argaiz, E.R.; Chavez-Canales, M.; Ostrosky-Frid, M.; Rodriguez-Gama, A.; Vazquez, N.; Gonzalez-Rodriguez, X.; Garcia-Valdes, J.; Hadchouel, J.; Ellison, D.; Gamba, G. Kidney-Specific Wnk1 Isoform (Ks-Wnk1) Is a Potent Activator of Wnk4 and Ncc. Am. J. Physiol. Renal Physiol. 2018, 315, 734–745. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; Vazquez, N.; Castaneda-Bueno, M.; de-Los-Heros, P.; Cortes-Gonzalez, C.; Moreno, E.; Meade, P.; Bobadilla, N.A.; Gamba, G. WNK3–SPAK interaction is required for the modulation of NCC and other members of the slc12 family. Cell. Physiol. Biochem. 2012, 29, 291–302. [Google Scholar] [CrossRef]
- Rinehart, J.; Kahle, K.T.; de Los Heros, P.; Vazquez, N.; Meade, P.; Wilson, F.H.; Hebert, S.C.; Gimenez, I.; Gamba, G.; Lifton, R.P. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc. Natl. Acad. Sci. USA 2005, 102, 16777–16782. [Google Scholar] [CrossRef]
- Oi, K.; Sohara, E.; Rai, T.; Misawa, M.; Chiga, M.; Alessi, D.R.; Sasaki, S.; Uchida, S. A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters In Vivo. Biol. Open 2012, 1, 120–127. [Google Scholar] [CrossRef]
- Glover, M.; Zuber, A.M.; O’Shaughnessy, K.M. Renal and brain isoforms of WNK3 have opposite effects on NCCT expression. J. Am. Soc. Nephrol. 2009, 20, 1314–1322. [Google Scholar] [CrossRef]
- Lalioti, M.D.; Zhang, J.; Volkman, H.M.; Kahle, K.T.; Hoffmann, K.E.; Toka, H.R.; Nelson-Williams, C.; Ellison, D.H.; Flavell, R.; Booth, C.J.; et al. WNK4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat. Genet. 2006, 38, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, M.; Mori, T.; Isobe, K.; Sohara, E.; Susa, K.; Araki, Y.; Chiga, M.; Kikuchi, E.; Nomura, N.; Mori, Y.; et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013, 3, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, X.; Wang, D.; Li, J.; Zhou, B.; Shi, Z.; Gu, D.; Denson, D.D.; Eaton, D.C.; Cai, H. WNK4 kinase inhibits Maxi K channel activity by a kinase-dependent mechanism. Am. J. Physiol. Renal Physiol. 2011, 301, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Leviel, F.; Hubner, C.A.; Houillier, P.; Morla, L.; El Moghrabi, S.; Brideau, G.; Hassan, H.; Parker, M.D.; Kurth, I.; Kougioumtzes, A.; et al. The Na.-dependent chloride bicarbonate exchanger SLC4A8 mediates an electroneutral Na. reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Invest. 2010, 120, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Bueno, M.; Cervantes-Perez, L.G.; Vazquez, N.; Uribe, N.; Kantesaria, S.; Morla, L.; Bobadilla, N.A.; Doucet, A.; Alessi, D.R.; Gamba, G. Activation of the renal Na.:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. USA 2012, 109, 7929–7934. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.A.; Nelson, J.H.; Yang, C.L.; Curry, J.N.; Ellison, D.H. Overexpression of the Sodium Chloride Cotransporter Is Not Sufficient to Cause Familial Hyperkalemic Hypertension. Hypertension 2011, 58, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.; Picard, N.; Miller, R.L.; Riemondy, K.A.; Houillier, P.; Sohet, F.; Ramakrishnan, S.K.; Büsst, C.J.; Jayat, M.; Cornière, N.; et al. Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J. Am. Soc. Nephrol. 2013, 24, 1104–1113. [Google Scholar] [CrossRef]
- Verlander, J.W.; Hassell, K.A.; Royaux, I.E.; Glapion, D.M.; Wang, M.E.; Everett, L.A.; Green, E.D.; Wall, S.M. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: Role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003, 42, 356–362. [Google Scholar] [CrossRef]
