Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis


Abstract
1. Introduction
2. Genetics of ALS
2.1. The Newly Identified ALS-Associated Genes (2018–2020)
2.2. The Old Genes with Important Contribution to ALS
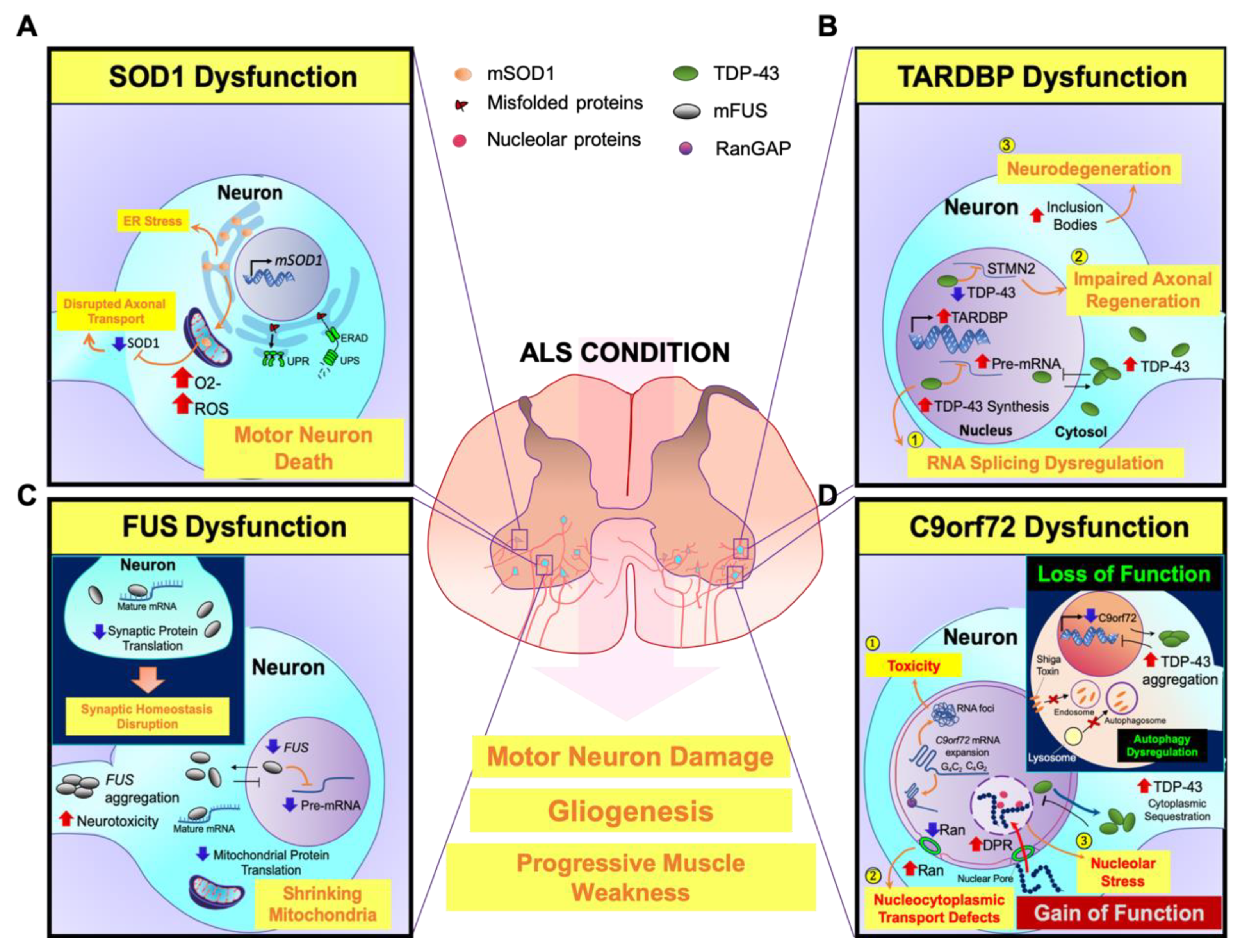
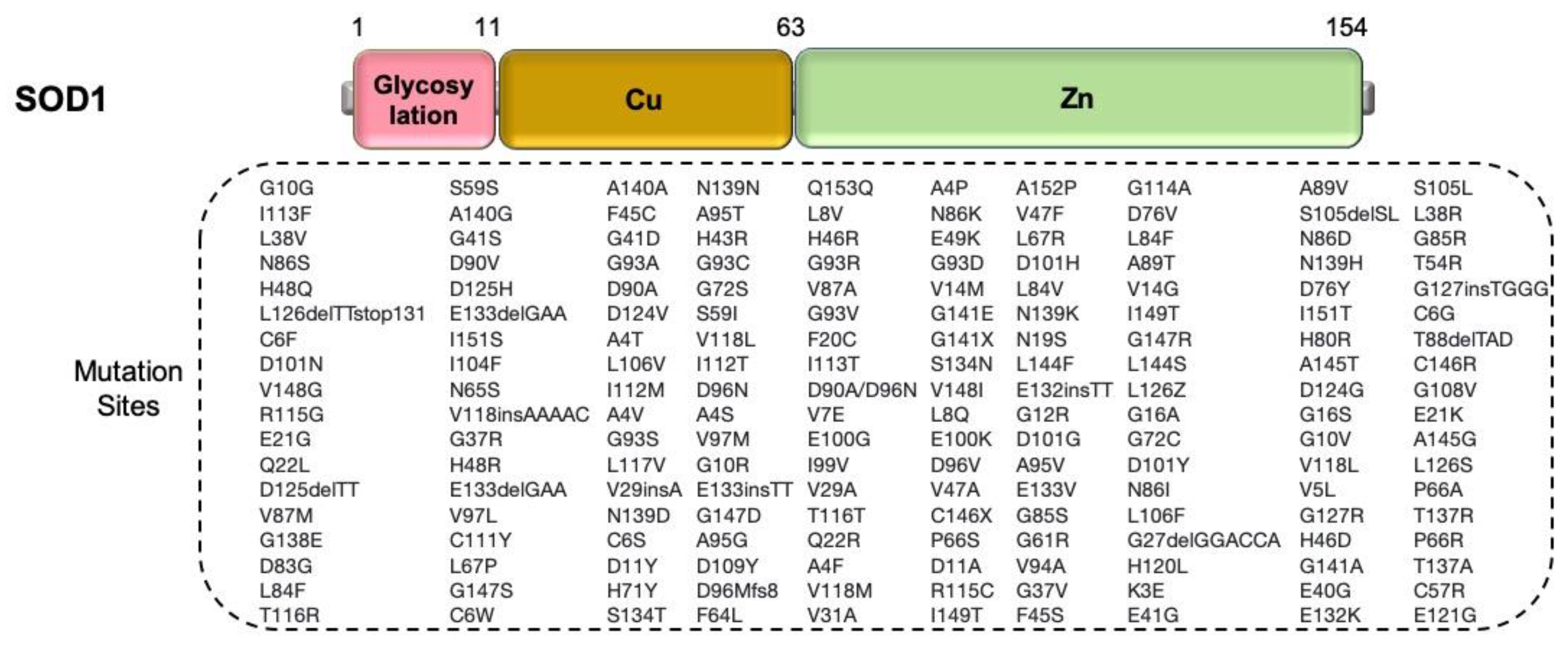
2.2.1. SOD1
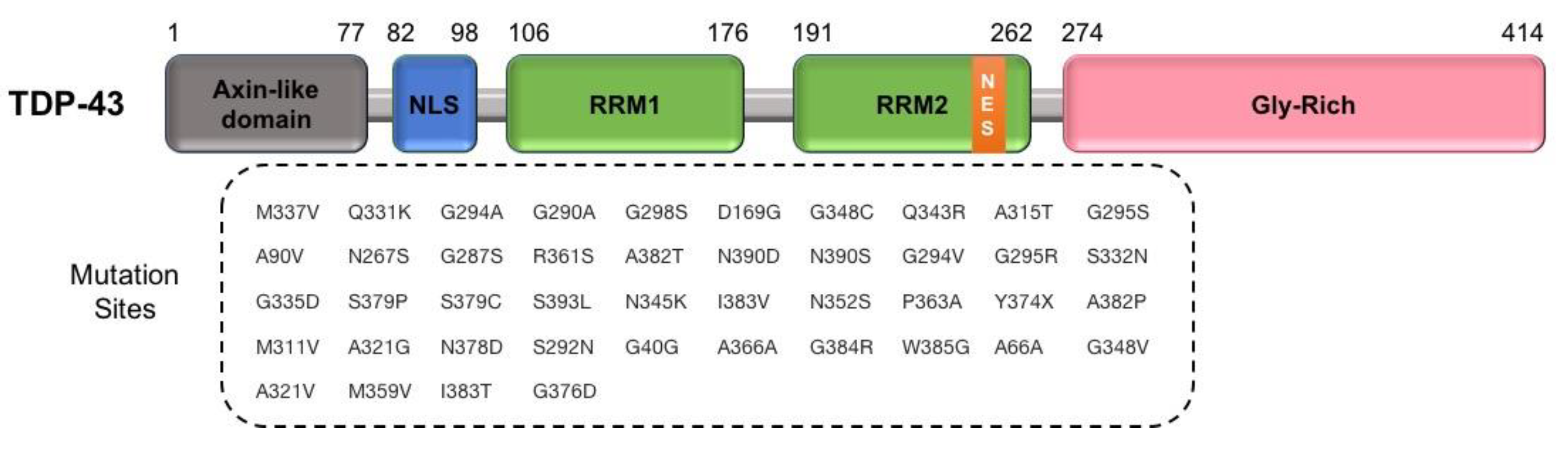
2.2.2. TARDBP
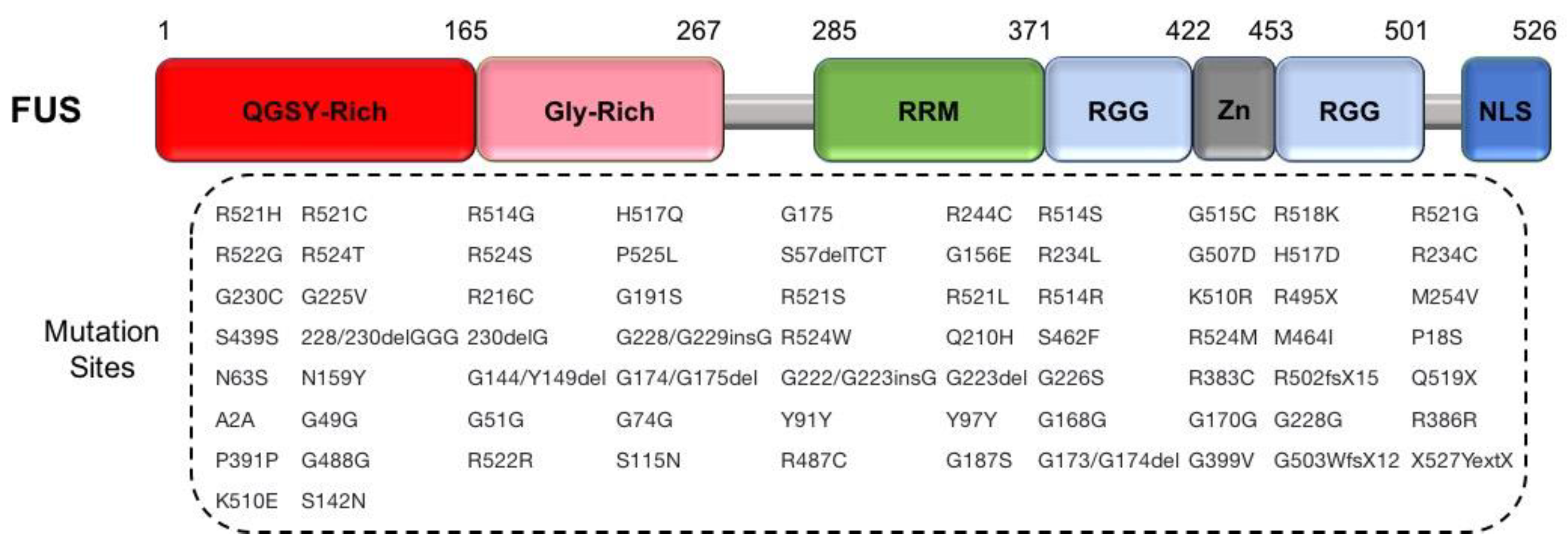
2.2.3. FUS
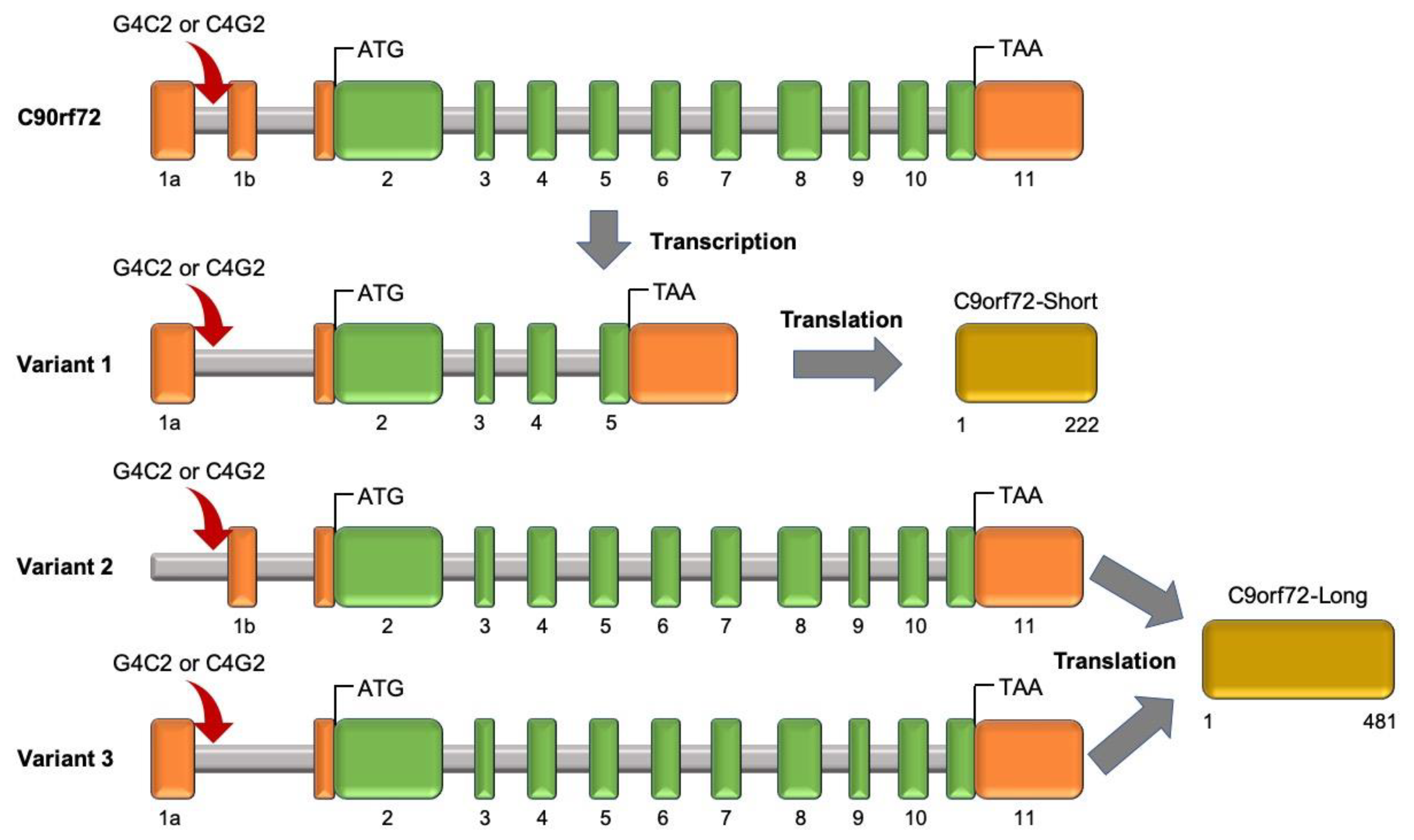
2.2.4. Chromosome 9 Open Reading Frame 72 (C9orf72)
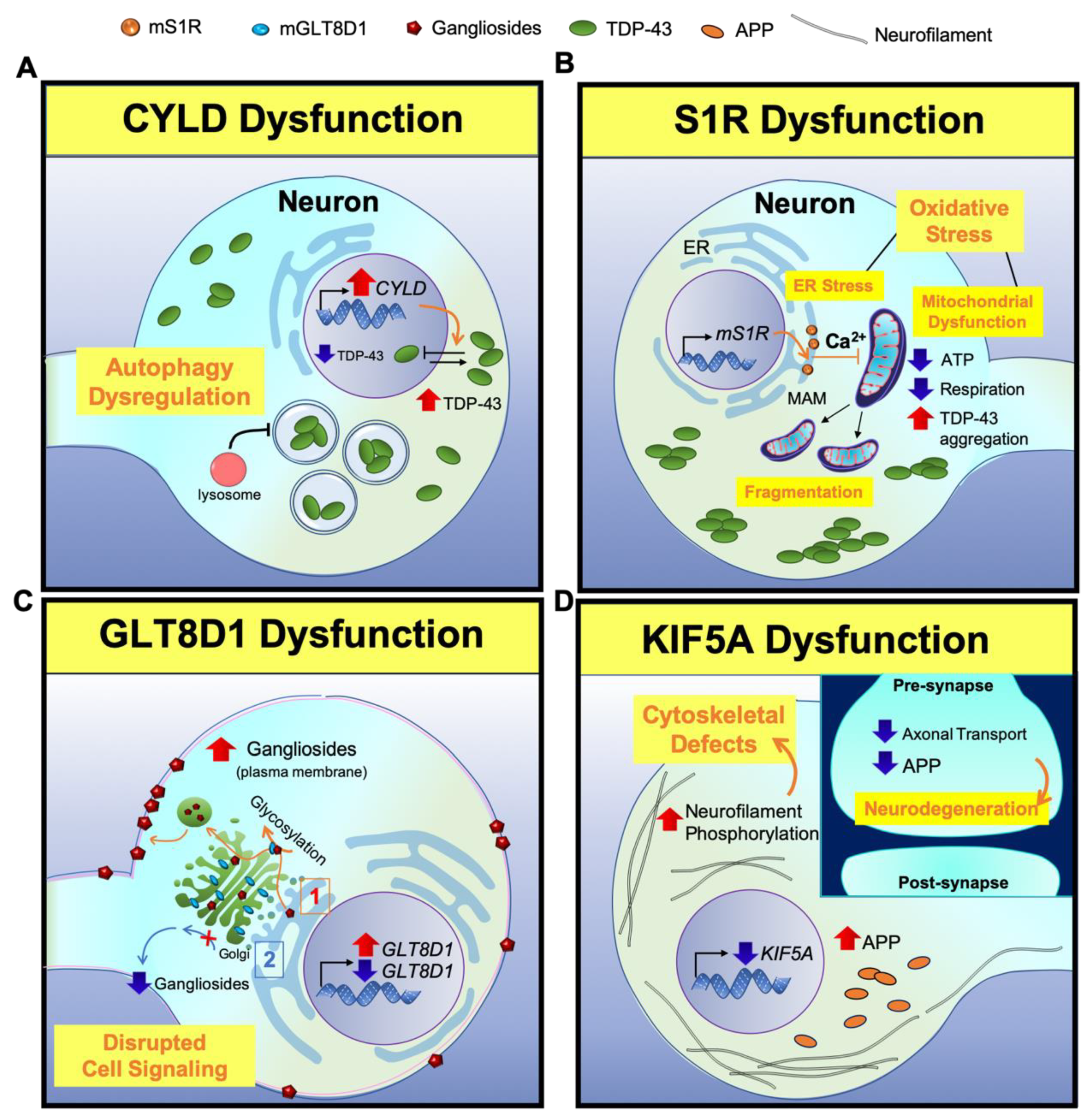
2.3. The Less Frequently Mutated Genes in ALS
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener 2013, 8, 28. [Google Scholar] [CrossRef]
- Iacoangeli, A.; Lin, T.; Al Khleifat, A.; Jones, A.R.; Opie-Martin, S.; Coleman, J.R.I.; Shatunov, A.; Sproviero, W.; Williams, K.L.; Garton, F.; et al. Genome-wide Meta-analysis Finds the ACSL5-ZDHHC6 Locus Is Associated with ALS and Links Weight Loss to the Disease Genetics. Cell Rep. 2020, 33, 108323. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e1266. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Moll, T.; Ramesh, T.; Castelli, L.; Beer, A.; Robins, H.; Fox, I.; Niedermoser, I.; Van Damme, P.; Moisse, M.; et al. Mutations in the Glycosyltransferase Domain of GLT8D1 Are Associated with Familial Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 2298–2306.e2295. [Google Scholar] [CrossRef]
- Couly, S.; Khalil, B.; Viguier, V.; Roussel, J.; Maurice, T.; Liévens, J.-C. Sigma-1 receptor is a key genetic modulator in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2019, 29, 529–540. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Howrigan, D.P.; Abbott, L.E.; Klim, J.R.; Topp, S.D.; Byrnes, A.E.; Churchhouse, C.; Phatnani, H.; Smith, B.N.; Rampersaud, E.; et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat. Neurosci. 2019, 22, 1966–1974. [Google Scholar] [CrossRef]
- Course, M.M.; Gudsnuk, K.; Smukowski, S.N.; Winston, K.; Desai, N.; Ross, J.P.; Sulovari, A.; Bourassa, C.V.; Spiegelman, D.; Couthouis, J.; et al. Evolution of a Human-Specific Tandem Repeat Associated with ALS. Am. J. Hum. Genet. 2020, 107, 445–460. [Google Scholar] [CrossRef]
- Tazelaar, G.H.P.; Boeynaems, S.; De Decker, M.; van Vugt, J.J.F.A.; Kool, L.; Goedee, H.S.; McLaughlin, R.L.; Sproviero, W.; Iacoangeli, A.; Moisse, M.; et al. ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization. Brain Commun. 2020, 2. [Google Scholar] [CrossRef]
