Abstract
The therapeutic landscape of malignant melanoma has been radically reformed in recent years, with novel treatments emerging in both the field of cancer immunotherapy and signalling pathway inhibition. Large-scale tumour genomic characterization has accurately classified malignant melanoma into four different genomic subtypes so far. Despite this, only somatic mutations in BRAF oncogene, as assessed in tumour biopsies, has so far become a validated predictive biomarker of treatment with small molecule inhibitors. The biology of tumour evolution and heterogeneity has uncovered the current limitations associated with decoding genomic drivers based only on a single-site tumour biopsy. There is an urgent need to develop minimally invasive biomarkers that accurately reflect the real-time evolution of melanoma and that allow for streamlined collection, analysis, and interpretation. These will enable us to face challenges with tumour tissue attainment and process and will fulfil the vision of utilizing “liquid biopsy” to guide clinical decisions, in a manner akin to how it is used in the management of haematological malignancies. In this review, we will summarize the most recent published evidence on the role of minimally invasive biomarkers in melanoma, commenting on their future potential to lead to practice-changing discoveries.
1. Introduction
The advent of novel cancer immunotherapies, as well as the discovery of driver signalling pathways in melanoma, such as the mitogen activate protein kinase (MAPK) pathway, have brought an unparalleled improvement in the survival of patients with advanced disease [1,2]. Immunotherapy, in the form of immune checkpoint inhibition, is associated with durable response rates and long-term survival but is also characterised by slow kinetics, when compared to targeted treatments. On the other hand, abrogating the MAPK pathway, with small molecule inhibitors, can elicit rapid response and swift symptomatic relief for the patients, which are however not as durable as those observed with immunotherapy and are invariably followed by emergence of resistance.
Melanoma is clinically characterised as a rapidly growing tumour that needs urgent decision making and close patient observation. Measures of response to existing melanoma treatments include, as in other tumour types, the traditional clinical response, as well as radiological response, both of which can contain inherent subjective biases. Specifically radiological responses, as measured by the RECIST 1.1 framework, have been found to poorly correlate with patient survival [3,4], and moreover, novel patterns of disease response emerging with current immunotherapy strategies might require more sensitive readouts. The phenomenon of pseudo progression observed with immunotherapy, for example, has no clear predictors and is an indication that molecularly intricate biomarkers will be required to accurately assess responses and to ensure continuation of treatment in cases when robust peritumoural inflammation is wrongly regarded as true disease progression [5]. Novel biomarkers such as measurement of circulating tumour DNA hold promise as a tool to aid distinction between pseudo progression and real disease progression [6,7,8,9].
Substantial efforts are directed nowadays towards minimally invasive biomarkers in oncology and specifically in melanoma. These can have considerable benefits for cancer patients, given the inherent reduced human risk involved in their collection and their rapid turnover. This could be particularly pertinent to melanoma, where access to tissue can be restricted due to the small actual size of the primary tissue. Additional aims throughout biomarker discovery should focus on the biomarker’s ability to represent a longitudinal surrogate of tumour burden, tumour clonal divergence and heterogeneity [10,11]. Herein, we provide an overview of circulating and other minimally invasive biomarkers for therapeutic monitoring of patients with melanoma. In particular, the use of nucleic acids, circulating tumour cells, exosomes and intestinal microbiota as surrogate markers for treatment response are explored (Figure 1). Not least, challenges in the application of these markers and opportunities for future work are described.
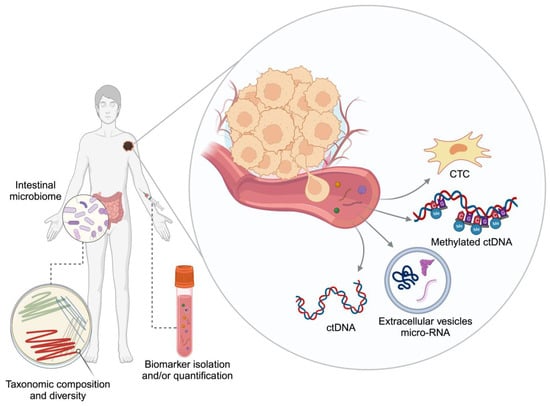
Figure 1.
Minimally invasive biomarkers. CTC: circulating tumour cell, ctDNA: circulating tumour DNA. Created with BioRender.com.
2. Circulating Surrogate Biomarkers in Melanoma before the Next Generation Sequencing (NGS) Era
Circulating molecules can be detected in patients with melanoma, either due to active cellular secretion or as a bioproduct of cell death. Multiple serological protein markers have been explored, with lactate dehydrogenase (LDH) currently being the only surrogate widely used in clinical practice. LDH has a significant role in disease stratification via its incorporation into the tumour, nodes and metastasis (TNM) staging [12]. There is strong evidence that LDH, a ubiquitous enzyme that is critical to anaerobic metabolism [13], correlates with prognosis and treatment response in melanoma patients treated with either targeted treatment or immunotherapy [14,15,16,17,18,19]. However, LDH is not consistently elevated in patients with high burden of disease, and its increase in the serum can simply be a consequence of any cellular necrosis. Therefore, LDH cannot be considered a specific marker for longitudinal treatment monitoring and is not always helpful in guiding treatment decisions in this context [10,20,21,22].
S100 is another important melanoma-specific marker, typically detected via immunohistochemistry to diagnose the presence of metastatic melanoma disease in the clinical setting [23,24]. The S100 protein family consists of 21 functionally different proteins that are aberrantly over-secreted [25], and their detectability in viable melanoma cells, as well as in blood circulation, make them promising markers for melanoma disease monitoring and prognosis [26]. Similarly to LDH, the detection of S100 has been found to correlate with disease activity and prognosis in both small- [24,27] and large-scale patient cohorts [28,29,30]. Therefore, although to a lesser degree than LDH [31], its use in the clinic has been adopted in certain parts of the world [32,33].
To date, no other widely validated, minimally invasive biomarkers are available for melanoma. The limited sensitivity and specificity of LDH and S100 highlight the need to discover molecular surrogates for melanoma biology and evolution. The advent of next-generation sequencing (NGS) and other novel technologies has bolstered the genomic characterisation of melanoma at a granular level and is currently continuously supporting the advancement of a new class of highly specific biomarkers (Table 1).

Table 1.
Melanoma studies registered on clinicaltrials.org involving liquid biopsy methodologies, as of 1 October 2023.
3. Mutational Landscape in Cutaneous and Non-Cutaneous Melanoma
Comprehensive genomic classification of cutaneous melanoma was achieved in 2015 via a systematic multi-platform characterization performed by the Cancer Genome Atlas Network [34], whereby the subdivision of cutaneous melanoma in the four types of mutant BRAF, mutant RAS (N/H/R), mutant NF1 and triple wild-type (triple WT) emerged. Hotspot mutations predominantly in the V600 codon of BRAF and the Q61 codon of NRAS, as well as loss-of-function events in NF1, led to a cascade of perturbations causing upregulation of the MAPK pathway, the most deregulated signalling pathway in cutaneous melanoma [35,36]. Triple WT tumours, which show a higher rate of somatic copy number alterations (CANs), can still exhibit constitutional activation of the MAPK pathway via alternative genomic aberrations like, for example, KIT mutations/amplifications [34]. A degree of cell cycle pathway disruption via mutations or deletions in cyclin-dependent kinase inhibitor 2A (CDKN2A) has been observed to overlap all four subtypes, whereas mutations of the telomerase reverse transcriptase (TERT) promoter are commonly found in the BRAF/RAS/NF1 mutated subtypes.
Uveal melanoma, which originates from melanocytes residing in the uveal track, has a molecular profile distinct from that of the cutaneous melanoma [37]. Here, we observe large genomic losses, including chromosomal losses in 1p, 6q, 8p and 16q; gains in 6p and 8q; and more importantly chromosome 3 monosomy [38,39,40]. Chromosome 3 aberrations are critically prognostic and, furthermore, contribute to BAP1 aberrancy via biallelic gene loss when superimposed BAP1 mutations are also present [37]. Additional mutations in EIF1AX and SF3B1 have been associated with a favourable prognosis subtype of uveal melanoma, whereas mutations in GNAQ/GNA11 are found in more than 90% of patients [37,41,42]. Lastly, mucosal melanoma also presents a unique molecular profile compared to cutaneous and uveal subtypes. The rate of NF1 mutations is comparable to that of cutaneous melanoma (~14%); however, BRAF/NRAS mutations rates are remarkably lower, and KIT mutations are observed in at least 13% of cases [35,43,44].
The growing knowledge on the mutational landscape of melanoma has contributed enormously to the development of some of the minimally invasive biomarkers explored in this review.
4. Circulating Tumour DNA in Melanoma
ctDNA introduction. Circulating tumour DNA (ctDNA) is the component of fragmented cell-free DNA (cfDNA) derived from cancer cells. Over the last decade or so, there has been a myriad of studies confirming that detection of ctDNA is readily possible and reproducible in patients with advanced malignancies [45,46,47,48,49,50]. ctDNA-based genomic profiles are highly concordant with those of tumour tissue, and ctDNA levels can directly correlate with tumour burden. Quantitative and digital polymerase chain reactions (like digital droplet PCR (ddPCR)) were the first ctDNA detection approaches for selective gene targets, but more recently, next-generation sequencing (NGS)-based techniques have increasingly been developed [47,48,49,51]. Both methods can be attractive in different contexts. ddPCR is suggested to have higher sensitivity of 0.001% when compared to NGS [52], and furthermore, it has an easy upstream preparation process and faster turnaround time and does not require highly skilled bioinformatics to facilitate analysis. The main drawback of ddPCR is that it can only test for known genomic aberrations, and multiplex biomarker detection is limited. In contrast, NGS allows for the detection of multiple mutations per patient and high sensitivity ctDNA tracking. NGS methods are applied in a “tumour-informed” manner and provide a signature of putative cancer-derived aberrations [53], and it may identify any type of genetic change in all the target regions screened, including novel mutations as well as chromosomal rearrangement and fusion genes. Although this is against the backdrop of being more costly and requiring more complex analytical methods compared to ddPCR [54], NGS methods certainly offer a more comprehensive and personalized approach to ctDNA detection. NGS can be performed via whole-genome sequencing (WGS), whole-exome sequencing (WES), targeted (TS) or candidate gene sequencing (CGS). The lower sensitivity encountered with NGS mandates a higher concentration of ctDNA, which can be challenging in patients with low disease burden [55,56].