- Wall, S.M.; Kim, Y.H.; Stanley, L.; Glapion, D.M.; Everett, L.A.; Green, E.D.; Verlander, J.W. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: Role in Cl- conservation. Hypertension 2004, 44, 982–987. [Google Scholar] [CrossRef]
- Schultheis, P.J.; Lorenz, J.N.; Meneton, P.; Nieman, M.L.; Riddle, T.M.; Flagella, M.; Duffy, J.J.; Doetschman, T.; Miller, M.L.; Shull, G.E. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na.-Cl- cotransporter of the distal convoluted tubule. J. Biol. Chem. 1998, 273, 29150–29155. [Google Scholar] [CrossRef]
- Amlal, H.; Petrovic, S.; Xu, J.; Wang, Z.; Sun, X.; Barone, S.; Soleimani, M. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(-)/HCO3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am. J. Physiol. Cell Physiol. 2010, 299, C33–C41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, X.; Shi, Y.; Shi, Z.; Niu, W.; Feng, X.; Gu, D.; Bao, H.F.; Ma, H.P.; Eaton, D.C.; et al. WNK1 activates large-conductance Ca2.activated K. channels through modulation of ERK1/2 signaling. J. Am. Soc. Nephrol. 2015, 26, 844–854. [Google Scholar] [CrossRef] [PubMed]
- López-Cayuqueo, K.I.; Chavez-Canales, M.; Pillot, A.; Houillier, P.; Jayat, M.; Baraka-Vidot, J.; Trepiccione, F.; Baudrie, V.; Büsst, C.; Soukaseum, C.; et al. A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int. 2018, 94, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, J.; Kobayashi, Y.; Umeda, T.; Vandewalle, A.; Takeda, K.; Ichijo, H.; Naguro, I. Osmotic stress induces the phosphorylation of WNK4 Ser575 via the p38MAPK-MK pathway. Sci. Rep. 2016, 6, 18710. [Google Scholar] [CrossRef]
- Wu, A.; Wolley, M.; Stowasser, M. The interplay of renal potassium and sodium handling in blood. J. Hum. Hypertens. 2019, 33, 508–523. [Google Scholar] [CrossRef]
- Boyd-Shiwarski, C.R.; Shiwarski, D.J.; Roy, A.; Namboodiri, H.N.; Nkashama, L.J.; Xie, J.; McClain, K.L.; Marciszyn, A.; Kleyman, T.R.; Tan, R.J.; et al. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol. Biol. Cell 2018, 29, 499–509. [Google Scholar] [CrossRef]
- Al-Qusairi, L.; Basquin, D.; Roy, A.; Stifanelli, M.; Rajaram, R.D.; Debonneville, A.; Nita, I.; Maillard, M.; Loffing, J.; Subramanya, A.R.; et al. Renal tubular SGK1 deficiency causes impaired K+ excretion via loss of regulation of NEDD4-2/WNK1 and ENaC. Am. J. Physiol. Renal Physiol. 2016, 311, F330–F342. [Google Scholar] [CrossRef]
- Melnikov, S.; Mayan, H.; Uchida, S.; Holtzman, E.J.; Farfel, Z. Cyclosporine metabolic side effects: Association with the WNK4 system. Eur. J. Clin. Invest. 2011, 41, 1113–1120. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Walsh, S.B.; McCormick, J.A.; Furstenberg, A.; Yang, C.L.; Roeschel, T.; Paliege, A.; Howie, A.J.; Conley, J.; Bachmann, S.; et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat. Med. 2011, 17, 1304–1309. [Google Scholar] [CrossRef]
- Van Angelen, A.A.; Glaudemans, B.; van der Kemp, A.W.; Hoenderop, J.G.; Bindels, R.J. Cisplatin-induced injury of the renal distal convoluted tubule is associated with hypomagnesaemia in mice. Nephrol. Dial. Transplant. 2013, 28, 879–889. [Google Scholar] [CrossRef]
- Tamari, M.; Daigo, Y.; Nakamura, Y. Isolation and characterization of a novel serine threonine kinase gene on chromosome 3p22–21.3. J. Hum. Genet. 1999, 44, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Piechotta, K.; Lu, J.; Delpire, E. Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J. Biol. Chem. 2002, 277, 50812–50819. [Google Scholar] [CrossRef] [PubMed]