- Nakamura, R.; Misawa, K.; Tohnai, G.; Nakatochi, M.; Furuhashi, S.; Atsuta, N.; Hayashi, N.; Yokoi, D.; Watanabe, H.; Watanabe, H.; et al. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol. 2020, 3, 526. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K. Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ’sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Project MinE ALS Sequencing Consortium. CHCHD10 variants in amyotrophic lateral sclerosis: Where is the evidence? Ann. Neurol. 2018, 84, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Luty, A.A.; Kwok, J.B.J.; Dobson-Stone, C.; Loy, C.T.; Coupland, K.G.; Karlström, H.; Sobow, T.; Tchorzewska, J.; Maruszak, A.; Barcikowska, M.; et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration–motor neuron disease. Ann. Neurol. 2010, 68, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious Variants of FIG4, a Phosphoinositide Phosphatase, in Patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA Helicase Gene Mutations in a Form of Juvenile Amyotrophic Lateral Sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156b, 285–290. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Andersen, P.M.; Nilsson, P.; Chioza, B.; Andersson, J.L.; Russ, C.; Shaw, C.E.; Powell, J.F.; Nigel Leigh, P. Deletions of the Heavy Neurofilament Subunit Tail in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 1999, 8, 157–164. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Gros-Louis, F.; Larivière, R.; Gowing, G.; Laurent, S.; Camu, W.; Bouchard, J.P.; Meininger, V.; Rouleau, G.A.; Julien, J.P. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J. Biol. Chem. 2004, 279, 45951–45956. [Google Scholar] [CrossRef]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef]
- Simpson, C.L.; Lemmens, R.; Miskiewicz, K.; Broom, W.J.; Hansen, V.K.; van Vught, P.W.J.; Landers, J.E.; Sapp, P.; Van Den Bosch, L.; Knight, J.; et al. Variants of the elongator protein 3 ( ELP3 ) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 2008, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.-J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Kaneb, H.M.; Folkmann, A.W.; Belzil, V.V.; Jao, L.-E.; Leblond, C.S.; Girard, S.L.; Daoud, H.; Noreau, A.; Rochefort, D.; Hince, P.; et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Dobson-Stone, C.; Luty, A.A.; Thompson, E.M.; Blumbergs, P.; Brooks, W.S.; Short, C.L.; Field, C.D.; Panegyres, P.K.; Hecker, J.; Solski, J.A.; et al. Frontotemporal dementia-amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1-q12.2: Genetic, clinical and neuropathological analysis. Acta Neuropathol. 2013, 125, 523–533. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Deng, Z.; Sheehan, P.; Chen, S.; Yue, Z. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol. Neurodegener. 2017, 12, 90. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and Its Impact on Neurodegenerative Diseases: New Roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Tesei, A.; Cortesi, M.; Zamagni, A.; Arienti, C.; Pignatta, S.; Zanoni, M.; Paolillo, M.; Curti, D.; Rui, M.; Rossi, D.; et al. Sigma Receptors as Endoplasmic Reticulum Stress "Gatekeepers" and their Modulators as Emerging New Weapons in the Fight Against Cancer. Front. Pharmacol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lim, L.; Dang, M.; Song, J. A novel mechanism for ATP to enhance the functional oligomerization of TDP-43 by specific binding. Biochem. Biophys. Res. Commun. 2019, 514, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Médard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Guo, L.-W.; Epstein, M.L.; Ruoho, A.E. Role of the Sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J. Pharmacol. Sci. 2015, 127, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Z.; Sun, W.; Yuan, Y.; Hu, Y.; Ni, J.; Jiao, B.; Fang, L.; Li, J.; Shen, L.; et al. Mutation analysis of GLT8D1 and ARPP21 genes in amyotrophic lateral sclerosis patients from mainland China. Neurobiol. Aging 2020, 85, 156.e151–156.e154. [Google Scholar] [CrossRef] [PubMed]
- d’Azzo, A.; Tessitore, A.; Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 2006, 13, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T. Functional Roles of Gangliosides in Neurodevelopment: An Overview of Recent Advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef]
- Moll, T.; Shaw, P.J.; Cooper-Knock, J. Disrupted glycosylation of lipids and proteins is a cause of neurodegeneration. Brain A J. Neurol. 2020, 143, 1332–1340. [Google Scholar] [CrossRef]
- Simone, M.; Trabacca, A.; Panzeri, E.; Losito, L.; Citterio, A.; Bassi, M.T. KIF5A and ALS2 Variants in a Family With Hereditary Spastic Paraplegia and Amyotrophic Lateral Sclerosis. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- Nakamura, R.; Tohnai, G.; Atsuta, N.; Nakatochi, M.; Hayashi, N.; Watanabe, H.; Yokoi, D.; Watanabe, H.; Katsuno, M.; Izumi, Y.; et al. Genetic and functional analysis of KIF5A variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2020. [Google Scholar] [CrossRef]