Detecting circulating DNA offers multiple opportunities in (a) early cancer diagnosis (b) assessment of minimal residual disease (MRD) (c) disease tracking during anticancer treatment—“molecular response” to therapy and (d) uncovering tumour heterogeneity and novel targets conferring treatment resistance [57,58,59,60,61,62]. Variant allele frequency (VAF) is the most common readout for a mutation detected in ctDNA and is defined as the fraction of cfDNA molecules sequenced at a particular locus that harbours the variant of interest. Several ctDNA assays have now entered clinical practice for various solid tumour types, with the bulk of the evidence stemming from lung cancer studies. In various contexts, such as lung, breast, colorectal and prostate cancer, plasma ctDNA detection assays can complement tissue genotyping and, in combination with tissue sequencing, increase the rate of driver mutation detection [63,64,65,66,67].
ctDNA in prognostication of advanced melanoma. A recent meta-analysis of 19 studies involving patients with metastatic melanoma summarized the role of ctDNA detection at baseline, before patients were exposed to systemic treatment [68]. These studies incorporated both ddPCR and non-ddPCR technologies, targeting mutations in BRAF, NRAS, KIT and TERT promoter. In more than 1000 patients tested with ddPCR methodology, detection of ctDNA at baseline was associated with a higher risk of disease progression, with HR = 2.10 (95% confidence interval [CI]: 1.71–2.59). It was also associated with higher risk of disease progression HR 2.15 (95% CI: 1.35–3.41) in 347 patients tested with non-ddPCR methodology. The prognostic value of ctDNA detection remains robust despite the type of treatment received, targeted or immunotherapy, for either detection method. The hazard ratio for death was 3.09 (95% CI 2.29–4.17) for immunotherapy-based treatment (n = 360) and HR 2.39 (95% CI 1.76–3.25) for targeted treatment (n = 666). The same meta-analysis confirmed that ctDNA positivity in metastatic melanoma is independently associated with higher disease staging, elevated LDH and visceral metastases, among other clinical characteristics that are historically linked with worse disease prognosis. Moreover, independent studies from McEvoy and Seremet and colleagues in a total of > 100 patients have demonstrated good correlation between ctDNA levels and 18F-fluoro-D-glucose positron emission tomography (FDG PET), an imaging tool that has become the standard of care in patients with advanced melanoma [69,70]. Therefore, ctDNA can be employed as companion test to inform disease prognostication alongside other widely used parameters.
ctDNA in melanoma minimal residual disease. The powerful role of ctDNA in reflecting residual microscopic disease was initially showed in 2008, in patients with colorectal cancer. Diehl and colleagues demonstrated that patients with incomplete tumour resections had lower rate of ctDNA clearance or rising ctDNA fractions postoperatively compared to patients with complete resections, [71]. In melanoma, the detection rate of ctDNA after complete surgical resection can be up to 25% when using highly sensitive NGS-based assays [59,72,73]. Admittedly, low ctDNA quantity and sequencing artifacts can restrict the use of large sequencing panels in the adjuvant setting, and methods to reduce background error rate will be necessary, in order to maximise analytical specificity [74].
Multiple small-scale studies have demonstrated a significant correlation between the presence of peri-operative ctDNA detection and inferior survival outcomes in resected melanoma [73,75] in patients with known mutation variants. More recently, tumour-informed ctDNA panels that encompass wide range of patient-specific mutations have also demonstrated that peri-operative detection of patient-customized genomic variants are possible and predictive of patient outcomes [76,77]. One of the most robust reports on the predictive value of ctDNA in resectable melanoma was conducted and reported as part of the CheckMate 915 study translational analysis. Using a tumour-informed, patient-guided panel of 200 variants, Long and colleagues observed that in a cohort of 1127 patients with resected stage IIIB-D/IV melanoma, ctDNA detection at baseline can be an efficient predictor of recurrence-free survival for patients undergoing ICI treatment [78]. When ctDNA detection was combined with the level of tumour mutational burden (TMB) and a tumour-derived interferon signature (including HLA-DRA, CXCL9/10/11, GZMA, PRF1, CCR5, IFNG, IDO1 and STAT1), the predictive value of the combined score was enhanced [79]. In a real-world study of 30 patients who were followed up after resection, in the adjuvant setting, tracking of molecular disease with a tumour-informed ctDNA assay showed a sensitivity of 83% for distant relapses, with a specificity of 96% [80]. Importantly, ctDNA analysis allowed for an average lead time of 3 months over detection of disease recurrence with standard radiological assessment.
The realisation that ctDNA can give us great insight into the molecular tumour burden inspired further questions into how we can take advantage of this intricate information and how to best incorporate it into clinical decisions. In early stage colorectal cancer (stage II), the DYNAMIC study showed that foregoing adjuvant chemotherapy in patents with undetectable ctDNA post-operatively does not compromise recurrence free survival [81], and hence it can spare unwarranted chemotherapy toxicity. There are ongoing longitudinal studies to elucidate the exact role of ctDNA in detecting early molecular disease (NCT05736523) in early stage melanoma. Moreover, efforts to incorporate ctDNA in the stratification of patients and escalation/de-escalation of systemic adjuvant treatment are also underway. In “Tiragolumab Plus Atezolizumab Versus Atezolizumab in the Treatment of Stage II Melanoma Patients Who Are ctDNA-positive Following Resection (NCT05060003)” [82], adjuvant treatment was titrated based on the ctDNA positive detection following definitive surgery for stage II melanoma.
ctDNA in advanced melanoma—molecular response to treatment. Molecular response and ctDNA kinetics refer to VAF dynamics during treatment, and it is worth mentioning that, besides tumour driver mutations, these can encompass the characterisation of neoantigens and/or chromosomal number aberrations [83,84,85]. Several studies and meta-analyses have attempted to highlight the robustness of ctDNA as a biomarker of treatment response, which can guide treatment duration or monitoring intensity in a pan-cancer fashion [59,62,68,86,87,88]. Across 16 tumour types, including melanoma, Zhang et al. confirmed that mean VAF pre-treatment with immune checkpoint inhibition was prognostic of overall survival [OS; stratified by the median; unadjusted HR, 0.58; 95% confidence interval (CI), 0.49–0.69; p < 0.0001]. Furthermore, on-treatment VAF dynamics correlated with overall response rate by radiological criteria [86]. This indicates that ctDNA is predictive of response to immunotherapy in the metastatic setting, and the authors concluded that a ratio-based molecular response metric can be used to guide clinical decisions. This ratio of on-treatment to baseline VAF demonstrated stronger association with RECIST response compared with on-treatment VAF alone (AUC = 0.82; 95% CI, 0.71–0.93 for the ratio and AUC = 0.73; 95% CI, 0.61–0.85 for on-treatment VAF). With the use of this score, patients could be stratified into “molecular responders”, and this molecular response could also predict future radiological response in patients who initially experienced radiologically stable disease, with a median lead time of 8 weeks. The meta-analysis from Zhang and colleagues included studies with either anti-PD1 or anti-PD1 plus anti-CTLA4 treatments [86].
In accordance with the above findings, in melanoma-specific studies, dynamic changes to quantified ctDNA demonstrate strong predictive value in estimating survival outcomes and response to immunotherapy in patients with metastatic disease [7,89,90,91]. With either immunotherapy, or targeted treatment, Varaljai and colleagues observed that a decrease in BRAF V600E ctDNA levels preceded radiologic detection of response in 80% of melanoma responders with an average lead-time window of 1.5 months (range, 0.023 to 3.45 months; p = 0.003). On the other hand, 86% of non-responders experienced an increase in ctDNA levels, which preceded radiologic progression, with an average lead-time window of 3.5 months (range, 0.23 to 18.86 months; p = 0.001) [92]. A similar trend was observed in the dynamics of NRAS and TERT promoter mutations. ctDNA decreases were more rapid with targeted treatment, which is in keeping with the symptomatic improvement that patients commonly experience when they start treatment with BRAF/MEK inhibitors in clinical practice. Interestingly, the same study demonstrated that NRASQ61 mutation can be detected with high-sensitivity ddPCR in the plasma of patients with tissue BRAF mutations, raising the hypothesis that NRASQ61 mutations might co-exist at low frequency in bulky tumours and suggests plasma ctDNA as a complementary genotyping tool too [92]. Nevertheless, anticipating disease response or progression is of paramount importance for the clinicians who, not infrequently, face discordance between clinical and radiological picture. Lee et al. observed that sensitivity of ctDNA for predicting pseudo-progression was 90% (95%CI, 68–99%) and specificity was 100% (95%CI, 60–100%) [8]. This indicates that ctDNA quantification is a valuable tool in distinguishing pseudo-progression from true progression on immune checkpoint treatment, an observation reported by others too [93] and in different cancer types [6,89]. Interestingly, ctDNA has also emerged as a biomarker during the novel treatment of tumour-infiltrating lymphocytes (TIL) in melanoma. Xi et al. reported the intriguing observation that patients with an early peak of ctDNA and subsequent clearance following TIL therapy experienced durable disease responses [94].
Overall, there is a multitude of studies confirming the validity of ctDNA as a biomarker of response to either targeted therapy or immunotherapy strategies [7,69,70,95,96,97,98,99,100,101,102,103,104,105,106]. A study that is strongly testing the ability of ctDNA measurement to aid clinical decision making in the metastatic setting is the CaCTUS trial (NCT03808441) [107]. In the CaCTUS study, patients with BRAF mutant melanoma on the intervention arm switch from targeted treatment to immunotherapy when there is evidence of molecular response, as defined by a decrease in mutant BRAF VAF of ≥80% by ddPCR. Results from this study will likely bolster ctDNA even more as a dynamic biomarker that can be incorporated into daily clinical practice of BRAF-mutated cutaneous melanoma. In a similar vein, the REPOSIT study attempted to investigate ctDNA mutational markers of resistance to targeted treatment with BRAF/MEK inhibition in unresectable stage IIIc/IV melanoma [108]. The study however was prematurely terminated due to slow accrual and the changing landscape of immunotherapy in advanced melanoma.
ctDNA in non-cutaneous melanoma—In uveal melanoma (UM), recurrent mutations affecting GNA11, GNAQ, PLCβ4 or CYSLTR2 are found in the majority of patients and, even if they are not determinants of prognosis, they can represent targetable mutations in the blood [37,109]. Using deep sequencing and hybridization capture to detect very low-frequency somatic alterations in exons and introns from 129 oncogenes (including GNAQ, GNA11, SF3B1 and EIF1AX), Francis and colleagues observed a ctDNA detection rate of 29% during the perioperative period for primary UM [110]. This was supported by another study that reported a detection rate of 26% in patients with non-metastatic primary UM [111]. Interestingly, at follow-up, patients with ctDNA that became detectable or had an increasing VAF were significantly more likely to develop metastatic disease compared to patients with no detectable ctDNA or with decreasing VAF. In a bigger study of 135 patients with primary UM, ctDNA sensitivity and specificity of detecting metastases were found to be 80% and 96%, respectively [112], with an average lead time of 5.7 months (range of 2–10 months). To add to this, at the time of metastasis development, the presence of ctDNA seemed to be a strong predictor of overall survival, as showed in a prospective study of 179 patients [113]. An interesting and promising approach to the detection of ctDNA in UM patients with low burden disease was introduced by Wong et al. In a small study of 11 patients, the authors attempted to develop a customized multi-modal approach of ctDNA characterisation using genome, fragmentome and methylome analyses with targeted sequencing (TS), shallow whole-genome sequencing (sWGS) and cell-free methylated DNA immunoprecipitation sequencing (cfMeDIPseq) [114]. Interestingly, they observed that fragmentomic analysis of sWGS data was more effective at detecting ctDNA abundance in patients preceding disease relapse when compared to TS. As will be discussed further below, cell-free DNA fragmentomics can carry useful and complementary information that is agnostic genetic alteration agnostic.
