Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation

 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines Culture
2.2. Tissue Samples
2.3. Chemicals
2.4. Lactate Supplementation
2.5. Conditioned Medium
2.6. SIRT Inhibition
2.7. MCT Inhibition
2.8. Extracellular Glucose and Lactate Quantification
2.9. RNA Extraction, cDNA Synthesis, and RT-qPCR
2.10. Western Blot and Coimmunoprecipitation
2.11. Immunofluorescence
2.12. Cell Proliferation Assay
2.13. Wound Healing Assay
2.14. Invasion Assay
2.15. Chicken Chorioallantoic Membrane (CAM) Assay
2.16. Immunohistochemistry
2.17. Statistical Analysis
3. Results
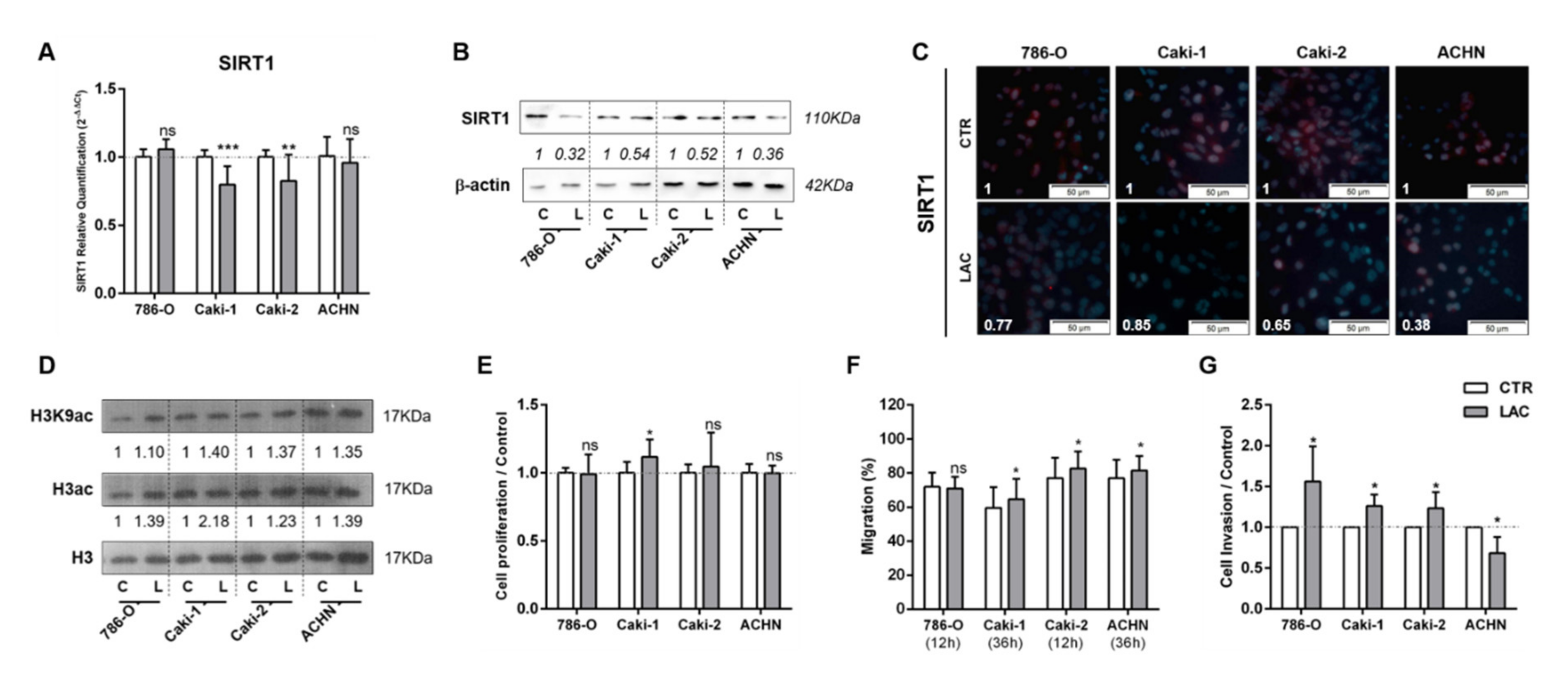
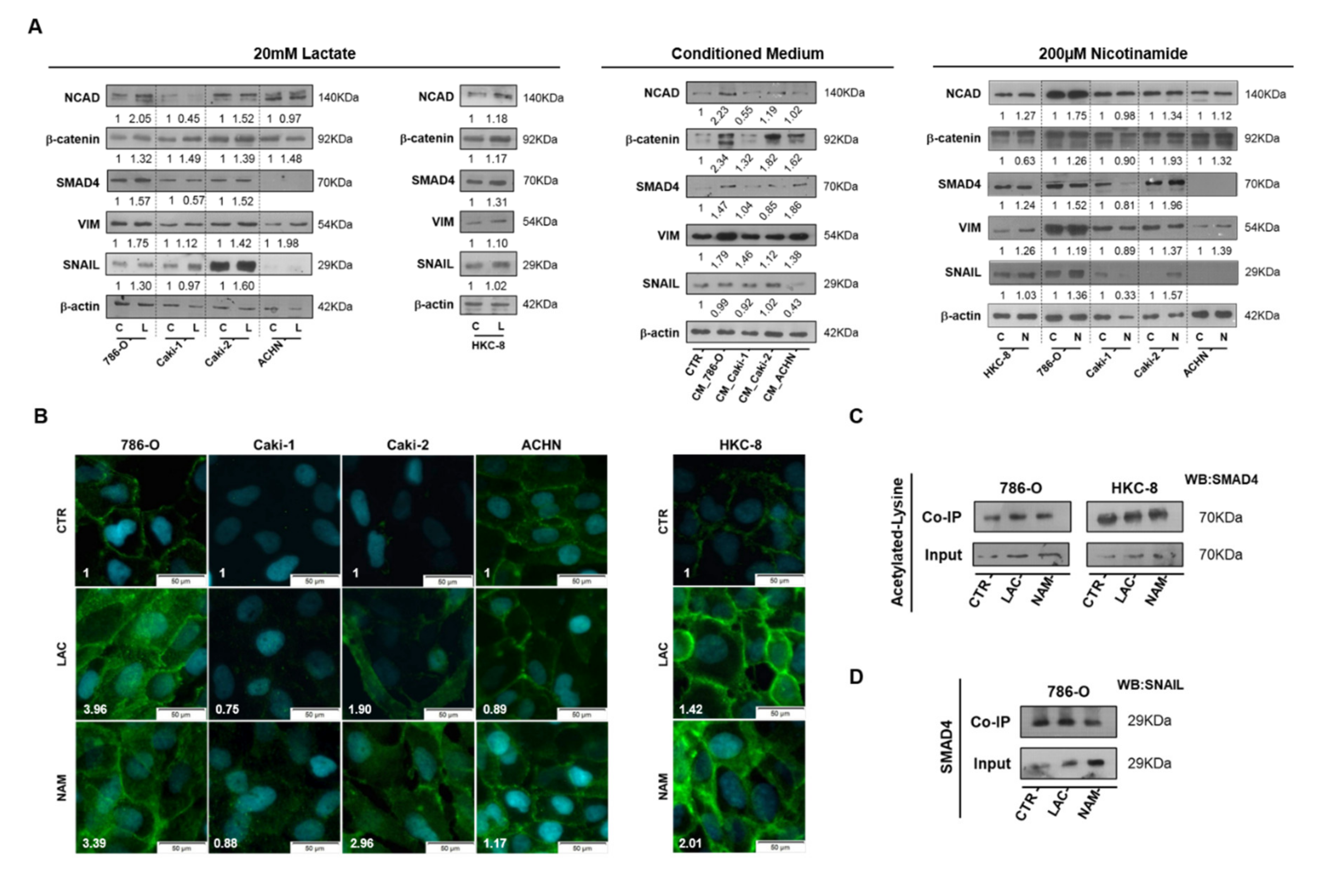
3.1. Lactate Decreased SIRT1 Expression, Increasing Cell Migration and Invasion in RCC
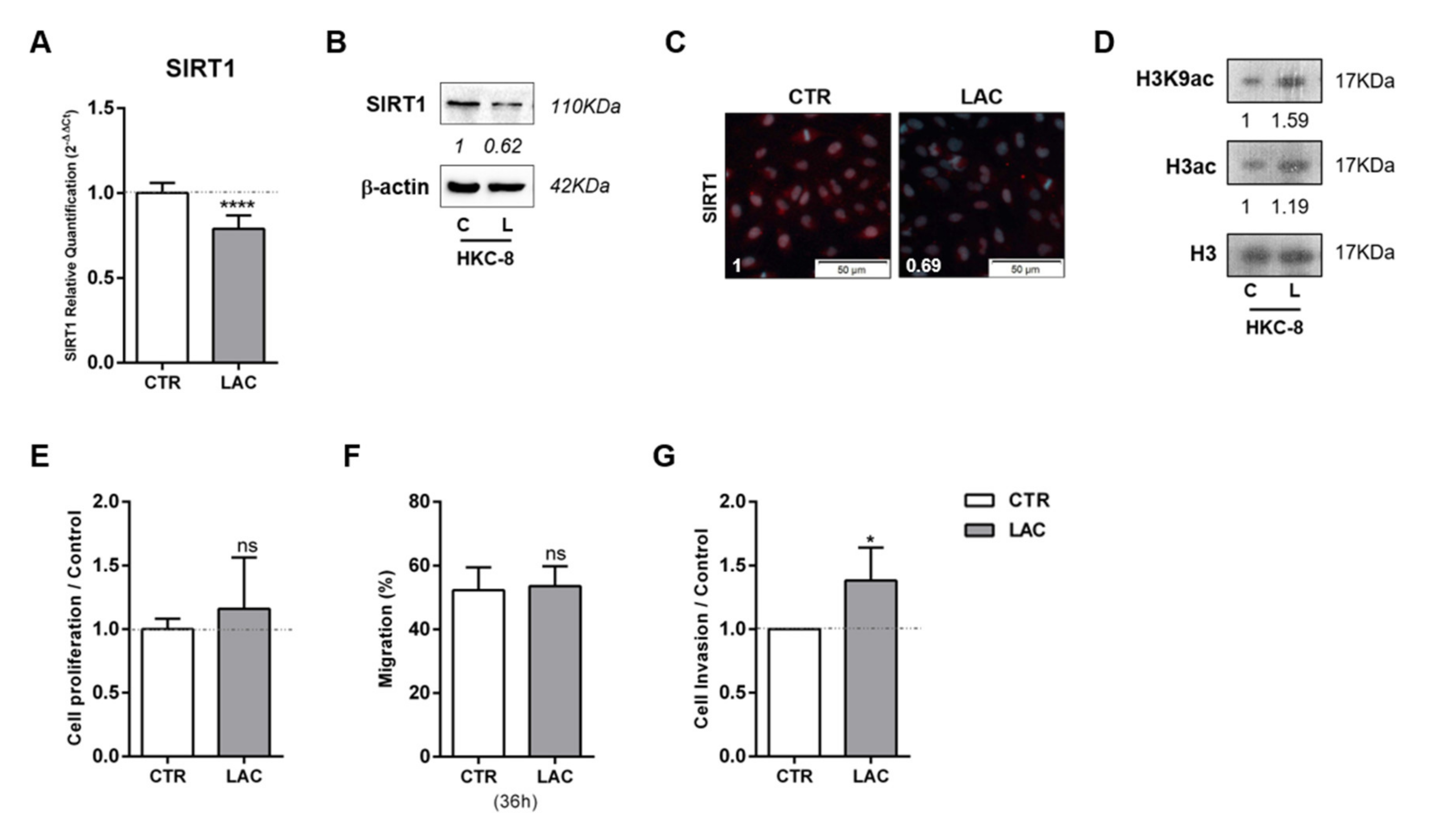
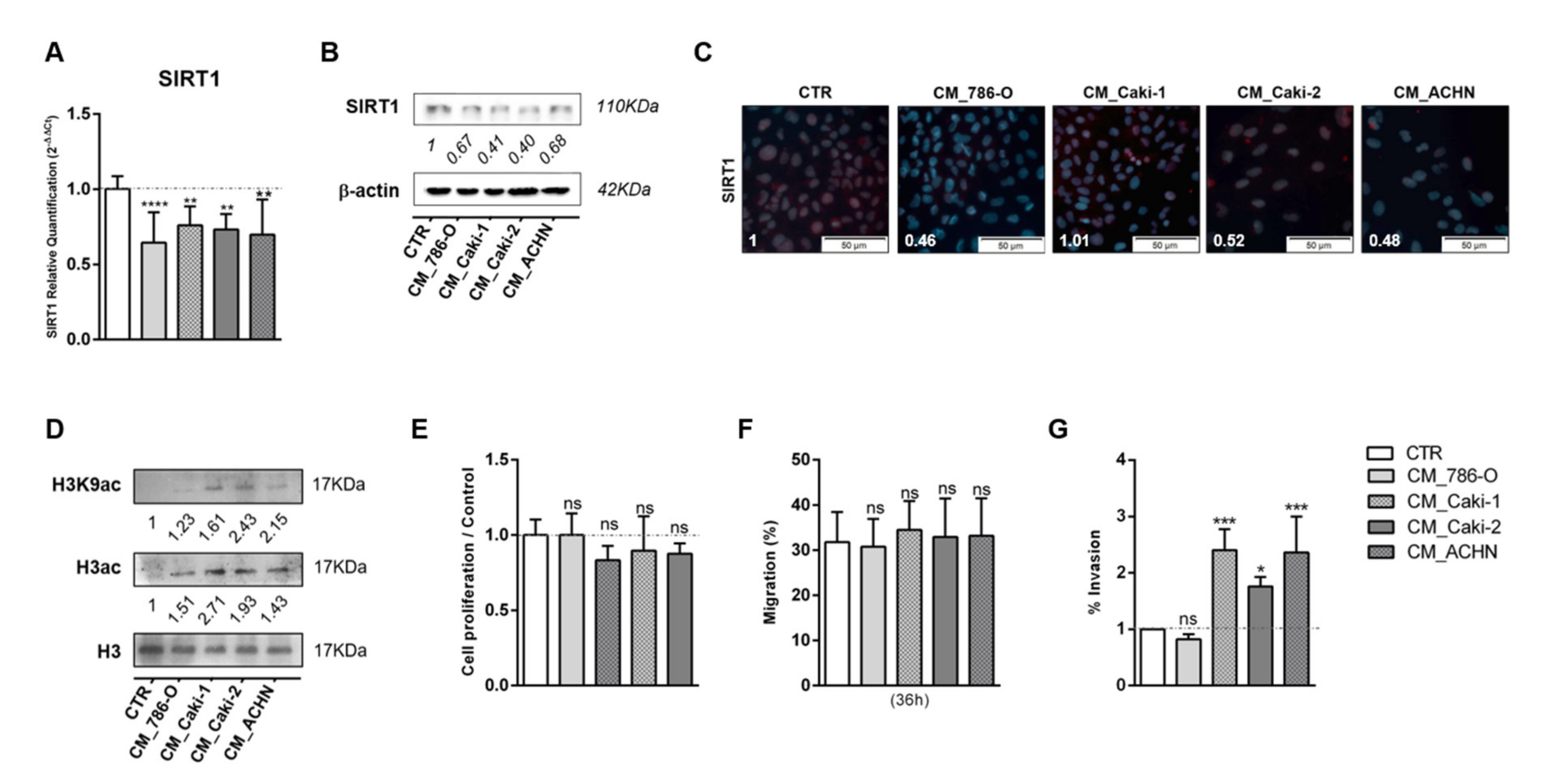
3.2. Tumor Metabolism Modulated Epigenetic Landscape of Normal Adjacent Cells
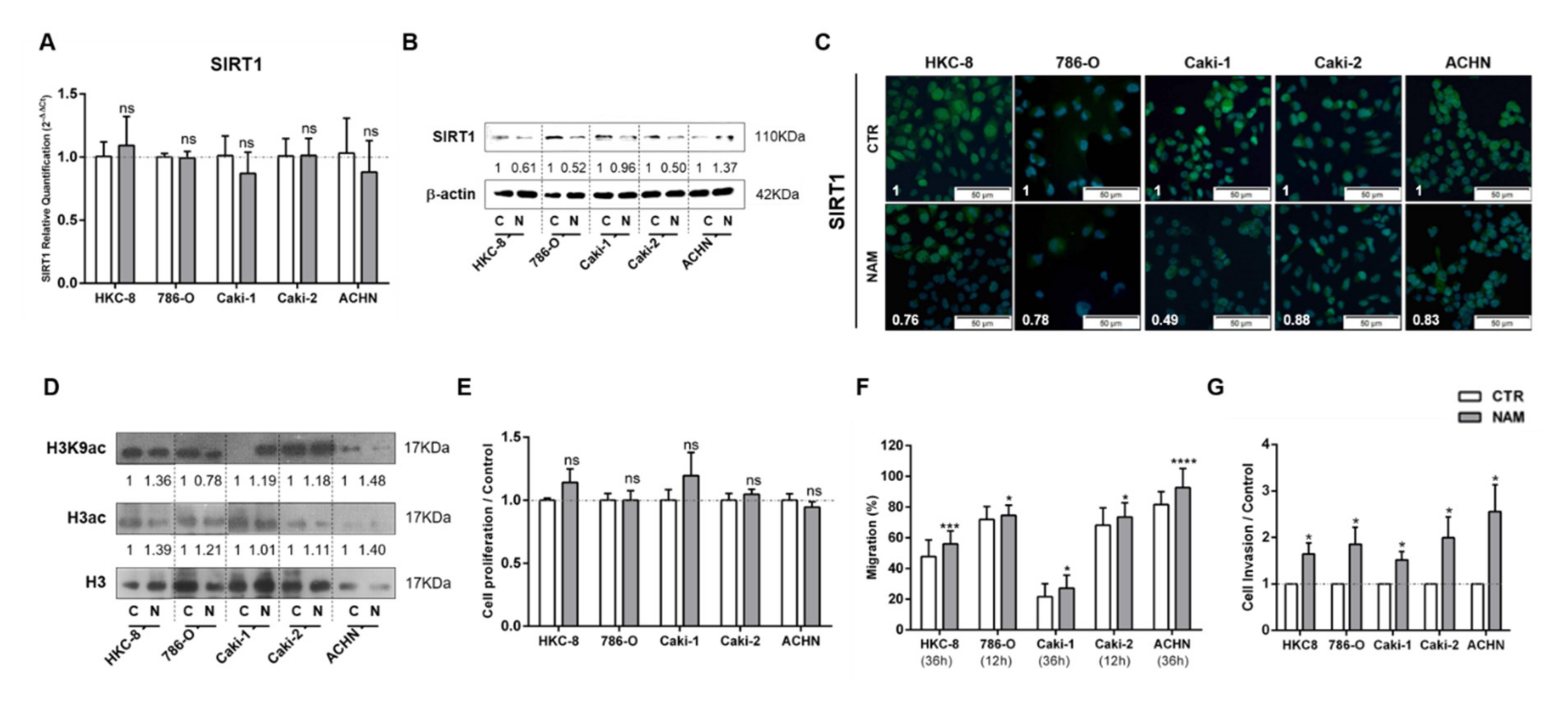
3.3. Treatment with SIRT1 Inhibitor, NAM, Mimicked the Effect of Lactate in Kidney Cells
3.4. Lactate Promoted EMT through SIRT1-Dependent SMAD4 Axis
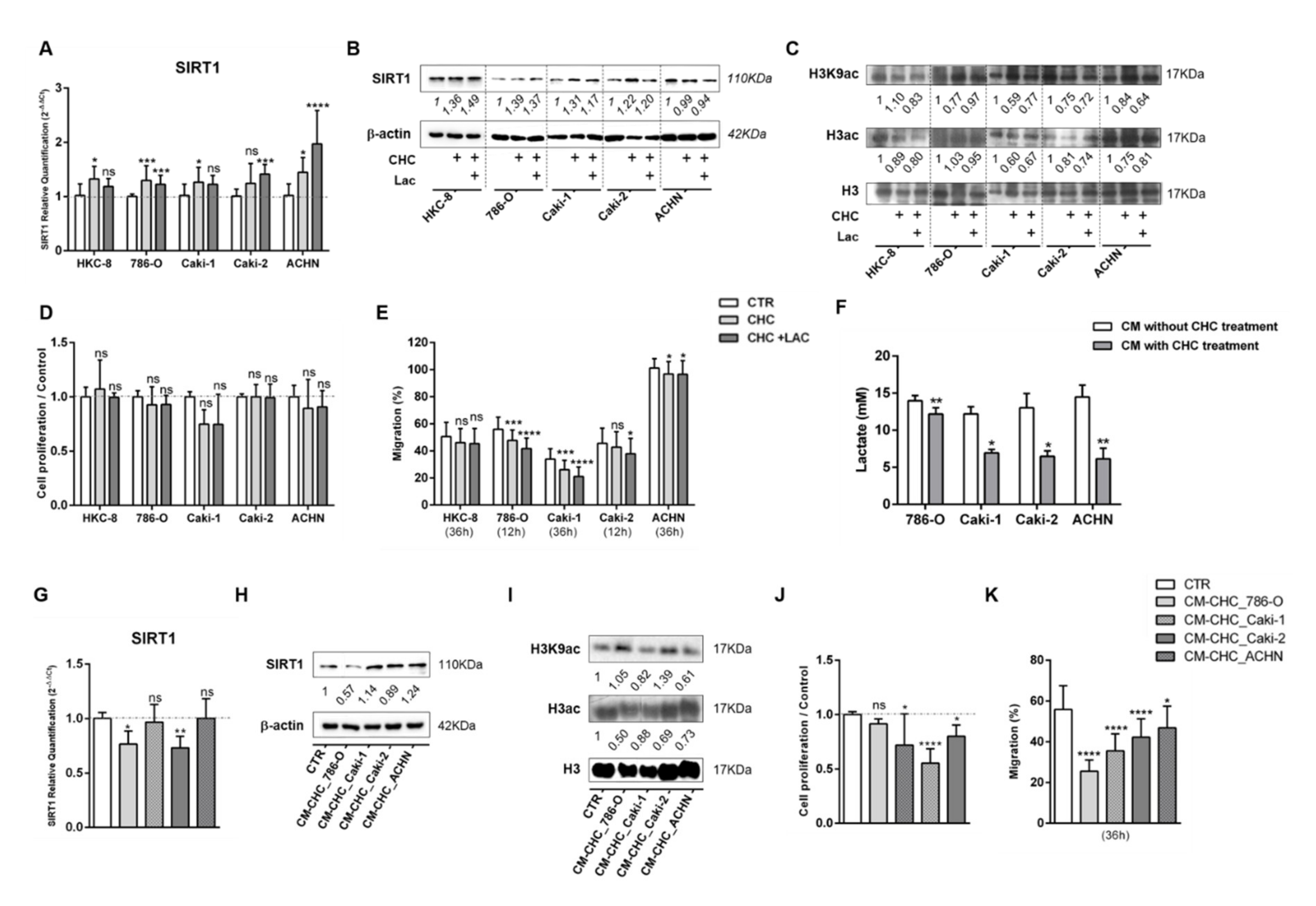