- Chevalier-Larsen, E.; Holzbaur, E.L.F. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin Transports RNA: Isolation and Characterization of an RNA-Transporting Granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Grüneberg, M.; Seelhöfer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Azadmanesh, J.; Borgstahl, G.E.O. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxidants 2018, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P. Amyotrophic lateral sclerosis. Unfolding the toxicity of the misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef]
- Hayashi, Y.; Homma, K.; Ichijo, H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul. 2016, 60, 95–104. [Google Scholar] [CrossRef]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.-P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: "Ambivalent" Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef]
- Mathis, S.; Couratier, P.; Julian, A.; Vallat, J.M.; Corcia, P.; Le Masson, G. Management and therapeutic perspectives in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2017, 17, 263–276. [Google Scholar] [CrossRef]
- Jain, K.K. Neuroprotection in Amyotrophic Lateral Sclerosis. In The Handbook of Neuroprotection; Jain, K.K., Ed.; Springer: New York, NY, USA, 2019. [Google Scholar]
- Andersen, P.M.; Nilsson, P.; Keranen, M.L.; Forsgren, L.; Hagglund, J.; Karlsborg, M.; Ronnevi, L.O.; Gredal, O.; Marklund, S.L. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain 1997, 120, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.N.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.E.; Gallo, J.M.; Weller, R.O.; Anderton, B.H. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 1991, 114, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L., 3rd; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol 2008, 63, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Yokoseki, A.; Shiga, A.; Tan, C.F.; Tagawa, A.; Kaneko, H.; Koyama, A.; Eguchi, H.; Tsujino, A.; Ikeuchi, T.; Kakita, A.; et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 2008, 63, 538–542. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef]
- Giordana, M.T.; Piccinini, M.; Grifoni, S.; De Marco, G.; Vercellino, M.; Magistrello, M.; Pellerino, A.; Buccinna, B.; Lupino, E.; Rinaudo, M.T. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Schipper, L.J.; Raaphorst, J.; Aronica, E.; Baas, F.; de Haan, R.; de Visser, M.; Troost, D. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review. Neuropathol. Appl. Neurobiol. 2016, 42, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, R.; Tada, M.; Shiga, A.; Toyoshima, Y.; Konno, T.; Sato, T.; Nozaki, H.; Kato, T.; Horie, M.; Shimizu, H.; et al. Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: Evidence for clinico-pathologic subtypes. Acta Neuropathol. Commun. 2016, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.Y. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef]