In the context of established metastatic disease, Mariani showed that patients with hepatic metastases and detectable ctDNA have inferior survival outcomes compared to patients with undetectable ctDNA after liver metastasectomy (median recurrence-free survival: 5.5 vs. 12.2 months; HR = 2.23, 95% CI: 1.06–4.69, p = 0.04) [115]. In an exploratory analysis of a multicentre single-arm phase 2 study evaluating tebentafusp efficacy in previously treated metastatic UM, the level of ctDNA was measured in 127 patients with a uveal melanoma customised panel of common genomic aberrations, in which ctDNA was amplified using multiplex PCR and analysed with next-generation sequencing (performed by Natera Inc.) [116]. Both baseline levels and on-treatment reduction of ctDNA were strongly associated with overall survival. Patients with ctDNA clearance at 9 weeks after initiation of tebentafusp had longer survival probability (HR 0.08; 95% CI 0.01–0.56, p < 0.05), with an overall one-year survival rate of 100% compared to 52% in patients with increasing ctDNA. Given the observed disconnect between radiological response and survival outcomes in UM patients treated with tebentafusp [117], and given the intense and laborious nature of this weekly treatment, an urgent effort to incorporate markers of molecular response is needed. Measurement of ctDNA appears to offer a solution to both prognostication and prediction of response, but it deserves further prospective validation [118].
Ongoing current studies investigating the role of ctDNA and other minimally invasive biomarkers are summarized in Table 1.
5. Methylated ctDNA as a Biomarker and Novel Approaches
DNA methylation. DNA hypermethylation at CpG islands and whole genome hypomethylation are both epigenetic hallmarks of melanoma [119,120,121]. A gradual gain of DNA hypermethylation status has been observed to occur in parallel with tumour aggressiveness. Characteristically, methylation of tumour suppressor genes’ promoter regions leads to their aberrant downregulation, which contributes to both oncogenesis and disease transformation/aggressiveness [122].
Recently, a lot of focus has been directed towards characterising ctDNA methylation status and the generation of methylation signatures that could function as disease biomarkers [123,124,125]. One of the candidate genes tested was tissue factor pathway inhibitor 2 (TFPI2), which was found to be methylated in the sera of patients with melanoma with the magnitude of methylation being more pronounced in the metastatic setting [126]. Hoon and colleagues examined the serum of patients with melanoma and discovered that the incidence of TSG hypermethylation increased during tumour progression and that specifically the hypermethylation of MGMT, RASSF1A and DAPK was significantly higher in metastatic tumours compared to primary melanoma tumours [127]. Moreover, others showed that a wider panel of genes can be interrogated via a ctDNA methylation analysis workflow using an amplicon-based NGS panel performed on bisulphite-treated DNA [56]. Diefenbach and colleagues confirmed the hypermethylation of seven genes (GJB2, HOXA9, MEOX2, OLIG3, PON3, RASSF1 and TFAP2B) known to be hypermethylated in patients with metastatic melanoma but not healthy controls. Moreover, they showed that this serum signature of methylated tumour-related genes can act as a predictive marker of response to treatment with chemotherapy or immunotherapy and predict survival outcomes [128]. Targeted hypermethylated genes in this study were RASSF1A and RAR-SS2. The studies above are limited to locus-specific analyses of known genes and involved the methodology of sodium bisulphite modification, which is notoriously laborious and lengthy.
Tissue-independent novel techniques. In an attempt to interrogate a larger part of the genome, Liu and colleagues developed an NGS-targeted methylation sequencing assay to measure the methylation status of more than 9000 CpG sites, selected according to TCGA data. This assay was used to predict tumour origin in a study including various tumour types, including melanoma. The methylation scores derived detected the presence of cancer in ~84% of the cases, and methylation-based signatures accurately classified the underlying cancer type in almost 79% of these [123]. Subsequently, further research revealed novel ways to dissect either large parts of or even the whole genome length, in order to discover signatures that could detect ctDNA with higher sensitivity and without the need for primary tissue. By testing the methylation status of >450,000 CpG islands of circulating free DNA (cfDNA), Moss and colleagues created a methylation atlas that can predict the presence of malignant DNA in circulating blood [129]. Additionally, the application of an immunoprecipitation-based protocol to sequence the entire cell-free DNA (cfMeDIPseq) detected large scale methylation patterns that are enriched for specific cancer types [130]. These novel techniques will undoubtedly be used in the near future for clinical validation in melanoma, where the challenges of primary tumour scarcity could be solved with the above tissue-independent assays. Moreover, additional epigenetic features of cfDNA, such as DNA fragment length are emerging as markers of underlying tumour biology and deserve further exploration in melanoma [131,132,133,134,135,136].
6. Limitations and Challenges with ctDNA
ctDNA in melanoma CNS metastases. One of the main limitations of ctDNA application particularly relevant to melanoma is the poor sensitivity in the context of exclusively intracranial disease [68,69,101,104,137]. Lee et al. showed what was essentially 0% detectability rate in patients with brain metastases only and a poor concordance of ctDNA response to intracranial disease response in patients who had mixed intra- and extracranial tumour burden [137]. In the same vein, Seremet and colleagues found that melanoma progression in the brain does not correlate with ctDNA detection, and similar observations have taken place in other tumour-type contexts too, making this a more universal phenomenon [70,138,139,140]. The known impact of the blood–brain barrier in hindering ctDNA from entering systemic circulation is a hypothetical culprit. A potential avenue for this challenge could be ctDNA measurement in cerebrospinal fluid (CSF), which has demonstrated improved sensitivity [141,142,143]. Due to the invasive nature of CSF sampling, there has been no large-scale study validating the wider applicability of CSF ctDNA measurement; nevertheless, case studies have showed that it reflects the disease dynamics in melanoma [144,145]. The low detectability of ctDNA in patients with brain metastatic disease could potentially be circumvented with the use of cfMeDIPseq. This novel technique that characterises methylome signatures in the blood was found to accurately discriminate between brain tumours based on their cell of origin, and based on their primary or metastatic nature, in a tissue-independent manner [146], and could potentially replace the use conventional ctDNA characterisation in patients with intracranial metastases from melanoma.
Technical challenges. In early-stage melanoma, where ctDNA shedding might be limited and specifically for the triple wild-type subtype, the need to optimise and validate DNA-based biomarkers with higher sensitivity is paramount [147]. Interpretation of results need to consider that in early disease, the ctDNA amount might not be enough to detect single nucleotide mutation that have been detected in tumour tissue [148]. In small primary melanoma tumours, there might not be enough tissue to implement a wide molecular genomic panel for full characterisation to begin with. Tissue-independent and agnostic methodologies, such as the methylation/fragmentomic signatures already mentioned, could provide some avenues for development. Moreover, exploring the tumour fraction profile of ctDNA, rather than targeting specific gene loci, also presents an interesting alternative [88]. Contamination of the results by DNA fragments derived from clonal haematopoiesis of indeterminate potential (CHIP) or non-neoplastic haematopoietic stem cells can lead to false-positive ctDNA results. This can be mitigated by advanced bioinformatic analysis or by filtering out mutations by sequencing matched tumour and leucocytes derived by the same sample [132,149]. Lastly, despite the myriad of techniques and technologies developed over the last few years for the detection of ctDNA, standardization regarding the wider use of a particular assay, streamlined analysis and even sampling timing are still not well defined. Recent intriguing evidence suggesting that intravasation of circulating tumour cells is circadian rhythm-dependent could signify that even the exact time of ctDNA sampling could affect the final result [150]. An overview of the potential application of ctDNA and other minimally invasive biomarkers are depicted in Figure 2.
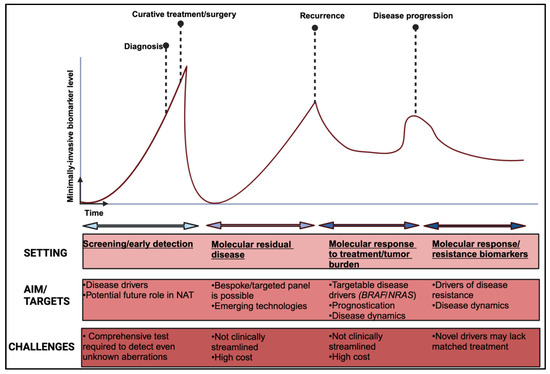
Figure 2.
Overview of non-invasive biomarkers applications in melanoma. NAT: neo-adjuvant treatment. Created with BioRender.com.
7. Circulating Melanoma Cells
Circulating tumour cells—the technique. Tumour progression results from a complex combination of genomic alterations that ultimately confer neoplastic cells with the capability to exit the primary site and migrate to distant tissues, primarily through haematogenous spread. During this process, primary tumour cells undergo genomic and structural alterations known as epithelial-to-mesenchymal transition, enabling them to enter the circulatory system as circulating tumour cells (CTCs). CTCs travel in the blood stream until permeation of the endothelium allows them to infiltrate distant tissues [151]. Minimally invasive blood sample collection, combined with novel isolation and purification techniques, presents CTCs as an appealing tool of liquid biopsy approaches in oncology. The methodologies for CTC acquisition have been evolving to ensure the capture of viable cells at the appropriate concentration, enabling the performance of downstream analyses across all levels of genomic, transcriptomic, proteomic and epigenomic readouts [152]. Among various techniques is the antibody-coated magnetic bead methodology directed towards surface proteins such as EpCAM, pan-CK and CD45. Additional approaches target specific physical features of CTCs, such as density-based separation [153]. Recent developments in techniques are grounded in the combination of physical and biological CTC features, such as fluid dynamics and membrane–cell superficial markers. Novel CTC chip devices exhibit the capability of efficiently capturing CTCs from whole blood samples within silicon micro-posts, integrating parameters such as cell flow velocity and cell attachment attributes within chambers housing anti-epithelial cell adhesion molecule (EpCAM) antibodies [152].