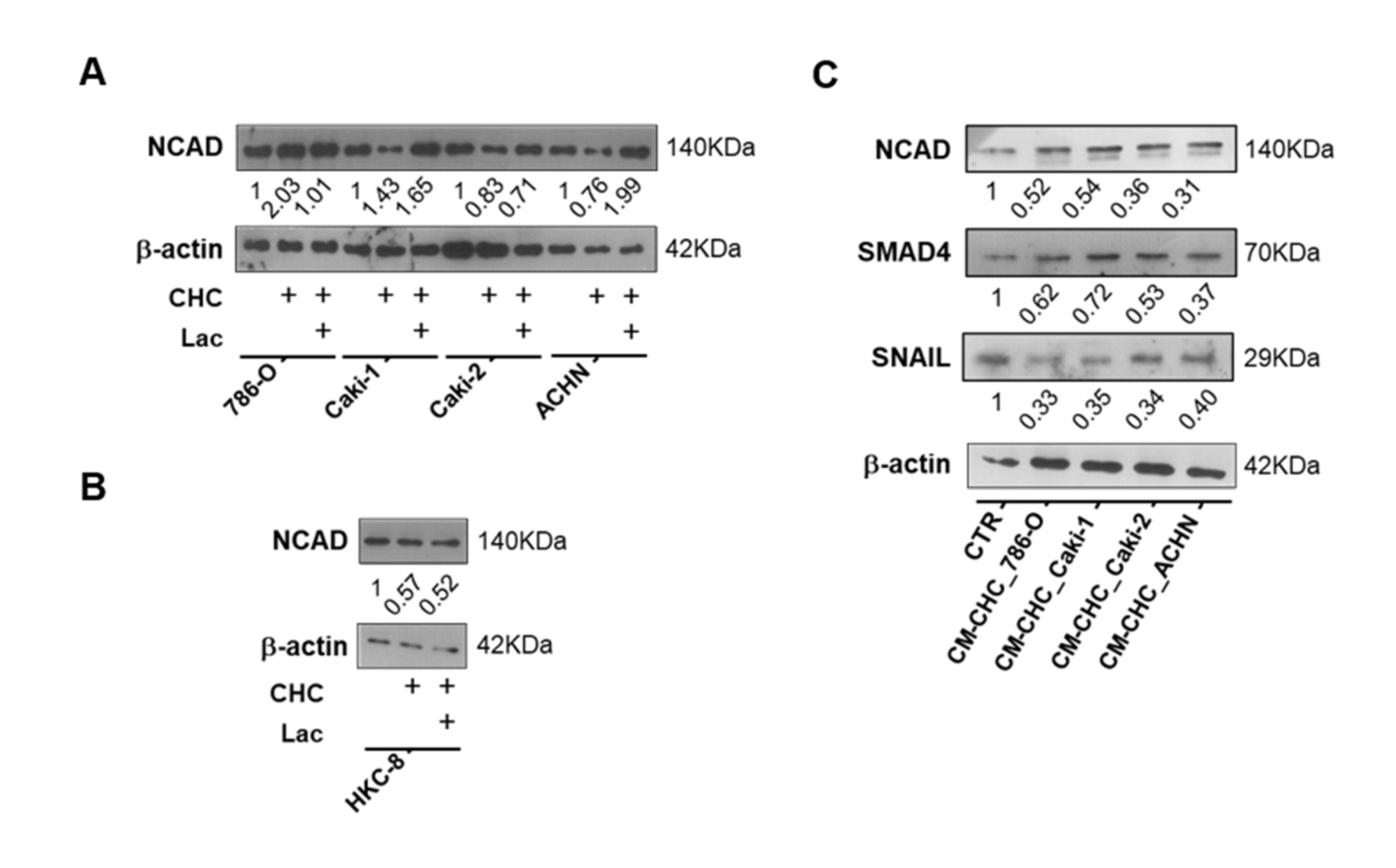
3.5. Lactate Transport Inhibition Increased SIRT1 Expression in RCC and Normal Kidney Cell Lines, Impacting Aggressiveness
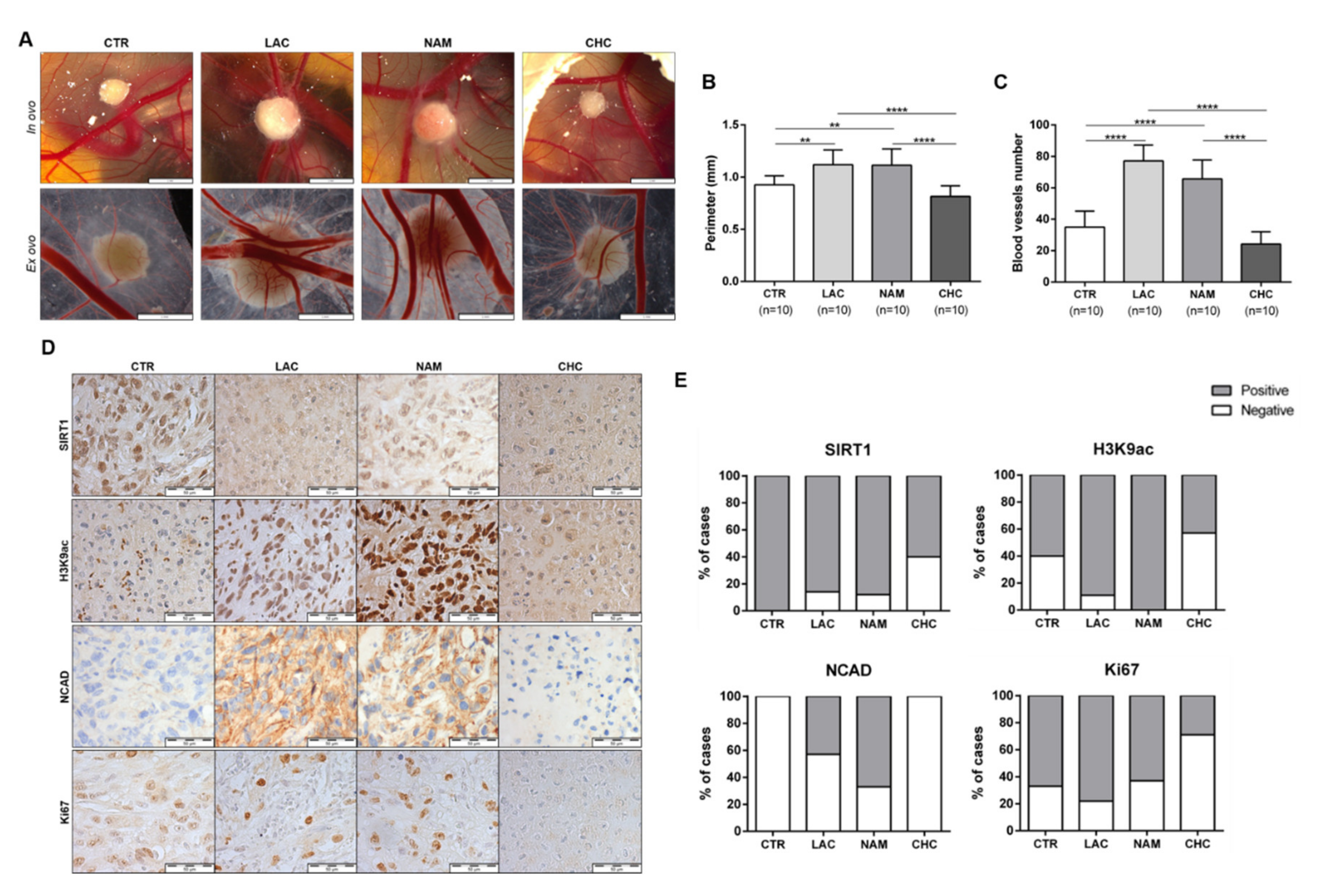
3.6. In Vivo Effect of Lactate, NAM, and CHC on RCC Cells
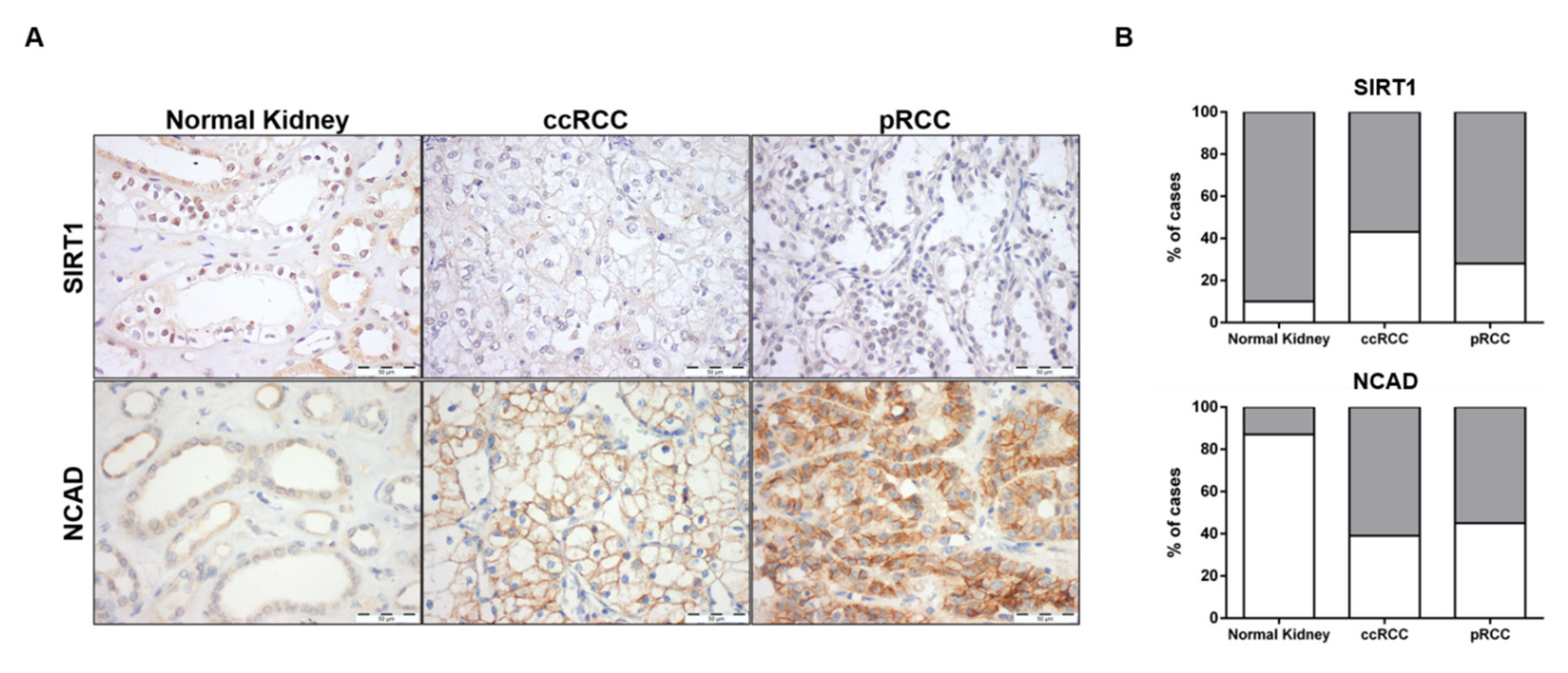
3.7. SIRT1 and N-Cadherin Protein Levels in Primary Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Bhayani, S.; Bro, W.P.; Chang, S.S.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; Fishman, M.; et al. Kidney cancer, version 2.2017, nccn clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 804–834. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P. Update on the biology and management of renal cell carcinoma. J. Investig. Med. 2019, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Feron, O. Pyruvate into lactate and back: From the warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Lameirinhas, A.; Henrique, R.; Baltazar, F.; Jeronimo, C. The metabolic landscape of urological cancers: New therapeutic perspectives. Cancer Lett. 2020, 477, 76–87. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Beaudet, A.L. Epigenetics and human disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a019497. [Google Scholar] [CrossRef]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bulow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Lameirinhas, A.; Henrique, R.; Jeronimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and d-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal. 2015, 13, 36. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Hagelkruys, A.; Sawicka, A.; Rennmayr, M.; Seiser, C. The biology of hdac in cancer: The nuclear and epigenetic components. Handb. Exp. Pharmacol. 2011, 206, 13–37. [Google Scholar] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual tumor suppressor and tumor promoter action of sirtuins in determining malignant phenotype. Front. Pharmacol. 2019, 10, 38. [Google Scholar] [CrossRef]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The roles of sirtuin family proteins in cancer progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef]
- Song, N.Y.; Surh, Y.J. Janus-faced role of sirt1 in tumorigenesis. Ann. N. Y. Acad. Sci. 2012, 1271, 10–19. [Google Scholar] [CrossRef]
- Palmirotta, R.; Cives, M.; Della-Morte, D.; Capuani, B.; Lauro, D.; Guadagni, F.; Silvestris, F. Sirtuins and cancer: Role in the epithelial-mesenchymal transition. Oxid. Med. Cell. Longev. 2016, 2016, 3031459. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of mct1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335–46353. [Google Scholar] [CrossRef]
- Costa-Pinheiro, P.; Ramalho-Carvalho, J.; Vieira, F.Q.; Torres-Ferreira, J.; Oliveira, J.; Goncalves, C.S.; Costa, B.M.; Henrique, R.; Jeronimo, C. Microrna-375 plays a dual role in prostate carcinogenesis. Clin. Epigenetics 2015, 7, 42. [Google Scholar] [CrossRef]
- Erman, M.; Benekli, M.; Basaran, M.; Bavbek, S.; Buyukberber, S.; Coskun, U.; Demir, G.; Karabulut, B.; Oksuzoglu, B.; Ozkan, M.; et al. Renal cell cancer: Overview of the current therapeutic landscape. Expert Rev. Anticancer Ther. 2016, 16, 955–968. [Google Scholar] [CrossRef]
- Pereira-Nunes, A.; Afonso, J.; Granja, S.; Baltazar, F. Lactate and lactate transporters as key players in the maintenance of the warburg effect. Adv. Exp. Med. Biol. 2020, 1219, 51–74. [Google Scholar]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Lameirinhas, A.; Miranda-Goncalves, V.; Henrique, R.; Jeronimo, C. The complex interplay between metabolic reprogramming and