- Strong, M.J.; Volkening, K.; Hammond, R.; Yang, W.; Strong, W.; Leystra-Lantz, C.; Shoesmith, C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell Neurosci. 2007, 35, 320–327. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, J.R.; van Bruggen, R.; Park, J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Misteli, T.; Baralle, F.E. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc. Natl. Acad. Sci. USA 2008, 105, 3785–3789. [Google Scholar] [CrossRef] [PubMed]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain A J. Neurol. 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef]
- Melamed, Z.E.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.-L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 2020, 130, 6080–6092. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Joyce, P.I.; Fratta, P.; Fisher, E.M.; Acevedo-Arozena, A. SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: Recent advances in understanding disease toward the development of clinical treatments. Mamm. Genome 2011, 22, 420–448. [Google Scholar] [CrossRef]
- Tsao, W.; Jeong, Y.H.; Lin, S.; Ling, J.; Price, D.L.; Chiang, P.M.; Wong, P.C. Rodent models of TDP-43: Recent advances. Brain Res. 2012, 1462, 26–39. [Google Scholar] [CrossRef]
- Xu, Z.S. Does a loss of TDP-43 function cause neurodegeneration? Mol. Neurodegener. 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, S.Y.; Yagudayeva, I.; Shneider, N.A. Mutant TDP-43 Causes Early-Stage Dose-Dependent Motor Neuron Degeneration in a TARDBP Knockin Mouse Model of ALS. Cell Rep. 2019, 26, 364–373.e364. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-linked TDP-43M337V knock-in mice exhibit splicing deregulation without neurodegeneration. Mol. Brain 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noel, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef]
- Waibel, S.; Neumann, M.; Rosenbohm, A.; Birve, A.; Volk, A.E.; Weishaupt, J.H.; Meyer, T.; Muller, U.; Andersen, P.M.; Ludolph, A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: A clinico-genetic study in Germany. Eur. J. Neurol. 2013, 20, 540–546. [Google Scholar] [CrossRef]
- Chio, A.; Calvo, A.; Mazzini, L.; Cantello, R.; Mora, G.; Moglia, C.; Corrado, L.; D’Alfonso, S.; Majounie, E.; Renton, A.; et al. Extensive genetics of ALS: A population-based study in Italy. Neurology 2012, 79, 1983–1989. [Google Scholar] [CrossRef]