CTC isolation in the blood can predate the conventional, radiological imaging-based detection of metastatic disease, allowing for earlier detection of advanced status [154]. In the clinic, this could guide treatment decisions into preventing symptoms development and clinical deterioration [155]. Notably, simultaneous assessment of these circulating components in blood samples may offer a more comprehensive reflection of the inherent tumour heterogeneity than compare to the conventional evaluation of a single-site tissue biopsy, with KRAS mutational profile representing an example [156,157]. CTC levels also hold predictive value, as demonstrated in a prospective non-inferiority phase III trial that utilized a CTC threshold (≥5 CTCs/7.5 mL) as a stratification factor between adjuvant chemotherapy or endocrine therapy alone in localized hormone receptor-positive HER2-negative breast cancer patients [158]. This study successfully reached its non-inferiority progression-free survival endpoint, showing that CTC count is a superior biomarker to the conventional stratification approaches, such as clinical and pathological tumour features [158]. Prospectively, the genomic analysis of prostate CTCs for the androgen receptor splice variant, AR-V7, could serve as a predictive tool for assessing the clinical benefits of enzalutamide or abiraterone in terms of prostatic surface antigen (PSA) and imaging responses, within the context of metastatic castration-resistant disease. Patients with CTCs expressing the AR-V7 variant experienced inferior outcomes in terms of PSA response rates, PSA progression-free survival, clinical and radiographic progression-free survival, and overall survival in comparison to those lacking the expression of this variant [159].
Circulating tumour cells in melanoma. Broadly, evidence suggests that melanoma cells spreading from primary tumours follow the same process described previously, supporting the presence of melanoma CTCs (mCTCs) in blood stream at all disease stages [160]. Although mCTCs lack the expression of membrane epithelial markers usually necessary for cellular isolation (e.g., EpCAM), they express other specific markers such as melanoma cell adhesion molecule (MCAM), paired box gene 3 d isoform (PAX3d), microphthalmia-associated transcription factor m isoform (MTIFm) and transforming growth factor-beta 2 isoform (TGF-β2) that can be used for diagnostic purposes, as well as prognostication in cases of advanced disease [160,161].
The potential role of mCTCs in uveal melanoma diagnosis was investigated in a small prospective study involving eight treatment-naïve patients and four healthy individuals with choroidal nevi. The detection of mCTCs was carried out via a semi-automatic CellSearch system (Veridex, Warren, NJ, USA) utilizing magnetic beads coated with anti-CD146 and anti-MEL (high-molecular-weight melanoma-associated antigen) from peripheral blood samples. In this study, a minimum of one mCTC/7.5 mL of blood was identified in four out of eight patients with uveal melanoma, while no mCTCs were detected in the control group. Notably, the sole patient who presented with three mCTCs/7.5 mL also exhibited an extra scleral extension of the primary tumour [162].
The assessment of minimal residual disease by identifying or quantifying mCTCs in the bloodstream may contribute to a more precise evaluation of the risk of recurrence, which, in cutaneous melanoma, has traditionally relied heavily on pathologic tumour features such as Breslow thickness, ulceration, lymph node involvement and others, particularly in the early stages of the disease. In an exploratory analysis of the phase III trial EORTC 18991, which compared adjuvant pegylated interferon-alpha-2b with surveillance in patients with resected stage III melanoma, the presence of mCTCs was verified using RT-PCR for tyrosinase and Mart-1/Melan-A transcripts. Patients who had detectable mCTCs during the follow-up period experienced a significantly increased risk of distant relapse (hazard ratio of 2.23; 95% confidence interval, 1.40–3.55; p < 0.001), regardless of whether they received adjuvant treatment or not [163]. This underscores the significance of mCTCs as a prognostic factor in this context. Similar findings were observed in the metastatic setting in a prospective study conducted by Khoja et al., which explored mCTC quantification using the previously described CellSearch system in 101 patients in the early era of clinical immunotherapy. In this study, a cutoff of ≥2 CTCs/7.5 mL at baseline was associated with significantly poorer overall survival compared to those with <2 CTCs/7.5 mL (median overall survival of 7.2 months vs. 2.6 months, respectively; hazard ratio of 0.43, 95% confidence interval, 0.22–0.81; p = 0.009). Even after adjusting for other factors such as BRAF status, treatment type and time to diagnosis of metastatic disease, the cutoff of ≥2 CTCs/7.5 mL remained a significant poor prognostic factor (p = 0.005) [164].
Using parameters such as receptor expression and cellular physical characteristics can enable the extraction of valuable information from mCTCs. In a cohort of 43 patients with metastatic melanoma, positive mCTC detection by a combination of droplet digital PCR (ddPCR), RT-PCRand immunocytochemistry had a statistically significant correlation with inferior overall survival (HR for death of 7.8) when compared to patients without mCTCs prior to systemic treatment with immunotherapy or targeted therapy. Furthermore, high transcriptomic mCTC scores (cut-off of more than 100 transcripts per ml of blood based on ddPCR analyses) were also associated with inferior survival in the same study [165]. In addition, a comprehensive examination involving mRNA analysis (employing qRT-PCR for MAGEA3, MLANA, B4GALNT1, PAX3 and DCT), ddPCR for BRAF V600R mutation, as well as lactate dehydrogenase levels, in stage III/IV mCTC patients undergoing immune checkpoint inhibitor therapy, demonstrated the capacity to classify patients into two distinct and cohesive risk categories utilizing a decision-tree methodology. Those identified as high-risk exhibited notably inferior disease-free and overall survival outcomes within this context [166]. When researchers specifically looked into response to anti-PD-1 inhibition, the expression of PD-L1 in circulating tumour cells (CTCs) holds potential as a predictive indicator of pembrolizumab’s efficacy within the metastatic context. Khattak et al. have reported that patients with positive PD-L1 status in mCTCs prior to treatment, as detected by flow cytometry, exhibit a progression-free survival of 26.6 months, in contrast to 5.5 months observed in PD-L1-negative mCTC cases. This difference was validated through multivariate analysis for the aforementioned endpoint [167].
The current absence of high-sensitivity standardized techniques for membrane markers associated to mCTCs, necessitates a significant volume of blood to facilitate a satisfactory isolation process. This presents a significant restricting factor in the development of CTCs’ clinical utility. Further challenges, such as the notably low success rates observed in ex vivo CTC cultures, also present considerable obstacles to their clinical translation [168]. On-going current studies investigating the role of mCTCs and other minimally invasive biomarkers are summarized in Table 1.
8. Melanoma Derived Extracellular Vesicles and Coding/Non-Coding RNA
Extracellular vesicles. Tumour cells are capable of intercellular communication via the secretion and packaging of intracellular signalling components (e.g., RNA, lipids, proteinsand DNA) into bilayer phospholipid membrane vesicles sized between 30 nm to 10 μm [169,170]. These extracellular vesicles (EV) can influence the local microenvironment or be exported to distant recipient tissues, ultimately fostering an environment that is conducive to tumour infiltration [171,172]. EVs are classified according to their size in exosomes (30 nm–150 nm) or shed microvesicles (50 nm–1.300 nm), with each exhibiting distinct biogenetic features [173]. The formation of exosomes is associated with the recycling processes of membrane multivesicular elements, as exemplified by endocytosis, wherein intracellular vesicles are generated through the invagination of the plasma membrane, subsequently undergoing loading and processing of the components of these vesicles for subsequent release into the extracellular environment [174]. On the other hand, shed microvesicles originate from the budding of cytoplasmic material surrounded by the plasma membrane through reorganization and contraction of the actin–myosin cytoskeleton near the cell periphery [175].
EV-mediated intercellular physiological communications are subverted by cancer cells in many steps involved in tumorigenesis. EVs transporting cargo could be selectively enriched with various types of coding and non-coding RNA capable of influencing transcript programs locally or in distant tissues [171]. Recent analyses have revealed that mRNA, miRNA and long non-coding RNA (lncRNA) represent the predominant nucleic acids within EVs. These can be assessed through methodologies such as quantitative polymerase chain reaction (qPCR), next-generation sequencing or microarray techniques [176]. Serum breast cancer associated exosomes contain-miRNAs that are capable of initiating tumorigenesis of normal epithelial cells trough modification of their transcriptomic program [177]. Similarly, patients’ culture-derived glioblastoma cells can secrete exosomes enriched in mRNA, miRNA and proteins capable of incorporation by brain endothelial cells and subsequent stimulation of angiogenesis. Moreover, EGFRvIII mRNA EV-cargo, a truncated form of epidermal growth factor receptor specifically expressed by glioblastoma cells, can be detected in patients’ serum, transferred to and merged with other glioma cancer cells lacking this marker and leading to upregulation of their oncogenic activity [178,179].
Interestingly, tumour-derived exosomes can exhibit tropism towards certain distant tissues owing to their encapsulation of tumour-specific adhesion molecules, such as integrins. Within these tissues, the cargo carried by EVs can instigate changes in the extracellular matrix, vasculature and intratumoural immune microenvironment. These modifications can bolster a microenvironment conducive to tumour colonization. The presence of these adhesion molecules could elucidate the predilection for metastatic localization in organs like the lungs and liver [180]. Aligned with the strong interaction between melanoma and the immune system, exosomes derived from the plasma of patients with melanoma exhibit a higher enrichment of immunosuppressive proteins when compared to healthy controls. These exosomes demonstrate the ability to hinder the activity of both CD8+ T cells and NK cells in vitro [181].
EV cargo is cell-specific and mirrors factors such as cellular metabolism and genomic profile [182,183]. Moreover, in vitro experiments have demonstrated that tumour cells exhibit increased amounts of EV compared to their normal tissue counterparts [184]. Interestingly, some tumour-associated EVs could be found in higher concentrations in different body fluids of cancer patients compared to healthy controls [185]. Exploring these characteristics allowed for the development of methods of measuring the EV-related components which could be used in clinical practice such as the evaluation of RNA contained-exosomes in urine samples for the diagnosis of high-grade prostate cancer [186,187,188]. These isolation techniques integrate physical EV features with cellular-origin specific markers to enhance the efficiency and purity of the isolation process [189]. For example, accurate characterization of surface EV proteins is essential to permit the production of specific monoclonal antibodies that could segregate EV subtypes for widespread differently clinical uses [171].
EVs in melanoma. Small-scale studies have revealed the clinical potential of melanoma-specific EV cargo measurement, as biomarkers for both early and advanced disease stages [190,191]. The exosomal miRNA plasma profile could potentially aid in the diagnosis of melanoma with a substantial degree of accuracy, as evidenced by the diminished levels of EV-miR-1180-3p observed in patients in comparison to healthy controls (AUC for the ROC curve of 0.729) [192]. Mechanistically, miR-1180-3p expression negatively regulates malignant cell traits, such as proliferation, invasion and migration, in an in vitro melanoma cell model, possibly through post-transcriptional inhibition of the melanoma related-gene ST3GAL4 expression [192]. In addition, elevated levels of serum melanoma exosome-associated proteins, namely S100 and MIA, demonstrated sensitivity in differentiating patients with metastatic disease against both disease-free and healthy individuals. Furthermore, heightened levels of serum MIA-associated exosomes proved to be a significant predictor of poorer survival outcomes [193].