epigenetic alterations in renal cell carcinoma. Genes 2019, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pastor, B.; Mostoslavsky, R. Sirtuins, metabolism, and cancer. Front. Pharmacol. 2012, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The critical role of the class iii histone deacetylase sirt1 in cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. Emt is associated with an epigenetic signature of ecm remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Miethe, S.; Schindler, V.; Alhamdan, F.; Garn, H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell Signal. 2020, 69, 109523. [Google Scholar] [CrossRef]
- Simic, P.; Williams, E.O.; Bell, E.L.; Gong, J.J.; Bonkowski, M.; Guarente, L. Sirt1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 2013, 3, 1175–1186. [Google Scholar] [CrossRef]
- Chen, I.C.; Chiang, W.F.; Huang, H.H.; Chen, P.F.; Shen, Y.Y.; Chiang, H.C. Role of sirt1 in regulation of epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis. Mol. Cancer 2014, 13, 254. [Google Scholar] [CrossRef]
- Sun, L.; Li, H.; Chen, J.; Dehennaut, V.; Zhao, Y.; Yang, Y.; Iwasaki, Y.; Kahn-Perles, B.; Leprince, D.; Chen, Q.; et al. A sumoylation-dependent pathway regulates sirt1 transcription and lung cancer metastasis. J. Natl. Cancer Inst. 2013, 105, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, H.; Chen, J.; Iwasaki, Y.; Kubota, T.; Matsuoka, M.; Shen, A.; Chen, Q.; Xu, Y. Piasy mediates hypoxia-induced sirt1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells. J. Cell Sci. 2013, 126, 3939–3947. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, C.; Meng, T.; Zhang, W.; Zhou, Q. Resveratrol attenuates renal injury and fibrosis by inhibiting transforming growth factor-beta pathway on matrix metalloproteinase 7. Exp. Biol. Med. 2016, 241, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, F.; Zha, S.; Cao, Q.; Sheng, J.; Chen, S. Sirt1 inhibits tgf-beta-induced endothelial-mesenchymal transition in human endothelial cells with smad4 deacetylation. J. Cell. Physiol. 2018, 233, 9007–9014. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin inhibition by nicotinamide: Altering the nad(+) cosubstrate specificity of a sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Audrito, V.; Vaisitti, T.; Rossi, D.; Gottardi, D.; D’Arena, G.; Laurenti, L.; Gaidano, G.; Malavasi, F.; Deaglio, S. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/mir-34a/sirt1 tumor suppressor network. Cancer Res. 2011, 71, 4473–4483. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of sirtuin histone deacetylase sirt1 in prostate cancer. A target for prostate cancer management via its inhibition? J. Biol. Chem. 2009, 284, 3823–3832. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (mcts) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1323, discussion 1323–1314. [Google Scholar] [CrossRef]
- Munoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queiros, O.; Preto, A.; Casal, M. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar] [PubMed]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Bezerra, F.; Costa-Almeida, R.; Freitas-Cunha, M.; Soares, R.; Martinho, O.; Reis, R.M.; Pinheiro, C.; Baltazar, F. Monocarboxylate transporter 1 is a key player in glioma-endothelial cell crosstalk. Mol. Carcinog. 2017, 56, 2630–2642. [Google Scholar] [CrossRef]
- Jeh, S.U.; Park, J.J.; Lee, J.S.; Kim, D.C.; Do, J.; Lee, S.W.; Choi, S.M.; Hyun, J.S.; Seo, D.H.; Lee, C.; et al. Differential expression of the sirtuin family in renal cell carcinoma: Aspects of carcinogenesis and prognostic significance. Urol. Oncol. 2017, 35, 675.e9–675.e15. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Gonçalves, V.; Lameirinhas, A.; Macedo-Silva, C.; Lobo, J.; C. Dias, P.; Ferreira, V.; Henrique, R.; Jerónimo, C. Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation. Cells 2020, 9, 1053. https://doi.org/10.3390/cells9041053
Miranda-Gonçalves V, Lameirinhas A, Macedo-Silva C, Lobo J, C. Dias P, Ferreira V, Henrique R, Jerónimo C. Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation. Cells. 2020; 9(4):1053. https://doi.org/10.3390/cells9041053
Chicago/Turabian StyleMiranda-Gonçalves, Vera, Ana Lameirinhas, Catarina Macedo-Silva, João Lobo, Paula C. Dias, Verónica Ferreira, Rui Henrique, and Carmen Jerónimo. 2020. "Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation" Cells 9, no. 4: 1053. https://doi.org/10.3390/cells9041053
APA StyleMiranda-Gonçalves, V., Lameirinhas, A., Macedo-Silva, C., Lobo, J., C. Dias, P., Ferreira, V., Henrique, R., & Jerónimo, C. (2020). Lactate Increases Renal Cell Carcinoma Aggressiveness through Sirtuin 1-Dependent Epithelial Mesenchymal Transition Axis Regulation. Cells, 9(4), 1053. https://doi.org/10.3390/cells9041053