- Hubers, A.; Just, W.; Rosenbohm, A.; Muller, K.; Marroquin, N.; Goebel, I.; Hogel, J.; Thiele, H.; Altmuller, J.; Nurnberg, P.; et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 2015, 36, 3117.e3111–3117.e3116. [Google Scholar] [CrossRef]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Belly, A.; Moreau-Gachelin, F.; Sadoul, R.; Goldberg, Y. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: Exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 2005, 379, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Zhang, H.; Loiselle, D.; Haystead, T.; Macara, I.G.; Mili, S. The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J. Cell. Biol. 2013, 203, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Tang, A.A.; Kulkarni, A.; West, J.; Brooks, M.; Stubblefield, J.J.; Liu, Y.; Zhang, M.Q.; Green, C.B.; Huber, K.M.; et al. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, E4769–E4778. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Akagi, T.; Hashikawa, T.; Doi, H.; Takumi, T.; Hicks, G.G.; et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2015, 3, 24. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.J.; Wang, D.; Wei, X.; Xia, X.G. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Deng, J.; Chen, X.; Ye, Y.; Zhu, L.; Liu, J.; Ye, H.; Shen, Y.; Li, Y.; et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2011, 2, 477–486. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897. [Google Scholar] [CrossRef]
- Nassif, M.; Woehlbier, U.; Manque, P.A. The Enigmatic Role of C9ORF72 in Autophagy. Front. Neurosci 2017, 11, 442. [Google Scholar] [CrossRef]
- Mizielinska, S.; Isaacs, A.M. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: Gain or loss of function? Curr. Opin. Neurol. 2014, 27, 515–523. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef] [PubMed]
- Hubers, A.; Marroquin, N.; Schmoll, B.; Vielhaber, S.; Just, M.; Mayer, B.; Hogel, J.; Dorst, J.; Mertens, T.; Just, W.; et al. Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexanucleotide repeat in different motor neuron diseases. Neurobiol. Aging 2014, 35, 1214.e1–1214.e6. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Devenney, E.; Hornberger, M.; Irish, M.; Mioshi, E.; Burrell, J.; Tan, R.; Kiernan, M.C.; Hodges, J.R. Frontotemporal dementia associated with the C9ORF72 mutation: A unique clinical profile. JAMA Neurol. 2014, 71, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.J.; Baumer, D.; East, S.; Neal, J.; Morris, H.R.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779.e5–1779.e13. [Google Scholar] [CrossRef]