In vitro, uveal melanoma (UM) cells demonstrated a greater propensity for EV shedding in comparison to normal choroidal melanocytes. This phenomenon extends to the clinical setting, wherein the mean concentration of EVs positive for melanoma markers such as CD63 and TSG101 was significantly higher in the vitreous fluid, aqueous fluid and plasma of UM patients when compared to a control group devoid of cancer [194]. Notably, it is of interest that the cargo content of these EVs remained consistent regardless of the origin of the fluid samples in UM patients, thereby underscoring their potential utility as a tool for disease monitoring [194]. In accordance with these findings, Wróblewska et al. demonstrated a distinct EV-associated miRNA profile in exosomes isolated from the bloodstream of UM patients when compared to that of healthy individuals. Employing a real-time qPCR approach, the authors observed a significantly elevated level of miRNAs associated with pivotal processes related to malignant acquisition traits in affected individuals [195]. Notably, the following miRNAs displayed heightened expression levels: hsa-miR-191-5p, hsa-miR-223-3p, hsa-miR-139-5p, hsa-miR-10b-5p, hsa-miR-483-5p, hsa-miR-203a and hsa-miR-122-5. Among these, hsa-miR-191-5p, hsa-miR-223-3p, hsa-miR-483-5p and hsa-miR-203a exhibited the most pronounced sensitivity for diagnosing UM, with an accuracy exceeding 80% for each. Moreover, hsa-miR-191-5p and hsa-miR-144-5p showed potential for discriminating between localized and metastatic diseases, with the former displaying lower and the latter higher expression levels. However, their accuracy in doing so was less robust, as indicated by area under the ROC curve values of 0.71 and 0.86, respectively [195]. Reinforcing the potential role of miRNAs as prognostic tools, Sun et al. developed an integrated risk signature for overall survival in UM-affected individuals. This signature was developed using the five highest-performing serum miRNA expression profiles (hsa-miR-513a-5p, miR-506-3p, miR-508-3p, miR-140-3pand miR-103a-2-5p), which were combined with clinical information and their respective target genes using Weighted Correlation Network Analysis (WGCNA). This risk signature consistently demonstrated the ability to differentiate between low and high-risk groups, as evidenced by Kaplan–Meier analysis (p < 0.001), ROC curve analysis (accuracy > 0.9) and its status as an independent prognostic factor according to multiple Cox regression analysis (hazard ratio for death of 3.799; 95% CI: 1.903–7.583; p < 0.001) [196].
Notably, these vesicular markers also hold promise for treatment monitoring. A correlation was observed between reduced plasma levels of four specific EV-melanoma membrane-bound proteins (MCSP, MCAM, ERBB3 and LNGFR) and response to treatment involving combination BRAF/MEK inhibitors in patients with metastatic disease harbouring the BRAF V600E mutation [197]. When examining soluble and exosomal PD-L1 in patients with melanoma, Cordonnier and colleagues highlighted the fact that exosomal PD-L1 was higher in concentration compared to soluble PD-L1 and that the exosomal form retained immunosuppressive properties. Intriguingly, diminished EV-PD-L1 plasma levels were prospectively linked to favourable responses to systemic treatment (specifically anti-PD1 inhibitors), in the context of metastatic disease, demonstrating strong accuracy (AUC of 0.867 for ROC curve) [198]. This raises hope that EV-PD-L1 could represent a reliable and dynamic biomarker of response to immunotherapy. Another potential avenue for use of EVs is treatment monitoring. Shi et al. showcased a robust concordance between plasma EVs transcriptomic profiles and those derived from bulk tumour biopsies. The assessment of EV transcriptomic profiles had the potential to delineate genomic signatures for predicting resistance to immune checkpoint inhibitors through dynamic evaluations before and during treatment, albeit with a marginally lower accuracy compared to that achieved through bulk tumour biopsies. Furthermore, the precision of these EV-genomic signatures can be enhanced through the application of a deconvolution model, allowing for a more accurate differentiation between tumoural EV-transcripts and those originating from non-tumoural sources [199].
Despite these promising prospects, employing EVs as clinical decision tools demands resolution of pivotal issues. These include the standardization of vesicle purification methodologies, the refinement of EV tumour-specific biomarkers not expressed by normal cells and their validation through large prospective studies (Table 1) [187].
Circulating free MicroRNA (miRNA) and long non-coding RNA in melanoma. Preliminary analysis has revealed abnormal expression profiles of miRNA in various tissue samples, including blood, from cancer patients compared to healthy individuals [200]. These findings have prompted further studies investigating the role of miRNA in carcinogenesis, as well as its potential utility as a diagnostic, prognostic and predictive tool in the field of melanoma [201]. In preclinical models, miR-221 plays a pro-oncogenic role in melanoma cells by downregulating the expression of the cellular cycle modulator p27Kip1/CDKN1B. This finding has been translated to clinical relevance as indicated by a positive association between serum miR-221 levels and more advanced disease stages as well as increased tumour thickness. Additionally, the concentration of miR-221 in serum may prove useful for disease monitoring following primary tumour resection [202]. A multicentre study, encompassing centres in Australia and Germany, utilizing a refined panel of 7 melanoma-related miRNAs (MELmir-7) measured in serum, has demonstrated a sensitivity of 93% and specificity of 82% for the detection of clinically occult metastatic disease. Additionally, it exhibited superior performance in detecting melanoma recurrences compared to serum measurements of LDH and S100B [203]. Aligned with these findings, Huber et al. illustrated a statistically significant elevation in serum levels of miRNAs associated with myeloid-derived suppressor cells (miR-146a, let-7e, miR-125aand miR-145b) in patients diagnosed with metastatic melanoma when compared to healthy controls. Moreover, the study demonstrated that elevated baseline serum levels of these miRNAs were correlated with diminished overall and progression-free survival in patients exposed to a combination of immunotherapy and targeted therapy in the metastatic setting [204].
Recent studies have brought to light the role of non-coding RNAs (ncRNAs), distinct from EV-associated miRNAs, in melanoma carcinogenesis, underscoring their potential as diagnostic tools and innovative therapeutic targets [205]. Previous analyses have unveiled heightened levels of SPRIGHTLY, a lncRNA, engaged in intracellular lipid regulation, within the plasma of melanoma patients as measured by qPCR, in contrast to healthy donors [206]. Furthermore, a cutoff of a 2.64-fold difference in cancerous/noncancerous SPRIGHTLY levels has been identified as a prognostic tool, achieving an ROC accuracy of 0.813 (p < 0.001) and correlating with diminished survival among high-expression melanoma patients, compared to their low-expression counterparts (38 vs. 51 months; p < 0.001). In concordance with these findings, RNA interference (RNAi) targeting SPRIGHTLY has been demonstrated to impede malignant behaviour, including migration and invasion and to diminish the viability of melanoma cells in vitro [207]. A pan-cancer RNA sequencing analysis, encompassing over 10,000 tumour specimens from the TCGA database, revealed a significant upregulation of Survival Associated Mitochondrial Melanoma Specific Oncogenic lncRNA (SAMMSON) in cutaneous and uveal melanoma (UM) samples compared to other tumour types. In UM patients, SAMMSON expression was also found to be associated with staging of metastatic disease. Biologically, SAMMSON expression appears to play a crucial role in melanoma cell viability, as demonstrated by its knockdown via antisense oligonucleotide (ASO), which resulted in apoptosis in various UM and cutaneous melanoma cell lines. Other analyses also revealed similar insights into circular RNAs (circRNAs), a specific type of ncRNA, in melanoma development. This was demonstrated by a distinct circRNA expression profile in tumour tissue samples when compared to adjacent normal tissue in six patients with lymph node-positive oral mucosal melanoma, as assessed by microarray analysis. The authors identified 58 upregulated and 32 downregulated circRNAs associated with melanoma samples, which were subsequently validated by qRT-PCR for the five circRNAs (upregulated: hsa_circ_0005320, hsa_circ_0067531, hsa_circ_0008042; downregulated: hsa_circ_0000869 and hsa_circ_0000853) among the top 10 altered ones, as compared with matched normal tissues. Additionally, bioinformatic analysis indicated that these three highly expressed circRNAs may be associated with tumourigenesis and metastatic facilitation by targeting specific microRNAs [208]. Similarly, Bian and colleagues demonstrated a high expression of hsa_circ_0025039 circRNA using a microarray approach, which was subsequently validated by qRT-PCR in melanoma tissue samples when compared to matched adjacent normal tissue samples [209]. The authors confirmed this expression profile in tumour samples from 43 patients with melanoma through qRT-PCR, comparing it to healthy skin samples. The authors also showed a negative prognostic value in patients with high expression compared to those with low expression (p < 0.05). Corroborating these findings, silencing hsa_circ_0025039 with siRNA was found to inhibit proliferation, impair cell invasionand reduce glucose consumption by melanoma cells in vitro. On-going current studies investigating the role of EVs and other minimally invasive biomarkers are summarized in Table 1.
9. Intestinal Microbiome as a Biomarker in Melanoma
Intestinal microbiota and their associated genome (microbiome) can modulate metabolism, antitumour activity and the safety of a broad variety of anticancer treatments, including immunotherapy [210,211,212,213,214]. We can infer the composition of the gut mucosal microbiome by examining the metagenomic landscape of the stool microbiome and this can give us invaluable information on how many different bacterial taxa are present in the host’s intestine i.e., on the taxonomic diversity. In a case control study, the gut microbiome diversity and composition were different between healthy controls, patients with early stage (stage I/II) and patients with late-stage melanoma (stage III/IV) [215]. Characteristically, α-diversity diminished in later stage melanoma.
An association between higher microbiota compositional diversity and superior benefit from anti-PD-1/PD-L1 immune checkpoint inhibition has been reported for several cancer types, including melanoma [216]. The abundance of specific bacteria populations such as Bifidobacterium and Ruminococcaceae has been detected in the gut microbiota of patients deriving benefit from immunotherapy [216,217]. On the other hand, the microbiome composition of non-responders is enriched for taxa such as Bacteroides and Clostridium species. Unsurprisingly, it has also emerged that antibiotic-induced gut microbiota dysbiosis adversely affects clinical outcomes during immune checkpoint inhibition [218,219,220,221,222]. So far, the mechanisms underlying the relationship between microbiota composition and response to immune checkpoint inhibitors suggest that microbiota can induce and activate multiple effector immune cells, such as NK, dendritic, CD4+ and CD8+ T cells, through interfering with the generation of several metabolites such as short-chain fatty acids, inosine and bile acids [223,224,225,226,227,228,229]. Furthermore, cross-reactivity between cancer and microbiota antigens can augment cancer immunogenicity and hence, promote immunotherapy efficacy [230,231]. Manipulation of the gut microbiome with faecal microbial transplantation can not only reverse the microbiome dysbiosis but also rescue anti-PD-1 resistance in patients with metastatic melanoma [224,232,233].