- Vitari, A.C.; Deak, M.; Morrice, N.A.; Alessi, D.R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome, phosphorylate and active SPAK and OSR1 protein kinases. Biochem. J. 2005, 391, 17–24. [Google Scholar] [CrossRef]
- Piechotta, K.; Garbarini, N.J.; England, R.; Delpire, E. Characterization of the interaction of the stress kinase SPAK with the Na+ –K+ –2Cl− cotransporter in the nervous system: Evidence for a scaffolding role of the kinase. J. Biol. Chem. 2003, 278, 52848–52856. [Google Scholar] [CrossRef]
- Xu, B.E.; Stippec, S.; Lazrak, A.; Huang, C.L.; Cobb, M.H. WNK1 activates SGK1 by a PI-3 kinase-dependent and non-catalytic mechanism. J. Biol. Chem. 2005, 280, 34218–34223. [Google Scholar] [CrossRef]
- Rafiqi, F.H.; Zuber, A.M.; Glover, M.; Richardson, C.; Fleming, S.; Jovanovic, S.; Jovanovic, A.; O’Shaughnessy, K.M.; Alessi, D.R. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2010, 2, 63–75. [Google Scholar] [CrossRef]
- McCormick, J.A.; Ellison, D.H. The WNKs: Atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011, 91, 177–219. [Google Scholar] [CrossRef]
- Yang, S.S.; Lo, Y.F.; Wu, C.C.; Lin, S.W.; Yeh, C.J.; Chu, P.; Sytwu, H.K.; Uchida, S.; Sasaki, S.; Lin, S.H. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010, 21, 1868–1877. [Google Scholar] [CrossRef]
- Zhang, J.; Siew, K.; Macartney, T.; O’Shaughnessy, K.M.; Alessi, D.R. Critical role of the SPAK protein kinase CCT domain in controlling blood pressure. Hum. Mol. Genet. 2015, 24, 4545–4558. [Google Scholar] [CrossRef]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015, 6, 39–50. [Google Scholar] [CrossRef]
- Grimm, P.R.; Taneja, T.K.; Liu, J.; Coleman, R.; Chen, Y.Y.; Delpire, E.; Wade, J.B.; Welling, P.A. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J. Biol. Chem. 2012, 2, 37673–37690. [Google Scholar] [CrossRef]
- Vidal-Petiot, E.; Cheval, L.; Faugeroux, J.; Malard, T.; Doucet, A.; Jeunemaitre, X.; Hadchouel, J. A New Methodology for Quantification of Alternatively Spliced Exons Reveals a Highly Tissue-Specific Expression Pattern of WNK1 Isoforms. PLoS ONE 2012, 7, e37751. [Google Scholar] [CrossRef] [PubMed]
- Ferdaus, M.Z.; Barber, K.W.; Lopez-Cayuqueo, K.I.; Terker, A.S.; Argaiz, E.R.; Gassaway, B.M.; Chambrey, R.; Gamba, G.; Rinehart, J.; McCormick, J.A. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J. Physiol. 2016, 594, 4945–4966. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Cheng, S.X.; Leng, Q.; Kahle, K.T.; Rinehart, J.; Lalioti, M.D.; Volkman, H.M.; Wilson, F.H.; Hebert, S.C.; Lifton, R.P. WNK4 regulates activity of the epithelial Na_ channel in vitro and In Vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 4020–4024. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.E.; Stippec, S.; Chu, P.Y.; Lazrak, A.; Li, X.J.; Lee, B.H.; English, J.M.; Ortega, B.; Huang, C.L.; Cobb, M.H. WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc. Natl. Acad. Sci. USA 2005, 102, 10315–10320. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.A.; Bhalla, V.; Pao, A.C.; Pearce, D. SGK1: A rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology 2005, 20, 134–139. [Google Scholar] [CrossRef]
- Yang, S.S.; Morimoto, T.; Rai, T.; Chiga, M.; Sohara, E.; Ohno, M.; Uchida, K.; Lin, S.H.; Moriguchi, T.; Shibuya, H.; et al. Molecular pathogenesis of pseudohypoaldosteronism type II: Generation and analysis of a Wnk4(D561A/_) knockin mouse model. Cell Metab. 2007, 5, 331–344. [Google Scholar] [CrossRef]