- Xiao, S.; MacNair, L.; McGoldrick, P.; McKeever, P.M.; McLean, J.R.; Zhang, M.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 78, 568–583. [Google Scholar] [CrossRef]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sa, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef]
- Foxe, D.; Elan, E.; Burrell, J.R.; Leslie, F.V.C.; Devenney, E.; Kwok, J.B.; Halliday, G.M.; Hodges, J.R.; Piguet, O. Intrafamilial Phenotypic Variability in the C9orf72 Gene Expansion: 2 Case Studies. Front. Psychol. 2018, 9, 1615. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881. [Google Scholar] [CrossRef]
- Yoon, Y.; Park, H.; Kim, S.; Nguyen, P.T.; Hyeon, S.J.; Chung, S.; Im, H.; Lee, J.; Lee, S.B.; Ryu, H. Genetic Ablation of EWS RNA Binding Protein 1 (EWSR1) Leads to Neuroanatomical Changes and Motor Dysfunction in Mice. Exp. Neurobiol. 2018, 27, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Pomponi, M.G.; Conte, A.; Marangi, G.; Bisogni, G.; Patanella, A.K.; Meleo, E.; Lunetta, C.; Riva, N.; Mosca, L.; et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 64, 157.e151–157.e155. [Google Scholar] [CrossRef] [PubMed]
- Aditi; Glass, L.; Dawson, T.R.; Wente, S.R. An amyotrophic lateral sclerosis-linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Adv. Biol. Regul. 2016, 62, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.S.; Mackenzie, I.R.; Cairns, N.J.; Swanson, E.; Boyer, P.J.; Drachman, D.A.; Jhaveri, B.S.; Karlawish, J.H.; Pestronk, A.; Smith, T.W.; et al. Novel Ubiquitin Neuropathology in Frontotemporal Dementia With Valosin-Containing Protein Gene Mutations. J. Neuropathol. Exp. Neurol. 2006, 65, 571–581. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Occhineri, P.; Cau, T.B.; Loi, D.; et al. TBK1 is associated with ALS and ALS-FTD in Sardinian patients. Neurobiol. Aging 2016, 43, 180. [Google Scholar] [CrossRef]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724. [Google Scholar] [CrossRef]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.-P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Deschauer, M.; Gaul, C.; Behrmann, C.; Prokisch, H.; Zierz, S.; Haack, T.B. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Ng, K.; Van Den Bos, M.; Byth, K.; Kiernan, M.C.; Vucic, S. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph Lateral Scler Front. Degener 2016, 17, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H., Jr.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ryu, H.; Keum, G.; Yoon, Y.J.; Kowall, N.W.; Ryu, H. Therapeutic Targeting of Epigenetic Components in Amyotrophic Lateral Sclerosis (ALS). Curr. Med. Chem. 2014, 21, 3576–3582. [Google Scholar] [CrossRef]