In addition to the role of gut microbiome in ICI responsiveness [217,233,234], imbalances in the composition or function of gut microbes has also been implicated in ICI toxicity, specifically in patients with melanoma [233]. Species belonging to Bacteroidetes, Clostridia, and Proteobacteria phyla have been linked to increased incidence and severity of immune-related adverse events (irAEs) [214,235,236]. Out of these events, ICI-related colitis has been most closely linked with the presence of members of the Firmicutes phyla (F. prausnitzi, G. formicilis, Ruminococcaceae spp.), during anti-CTLA-4 monotherapy and B. intestinalis (phylum Bacteroidetes) during combination CTLA-4/PD-1 immunotherapy [214]. Collectively, a microbial signature dominated by Streptococcus spp. is accompanied by worse clinical outcomes and a high frequency of distinct irAEs, such as immune-mediated arthropathy [233]. Intriguingly, a strong sequence and conformational homology exists between tumour-associated antigens and microbiome-derived peptides from species of the Firmicutes and Bacteroidetes phyla [237], suggesting molecular mimicry between tumour and microbiome epitopes as a cross-reactivity triggering event.
Notwithstanding all the interesting observations made in individual studies above, there is a notable lack of concordance and a common response-related microbial signature, when multiple studies are interrogated. Several factors may of course adversely influence the interpretation of data including small sample sizes, arbitrary definitions of clinical responders and non-responders and variations in bioinformatic analytic pipelines [238]. Importantly, there is widespread challenge in controlling for the confounding factors known to influence gut microbiome composition, such as medication intake, diet, geography and ethnicity [239,240]. In summary, given the complex and vast microcosmos of the gut microbiome, it might be significantly challenging to compile a unique and consistent compositional signature that would efficiently act as a biomarker of response or toxicity. It is conceivable that we would have to redirect our focus from compositional difference to differences in the overall functional capacity of an entire microbial community and discover a direct and quantifiable readout of this function, that could act as a biomarker.
10. Conclusions
For years, the oncology research community was entirely focused on the tumour microenvironment and the influence it exerts on its adjacent tissue. More recently however, it has become apparent that a lot of information can be extracted from circulating tumour products and their dynamics for both prognostication and treatment outcome prediction (Table 2). The initial excitement about circulating tumour cells seemed to have now been transferred to circulating tumour DNA, which is currently the most well-developed, minimally invasive biomarker. Tremendous effort has been directed towards its optimization and streamlining; and further on-going research is aiming at extracting all the possible information from circulating nucleic acid products, with the least possible nucleic acid volume required. Despite this, there are still on-going challenged pertaining to the complexity and cost of ctDNA and other minimally invasive biomarkers, indicating that their incorporation into clinical practice will require extensive work and validation (Figure 2). In the meantime, additional tumour products, such as extracellular vesicles and micro-RNAs can add further characterization and potentially work synergistically with ctDNA in create a complete molecular tumour profile. Last, but not least, the intestinal mucosal surface and its strong impact on modulating systemic immunity can also be exploited, with information residing into non-invasive stool samples. In keeping with biomarker development, standardization, harmonization, and cross validation will establish new discoveries into clinical practice. Serial monitoring of biomarkers in patients receiving treatment will inform escalation and de-escalation strategies. To achieve this, however, the incorporation of these biomarkers in large scale, national and international collaborative clinical studies, will be necessary.
Table 2.
List of emerging minimally invasive biomarkers in melanoma and their characteristics.
Author Contributions
Writing—original draft preparation, P.S. and C.D.H.L.; writing—review and editing, P.S., C.D.H.L. and A.S.; supervision, P.S. and A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Long, G.V.; Robert, C.; Tawbi, H.A.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized Phase III Trial Evaluating Spartalizumab Plus Dabrafenib and Trametinib for BRAF V600–Mutant Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, A.K.M.; Spindler, K.-L.G. ctDNA-Response evaluation criteria in solid tumors—A new measure in medical oncology. Eur. J. Cancer 2023, 180, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Haslam, A.; Hey, S.P.; Gill, J.; Prasad, V. A systematic review of trial-level meta-analyses measuring the strength of association between surrogate end-points and overall survival in oncology. Eur. J. Cancer 2019, 106, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Cindy Yang, S.Y.; Lien, S.C.; Wang, B.X.; Clouthier, D.L.; Hanna, Y.; Cirlan, I.; Zhu, K.; Bruce, J.P.; El Ghamrasni, S.; Iafolla, M.A.J.; et al. Pan-cancer analysis of longitudinal metastatic tumors reveals genomic alterations and immune landscape dynamics associated with pembrolizumab sensitivity. Nat. Commun. 2021, 12, 5137. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients with Metastatic Melanoma Treated with Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Huang, N.; Lee, K.J.; Stark, M.S. Current Trends in Circulating Biomarkers for Melanoma Detection. Front. Med. 2022, 9, 873728. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Forkasiewicz, A.; Dorociak, M.; Stach, K.; Szelachowski, P.; Tabola, R.; Augoff, K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell. Mol. Biol. Lett. 2020, 25, 35. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Grob, J.-J.; Nathan, P.; Ribas, A.; Robert, C.; Schadendorf, D.; Lane, S.R.; Mak, C.; Legenne, P.; Flaherty, K.T.; et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016, 17, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ferrucci, P.; Gonzalez, R.; Thomas, L.; Maio, M.; Hill, A. Efficacy of nivolumab (NIVO) plus ipilimumab (IPI) combination in patients with advanced melanoma (MEL) and elevated serum lactate dehydrogenase (LDH): A pooled analysis. In Proceedings of the Society for Melanoma Research 2016 Congress, Virtual, 28–31 October 2021; pp. 6–9. [Google Scholar]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]
- Egberts, F.; Kotthoff, E.M.; Gerdes, S.; Egberts, J.H.; Weichenthal, M.; Hauschild, A. Comparative study of YKL-40, S-100B and LDH as monitoring tools for Stage IV melanoma. Eur. J. Cancer 2012, 48, 695–702. [Google Scholar] [CrossRef]
- El-Sharkawi, D.; Basu, S.; Ocampo, C.; Qian, W.; D’Sa, S.; Hoskin, P.J.; Ardeshna, K.M. Elevated lactate dehydrogenase levels detected during routine follow-up do not predict relapse in patients with diffuse large B-cell lymphoma who achieve complete remission after primary treatment with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone-like immunochemotherapy. Leuk Lymphoma 2012, 53, 1949–1952. [Google Scholar]
- Kurzhals, J.K.; Klee, G.; Hagelstein, V.; Zillikens, D.; Terheyden, P.; Langan, E.A. Disease Recurrence during Adjuvant Immune Checkpoint Inhibitor Treatment in Metastatic Melanoma: Clinical, Laboratory, and Radiological Characteristics in Patients from a Single Tertiary Referral Center. Int. J. Mol. Sci. 2022, 23, 10723. [Google Scholar] [CrossRef] [PubMed]
- Szumera-Ciećkiewicz, A.; Bosisio, F.; Teterycz, P.; Antoranz, A.; Delogu, F.; Koljenović, S.; van de Wiel, B.A.; Blokx, W.; van Kempen, L.C.; Rutkowski, P.; et al. SOX10 is as specific as S100 protein in detecting metastases of melanoma in lymph nodes and is recommended for sentinel lymph node assessment. Eur. J. Cancer 2020, 137, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Harpio, R.; Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem. 2004, 37, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; De Roeck, A.; Garcia, C.; Stoebner, P.-E.; Fichel, F.; Garima, F.; Perriard, F.; Daures, J.-P.; Meunier, L.; Alix-Panabières, C. S100-EPISPOT: A New Tool to Detect Viable Circulating Melanoma Cells. Cells 2019, 8, 755. [Google Scholar] [CrossRef] [PubMed]
- Nikolin, B.; Djan, I.; Trifunovic, J.; Dugandzija, T.; Novkovic, D.; Djan, V.; Vucinic, N. MIA, S100 and LDH as important predictors of overall survival of patients with stage IIb and IIc melanoma. J. Buon 2016, 21, 691–697. [Google Scholar] [PubMed]
- Hauschild, A.; Engel, G.; Brenner, W.; Gläser, R.; Mönig, H.; Henze, E.; Christophers, E. S100B protein detection in serum is a significant prognostic factor in metastatic melanoma. Oncology 1999, 56, 338–344. [Google Scholar] [CrossRef]
- Kärnell, R.; von Schoultz, E.; Hansson, L.O.; Nilsson, B.; Arstrand, K.; Kågedal, B. S100B protein, 5-S-cysteinyldopa and 6-hydroxy-5-methoxyindole-2-carboxylic acid as biochemical markers for survival prognosis in patients with malignant melanoma. Melanoma Res. 1997, 7, 393–399. [Google Scholar] [CrossRef]
- Mocellin, S.; Zavagno, G.; Nitti, D. The prognostic value of serum S100B in patients with cutaneous melanoma: A meta-analysis. Int. J. Cancer 2008, 123, 2370–2376. [Google Scholar] [CrossRef]
- Alegre, E.; Sammamed, M.; Fernández-Landázuri, S.; Zubiri, L.; González, Á. Chapter Two—Circulating Biomarkers in Malignant Melanoma. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 69, pp. 47–89. [Google Scholar]
- Garbe, C.; Leiter, U.; Ellwanger, U.; Blaheta, H.-J.; Meier, F.; Rassner, G.; Schittek, B. Diagnostic value and prognostic significance of protein S-100β, melanoma-inhibitory activity, and tyrosinase/MART-1 reverse transcription-polymerase chain reaction in the follow-up of high-risk melanoma patients. Cancer 2003, 97, 1737–1745. [Google Scholar] [CrossRef]