- Take, C.; Ikeda, K.; Kurasawa, T.; Kurokawa, K. Increased chloride reabsorption as an inherited renal tubular defect in familial type II pseudohypoaldosteronism. N. Engl. J. Med. 1991, 324, 472–476. [Google Scholar] [CrossRef]
- Lazrak, A.; Liu, Z.; Huang, C.L. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc. Natl. Acad. Sci. USA 2006, 103, 1615–1620. [Google Scholar] [CrossRef]
- DeAizpurua, H.J.; Cram, D.S.; Naselli, G.; Devereux, L.; Dorow, D.S. Expression of mixed lineage kinase-1 in pancreatic β-cell lines at different stages of maturation and during embryonic pancreas development. J. Biol. Chem. 1997, 272, 16364–16373. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.; Murthy, M.; Golbang, A.P.; Hamad, A.; Liu, C.H.; Cuthbert, A.W.; O’Shaughnessy, K.M. WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J. Am. Soc. Nephrol. 2006, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Mayan, H.; Attar-Herzberg, D.; Shaharabany, M.; Holtzman, E.J.; Farfel, Z. Increased urinary Na-Cl cotransporter protein in familial hyperkalaemia and hypertension. Nephrol. Dial. Transplant. 2008, 2, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.B.; Bell, P.D. Cellular mechanisms of WNK4-mediated regulation of ion transport proteins in the distal tubule. Kidney Int. 2006, 69, 2116–2118. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef]
- Yamauchi, K.; Rai, T.; Kobayashi, K.; Sohara, E.; Suzuki, T.; Itoh, T.; Suda, S.; Hayama, A.; Sasaki, S.; Uchida, S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 2004, 101, 4690–4694. [Google Scholar] [CrossRef]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Furukawa, M.; He, Y.J.; Borchers, C.; Xiong, Y. Targeting of protein ubiquitination by BTB-Cullin3-Roc1 ubiquitin ligases. Nat. Cell Biol. 2003, 5, 1001–1007. [Google Scholar] [CrossRef]
- Stogios, P.J.; Downs, G.S.; Jauhal, J.J.; Nandra, S.K.; Prive, G.G. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005, 6, R82. [Google Scholar] [CrossRef]
- Hudson, A.M.; Cooley, L. Phylogenetic, structural and functional relatioships between WD and Kelch repeat proteins. Subcell. Biochem. 2008, 48, 6–19. [Google Scholar]
- Zimmerman, E.S.; Schulman, B.A.; Zheng, N. Structural assembly of Cullin-ring ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2010, 20, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Schumacher, F.-R.; Mehellou, Y.; Johnson, C.; Knebel, A.; Macartney, T.J.; Wood, N.T.; Alessi, D.R.; Kurz, T. The CUL3–KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem. J. 2013, 451, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Susa, K.; Sohara, E.; Rai, T.; Zeniya, M.; Mori, Y.; Mori, T.; Chiga, M.; Nomura, N.; Nishida, H.; Takahashi, D.; et al. Impaired degradation of WNK1 and WNK4 kinases causes PHAII in mutant KLHL3 knock-in mice. Hum. Mol. Genet. 2014, 23, 5052–5060. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Susa, K.; Mori, T.; Isobe, K.; Araki, Y.; Inoue, Y.; Yoshizaki, Y.; Ando, F.; Mori, Y.; Mandai, S.; et al. KLHL3 Knockout Mice Reveal the Physiological Role of KLHL3 and the Pathophysiology of Pseudohypoaldosteronism Type II Caused by Mutant KLHL3. Mol. Cell. Biol. 2017, 37, e00508–e00516. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, C.; Cai, R.; Chen, X.Z.; Peng, J.B. Unveiling the Distinct Mechanisms by which Disease-Causing Mutations in the Kelch Domain of KLHL3 Disrupt the Interaction with the Acidic Motif of WNK4 through Molecular Dynamics Simulation. Biochemistry 2019, 58, 2105–2115. [Google Scholar] [CrossRef]