- Lee, J.; Ryu, H. Epigenetic modification is linked to Alzheimer’s disease: Is it a maker or a marker? BMB Rep. 2010, 43, 649–655. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
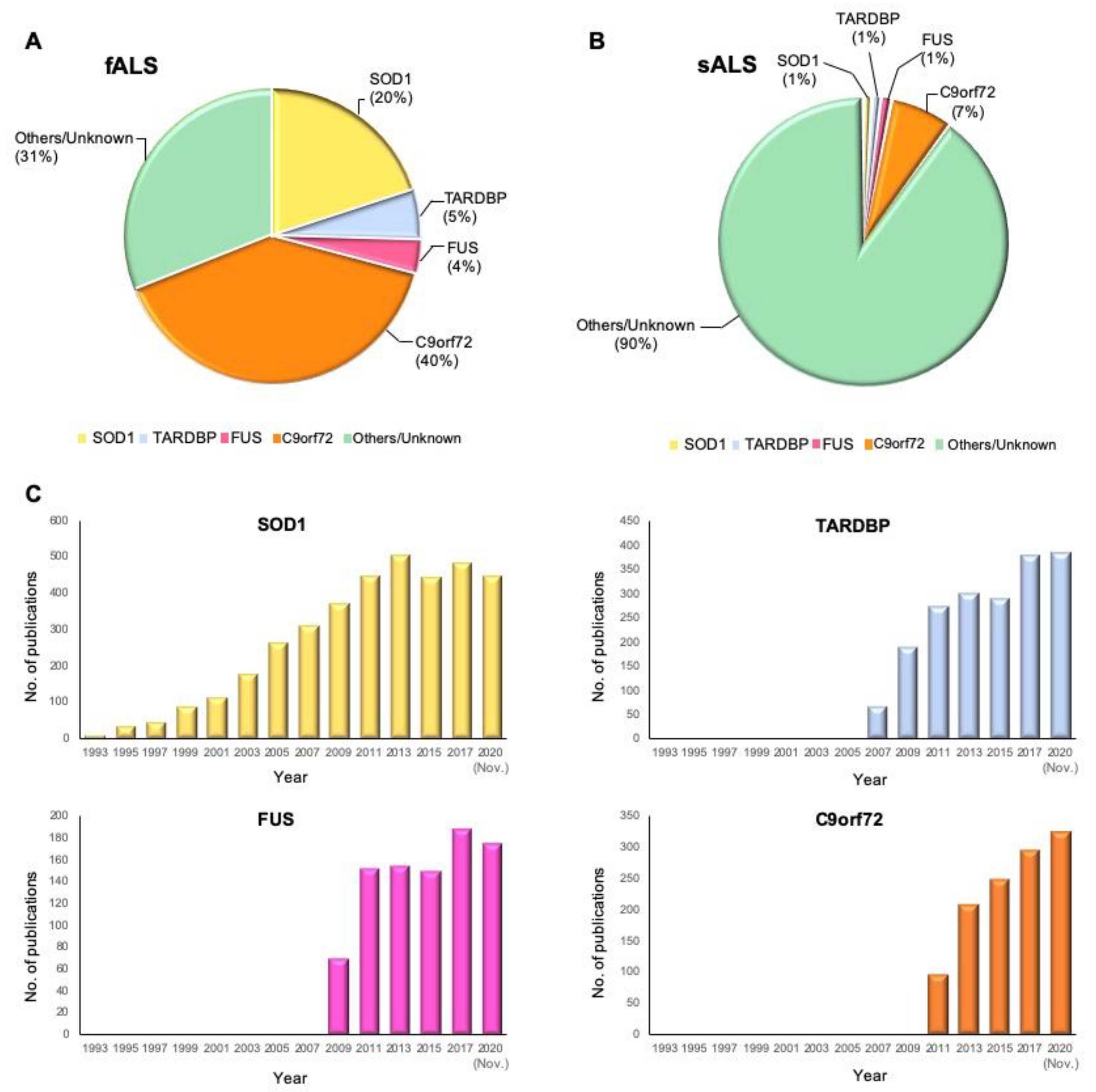
| Gene | Cases/Control | Frequency (Familial) | Frequency (Sporadic) | Reference (s) |
|---|---|---|---|---|
| SOD1 | 2874/6405 | 20% | 1% | [25] |
| TARDBP | 526/1262 | 4% | 1% | [26] |
| OPTN | 673/781 | <1% | <1% | [21] |
| SPG11 | 3056/900 | <1% | <1% | [27] |
| VCP | 288/1569 | 1% | 1% | [28] |
| hnRNPA1 | 517/625 | <1% | <1% | [29] |
| ATXN2 | 915/980 | <1% | <1% | [30] |
| ANG | 1629/1264 | <1% | <1% | [31] |
| CHCHD10 | 4853/1991 | <1% | <1% | [32] |
| SIGMAR1 | 158/1100 | <1% | <1% | [33,34] |
| FIG4 | 473/558 | <1% | <1% | [35] |
| SETX | 49/100 | <1% | <1% | [36] |
| SQSTM1 | 546/738 | 1% | <1% | [37] |
| TBK1 | 252/827 | 1% | <1% | [38] |
| NEK1 | 1022/7315 | 1% | 1% | [39] |
| TAF15 | 357/1100 | <1% | <1% | [40] |
| FUS | 583/1446 | 4% | 1% | [41] |
| ALS2 | 42/533 | <1% | <1% | [42] |
| VAPB | 24/400 | <1% | <1% | [43] |
| NEFH | 530/447 | <1% | <1% | [44] |
| C9orf72 | 696/909 | 40% | 7% | [45] |
| CHMP2B | 400/640 | <1% | <1% | [46] |
| MATR3 | 108/1051 | <1% | <1% | [47] |
| PFN1 | 274/1089 | <1% | <1% | [48] |
| PRPH | 189/380 | <1% | <1% | [49] |
| TUBA4A | 363/5510 | 1% | <1% | [50] |
| ELP3 | 781/702 | <1% | <1% | [51] |
| DCTN1 | 333/200 | <1% | <1% | [52] |
| EWSR1 | 817/1082 | <1% | <1% | [53] |
| GLE1 | 933/190 | <1% | <1% | [54] |
| UBQLN2 | 200/928 | <1% | <1% | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousefian-Jazi, A.; Seol, Y.; Kim, J.; Ryu, H.L.; Lee, J.; Ryu, H. Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 2687. https://doi.org/10.3390/cells9122687
Yousefian-Jazi A, Seol Y, Kim J, Ryu HL, Lee J, Ryu H. Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis. Cells. 2020; 9(12):2687. https://doi.org/10.3390/cells9122687
Chicago/Turabian StyleYousefian-Jazi, Ali, YunHee Seol, Jieun Kim, Hannah L. Ryu, Junghee Lee, and Hoon Ryu. 2020. "Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis" Cells 9, no. 12: 2687. https://doi.org/10.3390/cells9122687
APA StyleYousefian-Jazi, A., Seol, Y., Kim, J., Ryu, H. L., Lee, J., & Ryu, H. (2020). Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis. Cells, 9(12), 2687. https://doi.org/10.3390/cells9122687