- Dummer, R.; Panizzon, R.; Bloch, P.H.; Burg, G. Updated Swiss guidelines for the treatment and follow-up of cutaneous melanoma. Dermatology 2005, 210, 39–44. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Kwong, L.N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.; Dopierala, J.A.; Coupland, S.E. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin. Cancer Res. 2010, 16, 6083–6092. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Ganguly, A.; Bianciotto, C.G.; Turaka, K.; Tavallali, A.; Shields, J.A. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology 2011, 118, 396–401. [Google Scholar] [CrossRef]
- Coupland, S.E.; Damato, B.E. Molecular analysis of uveal melanoma. Ophthalmology 2013, 120, e50. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Nassar, K.W.; Tan, A.C. The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 2020, 61, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef]
- Perkins, G.; Lu, H.; Garlan, F.; Taly, V. Droplet-Based Digital PCR: Application in Cancer Research. Adv. Clin. Chem. 2017, 79, 43–91. [Google Scholar]
- Anagnostou, V.; Forde, P.M.; White, J.R.; Niknafs, N.; Hruban, C.; Naidoo, J.; Marrone, K.; Sivakumar, I.K.A.; Bruhm, D.C.; Rosner, S.; et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Res. 2019, 79, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.N.; Becker, T.; Bray, V.; Chua, W.; Ma, Y.; Xu, B.; Lynch, D.; de Souza, P.; Roberts, T. Plasma next generation sequencing and droplet digital PCR-based detection of epidermal growth factor receptor (EGFR) mutations in patients with advanced lung cancer treated with subsequent-line osimertinib. Thorac. Cancer 2019, 10, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Strbenac, D.; Yang, J.Y.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Spillane, A.J.; Stretch, J.R.; Saw, R.P.M.; et al. Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma. Cancers 2019, 11, 1905. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Siravegna, G. How to use liquid biopsies to treat patients with cancer. ESMO Open 2021, 6, 100060. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Stadler, J.C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Fribbens, C.; Garcia Murillas, I.; Beaney, M.; Hrebien, S.; O’Leary, B.; Kilburn, L.; Howarth, K.; Epstein, M.; Green, E.; Rosenfeld, N.; et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann. Oncol. 2018, 29, 145–153. [Google Scholar] [CrossRef]
- Laith, A.-S.; Brooke, W.; Faris, T.; Consolacion, M.; Abhenil, M.; David, W.C.; Eitan, A. Changes in circulating tumor DNA and outcomes in solid tumors treated with immune checkpoint inhibitors: A systematic review. J. Immunother. Cancer 2023, 11, e005854. [Google Scholar]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Gracie, L.; Pan, Y.; Atenafu, E.G.; Ward, D.G.; Teng, M.; Pallan, L.; Stevens, N.M.; Khoja, L. Circulating tumour DNA (ctDNA) in metastatic melanoma, a systematic review and meta-analysis. Eur. J. Cancer 2021, 158, 191–207. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- McEvoy, A.C.; Pereira, M.R.; Reid, A.; Pearce, R.; Cowell, L.; Al-Ogaili, Z.; Khattak, M.A.; Millward, M.; Meniawy, T.M.; Gray, E.S.; et al. Monitoring melanoma recurrence with circulating tumor DNA: A proof of concept from three case studies. Oncotarget 2019, 10, 113–122. [Google Scholar] [CrossRef][Green Version]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef]
- Genta, S.; Araujo, D.V.; Keshavarzi, S.; Pimentel Muniz, T.; Saeed Kamil, Z.; Howarth, K.; Terrell, S.; Joad, A.; Ventura, R.; Covelli, A.; et al. Leveraging personalized circulating tumor DNA (ctDNA) for detection and monitoring of molecular residual disease in high-risk melanoma. J. Clin. Oncol. 2022, 40, 9579. [Google Scholar] [CrossRef]
- Kaneko, A.; Kanemaru, H.; Kajihara, I.; Mijiddorj, T.; Miyauchi, H.; Kuriyama, H.; Kimura, T.; Sawamura, S.; Makino, K.; Miyashita, A.; et al. Liquid biopsy-based analysis by ddPCR and CAPP-Seq in melanoma patients. J. Dermatol. Sci. 2021, 102, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Desai, K.; Tang, T.; Weber, J.S.; Dolfi, S.; Ritchings, C.; Huang, S.-P.; Bolisetty, M.; Sausen, M.; Del Vecchio, M.; et al. Association of pre-treatment ctDNA with disease recurrence and clinical and translational factors in patients with stage IIIBD/IV melanoma treated with adjuvant immunotherapy (CheckMate 915). Ann. Oncol. 2022, 33, S904. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Krinshpun, S.; Kalashnikova, E.; Sudhaman, S.; Ozturk Topcu, T.; Nichols, M.; Martin, J.; Bui, K.M.; Palsuledesai, C.C.; Malhotra, M.; et al. Circulating tumor DNA-based molecular residual disease detection for treatment monitoring in advanced melanoma patients. Cancer 2023, 129, 1723–1734. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Tiragolumab Plus Atezolizumab versus Atezolizumab in the Treatment of Stage II Melanoma Patients Who Are ctDNA-Positive Following Resection 2023. Available online: https://clinicaltrials.gov/study/NCT05060003?cond=melanoma&term=ctDNA&rank=3 (accessed on 8 March 2022).
- Jia, Q.; Chiu, L.; Wu, S.; Bai, J.; Peng, L.; Zheng, L.; Zang, R.; Li, X.; Yuan, B.; Gao, Y.; et al. Tracking Neoantigens by Personalized Circulating Tumor DNA Sequencing during Checkpoint Blockade Immunotherapy in Non-Small Cell Lung Cancer. Adv. Sci. 2020, 7, 1903410. [Google Scholar] [CrossRef]
- Weiss, G.J.; Beck, J.; Braun, D.P.; Bornemann-Kolatzki, K.; Barilla, H.; Cubello, R.; Quan, W., Jr.; Sangal, A.; Khemka, V.; Waypa, J.; et al. Tumor Cell-Free DNA Copy Number Instability Predicts Therapeutic Response to Immunotherapy. Clin. Cancer Res. 2017, 23, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Goodman, A.M.; Kato, S.; Ellison, C.K.; Daniels, G.A.; Kim, L.; Nakashe, P.; McCarthy, E.; Mazloom, A.R.; McLennan, G.; et al. Genome-Wide Sequencing of Cell-Free DNA Identifies Copy-Number Alterations That Can Be Used for Monitoring Response to Immunotherapy in Cancer Patients. Mol. Cancer Ther. 2019, 18, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Proudhon, C.; Romano, E.; Girard, N.; Lantz, O.; Stern, M.-H.; Pierga, J.-Y.; Bidard, F.-C. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2018, 15, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Reichert, Z.R.; Morgan, T.M.; Li, G.; Castellanos, E.; Snow, T.; Dall’Olio, F.G.; Madison, R.W.; Fine, A.D.; Oxnard, G.R.; Graf, R.P.; et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: A real-world outcomes study. Ann. Oncol. 2023, 34, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Bratman, S.V.; Yang, S.Y.C.; Iafolla, M.A.J.; Liu, Z.; Hansen, A.R.; Bedard, P.L.; Lheureux, S.; Spreafico, A.; Razak, A.A.; Shchegrova, S.; et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat. Cancer 2020, 1, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating Tumor DNA Predicts Outcome from First-, but not Second-line Treatment and Identifies Melanoma Patients Who May Benefit from Combination Immunotherapy. Clin. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef]
- Váraljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.-M.; Horn, P.A.; von Neuhoff, N.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Takai, E.; Omata, W.; Totoki, Y.; Nakamura, H.; Shiba, S.; Takahashi, A.; Namikawa, K.; Mori, T.; Yamazaki, N.; Shibata, T.; et al. Clonal dynamics of circulating tumor DNA during immune checkpoint blockade therapy for melanoma. Cancer Sci. 2021, 112, 4748–4757. [Google Scholar] [CrossRef]
- Xi, L.; Pham, T.H.; Payabyab, E.C.; Sherry, R.M.; Rosenberg, S.A.; Raffeld, M. Circulating Tumor DNA as an Early Indicator of Response to T-cell Transfer Immunotherapy in Metastatic Melanoma. Clin. Cancer Res. 2016, 22, 5480–5486. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Herbreteau, G.; Vallée, A.; Knol, A.-C.; Théoleyre, S.; Quéreux, G.; Varey, E.; Khammari, A.; Dréno, B.; Denis, M.G. Quantitative monitoring of circulating tumor DNA predicts response of cutaneous metastatic melanoma to anti-PD1 immunotherapy. Oncotarget 2018, 9, 25265–25276. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Rozeman, E.A.; Mason, R.; Goldinger, S.M.; Geukes Foppen, M.H.; Hoejberg, L.; Schmidt, H.; van Thienen, J.V.; Haanen, J.; Tiainen, L.; et al. Discontinuation of anti-PD-1 antibody therapy in the absence of disease progression or treatment limiting toxicity: Clinical outcomes in advanced melanoma. Ann. Oncol. 2019, 30, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.J.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef]
- Forthun, R.B.; Hovland, R.; Schuster, C.; Puntervoll, H.; Brodal, H.P.; Namløs, H.M.; Aasheim, L.B.; Meza-Zepeda, L.A.; Gjertsen, B.T.; Knappskog, S.; et al. ctDNA detected by ddPCR reveals changes in tumour load in metastatic malignant melanoma treated with bevacizumab. Sci. Rep. 2019, 9, 17471. [Google Scholar] [CrossRef]
- Kozak, K.; Kowalik, A.; Gos, A.; Wasag, B.; Lugowska, I.; Jurkowska, M.; Krawczynska, N.; Kosela-Paterczyk, H.; Switaj, T.; Teterycz, P.; et al. Cell-free DNA BRAF V600E measurements during BRAF inhibitor therapy of metastatic melanoma: Long-term analysis. Tumori J. 2020, 106, 241–248. [Google Scholar] [CrossRef]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Herbreteau, G.; Vallée, A.; Knol, A.-C.; Théoleyre, S.; Quéreux, G.; Varey, E.; Khammari, A.; Dréno, B.; Denis, M.G. Circulating Tumor DNA Early Kinetics Predict Response of Metastatic Melanoma to Anti-PD1 Immunotherapy: Validation Study. Cancers 2021, 13, 1826. [Google Scholar] [CrossRef]
- Garlan, F.; Blanchet, B.; Kramkimel, N.; Puszkiel, A.; Golmard, J.-L.; Noe, G.; Dupin, N.; Laurent-Puig, P.; Vidal, M.; Taly, V.; et al. Circulating Tumor DNA Measurement by Picoliter Droplet-Based Digital PCR and Vemurafenib Plasma Concentrations in Patients with Advanced BRAF-Mutated Melanoma. Target. Oncol. 2017, 12, 365–371. [Google Scholar] [CrossRef]
- Haselmann, V.; Gebhardt, C.; Brechtel, I.; Duda, A.; Czerwinski, C.; Sucker, A.; Holland-Letz, T.; Utikal, J.; Schadendorf, D.; Neumaier, M. Liquid Profiling of Circulating Tumor DNA in Plasma of Melanoma Patients for Companion Diagnostics and Monitoring of BRAF Inhibitor Therapy. Clin. Chem. 2018, 64, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative Cell-Free Circulating BRAFV600E Mutation Analysis by Use of Droplet Digital PCR in the Follow-up of Patients with Melanoma Being Treated with BRAF Inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Schreuer, M.; Meersseman, G.; Van Den Herrewegen, S.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Rothwell, D.G.; Chow, S.; Shaw, H.M.; Turajlic, S.; Smith, N.; Clipson, A.; Clarke, H.; Kelso, N.; Mitchell, J.; et al. CAcTUS: A parallel arm, biomarker driven, phase II feasibility trial to determine the role of circulating tumor DNA in guiding a switch between targeted therapy and immune therapy in patients with advanced cutaneous melanoma. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS9587. [Google Scholar] [CrossRef]