- Lin, C.M.; Cheng, C.J.; Yang, S.S.; Tseng, M.H.; Yen, M.T.; Sung, C.C.; Lin, S.H. Generation and analysis of a mouse model of pseudohypoaldosteronism type II caused by KLHL3 mutation in BTB domain. FASEB J. 2019, 33, 1051–1061. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- McCormick, J.A.; Yang, C.L.; Zhang, C.; Davidge, B.; Blankenstein, K.I.; Terker, A.S.; Yarbrough, B.; Meermeier, N.P.; Park, H.J.; McCully, B.; et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J. Clin. Invest. 2014, 124, 4723–4736. [Google Scholar] [CrossRef]
- Abdel Khalek, W.; Rafael, C.; Loisel-Ferreira, I.; Kouranti, I.; Clauser, E.; Hadchouel, J.; Jeunemaitre, X. Severe Arterial Hypertension from Cullin 3 Mutations Is Caused by Both Renal and Vascular Effects. J. Am. Soc. Nephrol. 2019, 30, 811–823. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WNK1 | WNK4 | KLHL3 | CUL3 | |
|---|---|---|---|---|
| Hypertension | Least severe phenotype and metabolic disorder often precedes hypertension | Metabolic disorder often precedes hypertension | Recessive mutations are more severe and diagnosed at an earlier age than dominant mutations | Most severe phenotype. Presents at youngest age (>90% had hypertension <age 18. |
| Hyperkalaemia | Least severe | Yes | Dominant mutations had significantly higher serum K+ than recessive mutations | Most severe Presents at youngest age |
| Metabolic Acidosis | Least severe | Yes | Yes | Most severe |
| Other features | Hypercalciuria Hypocalcaemia Decreased bone mineral density Renal calcium stones | Fertility likely affected in de novo mutations. Growth impairment most likely |
| Diagnosis | Genes/Loci | Gene Product | Inheritance | Reason for Hypertension | Other Features |
|---|---|---|---|---|---|
| Gordon Syndrome | 1q31-q42 (Unknown gene) 17p11-q21 (WNK4) 12p13 (WNK1) 5q31 (KLHL3) 2q36 (CUL3) | Mutant WNK-kinase 1, WNK-kinase 4, Kelch-like 3 or Cullin 3 | Dominant (can be recessive or de novo) | Excessive sodium reabsorption via NCC |
|
| Liddle Syndrome | 16p12.1 (SCNN1B and SCNN1G) 12p13.31 (SCNN1A) | Faulty ENaC (β subunit SCNN1B, γ subunit SCNN1G or α subunit SCNN1A) | Dominant | Excessive sodium reabsorption by ENaC |
|
| Congenital Adrenal Hyperplasia | 1p12 (HSD3B2) 6p21 (CYP21A2) | 3-β-hydroxysteroid dehydrogenase 2 deficiency 21-hydroxylase deficiency | Recessive | Excessive ACTH to try to maintain cortisol which causes excess mineralocorticoid-like hormones which cross react with mineralocorticoid receptors |
|
| Glucocorticoid Remedial Hyperaldosteronism | 8q24 (CYP11B1 & CYP11B2) | 18-hydroxylase, 11-β hydroxylase hybrid gene causing aldosterone to be under ACTH control | Dominant | Inappropriately high aldosterone levels |
|
| Syndrome of Apparent Mineralocorticoid Excess | 16q22 (HSD11B2) | 11-β hydroxysteroid dehydrogenase (type 2) deficiency | Recessive | Defect in ability to metabolise cortisol to cortisone resulting in cortisol cross reactivity with the mineralocorticoid receptor |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabillard, H.; Sayer, J.A. The Molecular Genetics of Gordon Syndrome. Genes 2019, 10, 986. https://doi.org/10.3390/genes10120986
Mabillard H, Sayer JA. The Molecular Genetics of Gordon Syndrome. Genes. 2019; 10(12):986. https://doi.org/10.3390/genes10120986
Chicago/Turabian StyleMabillard, Holly, and John A. Sayer. 2019. "The Molecular Genetics of Gordon Syndrome" Genes 10, no. 12: 986. https://doi.org/10.3390/genes10120986
APA StyleMabillard, H., & Sayer, J. A. (2019). The Molecular Genetics of Gordon Syndrome. Genes, 10(12), 986. https://doi.org/10.3390/genes10120986