- Van der Hiel, B.; Haanen, J.B.A.G.; Stokkel, M.P.M.; Peeper, D.S.; Jimenez, C.R.; Beijnen, J.H.; van de Wiel, B.A.; Boellaard, R.; van den Eertwegh, A.J.M.; REPOSIT Study Group. Vemurafenib plus cobimetinib in unresectable stage IIIc or stage IV melanoma: Response monitoring and resistance prediction with positron emission tomography and tumor characteristics (REPOSIT): Study protocol of a phase II, open-label, multicenter study. BMC Cancer 2017, 17, 649. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Chen, R. The DecisionDx-UM Gene Expression Profile Test Provides Risk Stratification and Individualized Patient Care in Uveal Melanoma. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Francis, J.H.; Barker, C.A.; Brannon, A.R.; Canestraro, J.; Robbins, M.; Swartzwelder, C.E.; Levine, S.; Law, C.; Berger, M.F.; Shoushtari, A.; et al. Detectability of Plasma-Derived Circulating Tumor DNA Panel in Patients Undergoing Primary Treatment for Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2022, 63, 17. [Google Scholar] [CrossRef]
- Beasley, A.; Isaacs, T.; Khattak, M.A.; Freeman, J.B.; Allcock, R.; Chen, F.K.; Pereira, M.R.; Yau, K.; Bentel, J.; Vermeulen, T.; et al. Clinical Application of Circulating Tumor Cells and Circulating Tumor DNA in Uveal Melanoma. JCO Precis Oncol. 2018, 2, 279. [Google Scholar] [CrossRef]
- Le Guin, C.H.D.; Bornfeld, N.; Bechrakis, N.E.; Jabbarli, L.; Richly, H.; Lohmann, D.R.; Zeschnigk, M. Early detection of metastatic uveal melanoma by the analysis of tumor-specific mutations in cell-free plasma DNA. Cancer Med. 2021, 10, 5974–5982. [Google Scholar] [CrossRef]
- Beasley, A.B.; de Bruyn, D.P.; Calapre, L.; Al-Ogaili, Z.; Isaacs, T.W.; Bentel, J.; Reid, A.L.; Dwarkasing, R.S.; Pereira, M.R.; Khattak, M.A.; et al. Detection of metastases using circulating tumour DNA in uveal melanoma. J. Cancer Res. Clin. Oncol. 2023, 149, 14953–14963. [Google Scholar] [CrossRef]
- Wong, D.; Luo, P.; Znassi, N.; Arteaga, D.P.; Gray, D.; Danesh, A.; Han, M.; Zhao, E.Y.; Pedersen, S.; Prokopec, S.; et al. Integrated, Longitudinal Analysis of Cell-free DNA in Uveal Melanoma. Cancer Res. Commun. 2023, 3, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Mariani, P.; Bidard, F.C.; Rampanou, A.; Houy, A.; Servois, V.; Ramtohul, T.; Pierron, G.; Chevrier, M.; Renouf, B.; Lantz, O.; et al. Circulating Tumor DNA as a Prognostic Factor in Patients with Resectable Hepatic Metastases of Uveal Melanoma. Ann. Surg. 2023, 278, e827–e834. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Butler, M.O.; Shoushtari, A.N.; Hassel, J.C.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Piulats, J.M.; Rioth, M.; et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: A phase 2 trial. Nat. Med. 2022, 28, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Beasley, A.B.; Chen, F.K.; Isaacs, T.W.; Gray, E.S. Future perspectives of uveal melanoma blood based biomarkers. Br. J. Cancer 2022, 126, 1511–1528. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Mo, Y.; Yu, Y.; Amiri, K.; Sosman, J.; Greally, J.; Verma, A. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010, 20, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Dobre, E.G.; Constantin, C.; Costache, M.; Neagu, M. Interrogating Epigenome toward Personalized Approach in Cutaneous Melanoma. J. Pers. Med. 2021, 11, 901. [Google Scholar] [CrossRef]
- Howell, P.M.; Liu, S.; Ren, S.; Behlen, C.; Fodstad, O.; Riker, A.I. Epigenetics in Human Melanoma. Cancer Control 2009, 16, 200–218. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Salvianti, F.; Orlando, C.; Massi, D.; De Giorgi, V.; Grazzini, M.; Pazzagli, M.; Pinzani, P. Tumor-Related Methylated Cell-Free DNA and Circulating Tumor Cells in Melanoma. Front. Mol. Biosci. 2016, 2, 76. [Google Scholar] [CrossRef]
- Nigro, C.L.; Wang, H.; McHugh, A.; Lattanzio, L.; Matin, R.; Harwood, C.; Syed, N.; Hatzimichael, E.; Briasoulis, E.; Merlano, M.; et al. Methylated Tissue Factor Pathway Inhibitor 2 (TFPI2) DNA in Serum Is a Biomarker of Metastatic Melanoma. J. Investig. Dermatol. 2013, 133, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Hoon, D.S.B.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004, 23, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; O’Day, S.J.; Umetani, N.; Martinez, S.R.; Kitago, M.; Koyanagi, K.; Kuo, C.; Takeshima, T.-L.; Milford, R.; Wang, H.-J.; et al. Predictive Utility of Circulating Methylated DNA in Serum of Melanoma Patients Receiving Biochemotherapy. J. Clin. Oncol. 2005, 23, 9351–9358. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Saw, R.P.M.; Howle, J.R.; Spillane, A.J.; Scolyer, R.A.; Kefford, R.F.; et al. Design and Testing of a Custom Melanoma Next Generation Sequencing Panel for Analysis of Circulating Tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Doebley, A.-L.; Ko, M.; Liao, H.; Cruikshank, A.E.; Kikawa, C.; Santos, K.; Hiatt, J.; Patton, R.D.; De Sarkar, N.; Hoge, A.C.H.; et al. Griffin: Framework for clinical cancer subtyping from nucleosome profiling of cell-free DNA. medRxiv 2021. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Heitzer, E.; Lo, Y.M.D.; Mouliere, F.; Tsui, D.W.Y. Cell-Free DNA Fragmentomics: The New “Omics” on the Block. Clin. Chem. 2020, 66, 1480–1484. [Google Scholar] [CrossRef]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of ctDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef]
- Singh, A.P.; Cheng, H.; Guo, X.; Levy, B.; Halmos, B. Circulating Tumor DNA in Non–Small-Cell Lung Cancer: A Primer for the Clinician. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Huang, W.-T.; Lu, N.-M.; Hsu, W.-Y.; Chang, S.-E.; Atkins, A.; Mei, R.; Javey, M. CSF-ctDNA SMSEQ Analysis to Tailor the Treatment of a Patient with Brain Metastases: A Case Report. Case Rep. Oncol. 2018, 11, 68–74. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L. Liquid biopsy for HER2-positive breast cancer brain metastasis: The role of the cerebrospinal fluid. ESMO Open 2017, 2, e000270. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Momtaz, P.; Pentsova, E.; Abdel-Wahab, O.; Diamond, E.; Hyman, D.; Merghoub, T.; You, D.; Gasmi, B.; Viale, A.; Chapman, P.B. Quantification of tumor-derived cell free DNA(cfDNA) by digital PCR (DigPCR) in cerebrospinal fluid of patients with BRAFV600 mutated malignancies. Oncotarget 2016, 7, 85430–85436. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Glitza Oliva, I.C.; Douse, D.Y.; Chen, M.M.; Lan, C.; Haydu, L.E.; Huse, J.T.; Roy-Chowdhuri, S.; Luthra, R.; Wistuba, I.I.; et al. Evaluating Circulating Tumor DNA From the Cerebrospinal Fluid of Patients with Melanoma and Leptomeningeal Disease. J. Neuropathol. Exp. Neurol. 2018, 77, 628–635. [Google Scholar] [CrossRef]
- Melms, J.C.; Ho, K.W.; Thummalapalli, R.; Tyler, J.; Brinker, T.J.; Singh, V.; Sengupta, S.; Mier, J.; Izar, B. Implementation of cell-free tumor DNA sequencing from the cerebrospinal fluid to guide treatment in a patient with primary leptomeningeal melanoma: A case report. Mol. Clin. Oncol. 2018, 9, 58–61. [Google Scholar] [CrossRef]
- Li, Y.; Pan, W.; Connolly, I.D.; Reddy, S.; Nagpal, S.; Quake, S.; Gephart, M.H. Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J. Neurooncol. 2016, 128, 93–100. [Google Scholar] [CrossRef]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, R.; Yang, L.; Shi, W.; Zhang, Y.; Liu, J.; Li, X.; Zhou, J.; Bing, P. Minimal residual disease (MRD) detection in solid tumors using circulating tumor DNA: A systematic review. Front. Genet. 2023, 14, 1172108. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Diamantopoulou, Z.; Castro-Giner, F.; Schwab, F.D.; Foerster, C.; Saini, M.; Budinjas, S.; Strittmatter, K.; Krol, I.; Seifert, B.; Heinzelmann-Schwarz, V.; et al. The metastatic spread of breast cancer accelerates during sleep. Nature 2022, 607, 156–162. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.-i. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694–710. [Google Scholar] [CrossRef]
- Hosseini, H.; Obradović, M.M.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef]
- Liu, M.C.; Shields, P.G.; Warren, R.D.; Cohen, P.; Wilkinson, M.; Ottaviano, Y.L.; Rao, S.B.; Eng-Wong, J.; Seillier-Moiseiwitsch, F.; Noone, A.-M. Circulating tumor cells: A useful predictor of treatment efficacy in metastatic breast cancer. J. Clin. Oncol. 2009, 27, 5153. [Google Scholar] [CrossRef]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef]
- Kondo, Y.; Hayashi, K.; Kawakami, K.; Miwa, Y.; Hayashi, H.; Yamamoto, M. KRAS mutation analysis of single circulating tumor cells from patients with metastatic colorectal cancer. BMC Cancer 2017, 17, 311. [Google Scholar] [CrossRef]
- Bidard, F.-C.; Jacot, W.; Kiavue, N.; Dureau, S.; Kadi, A.; Brain, E.; Bachelot, T.; Bourgeois, H.; Gonçalves, A.; Ladoire, S. Efficacy of circulating tumor cell count–driven vs clinician-driven first-line therapy choice in hormone receptor–positive, ERBB2-negative metastatic breast cancer: The STIC CTC randomized clinical trial. JAMA Oncol. 2021, 7, 34–41. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Rapanotti, M.C.; Campione, E.; Spallone, G.; Orlandi, A.; Bernardini, S.; Bianchi, L. Minimal residual disease in melanoma: Circulating melanoma cells and predictive role of MCAM/MUC18/MelCAM/CD146. Cell Death Discov. 2017, 3, 17005. [Google Scholar] [CrossRef]
- Vendittelli, F.; Paolillo, C.; Autilio, C.; Lavieri, M.M.; Silveri, S.L.; Capizzi, R.; Capoluongo, E. Absolute quantitative PCR for detection of molecular biomarkers in melanoma patients: A preliminary report. Clin. Chim. Acta 2015, 444, 242–249. [Google Scholar] [CrossRef]
- Bande, M.F.; Santiago, M.; Muinelo-Romay, L.; Blanco, M.J.; Mera, P.; Capeans, C.; Pardo, M.; Piñeiro, A. Detection of circulating melanoma cells in choroidal melanocytic lesions. BMC Res. Notes 2015, 8, 452. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Suciu, S.; Santinami, M.; Testori, A.; Kruit, W.H.; Marsden, J.; Punt, C.J.; Salès, F.; Gore, M.; MacKie, R.; et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: Final results of EORTC 18991, a randomised phase III trial. Lancet 2008, 372, 117–126. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker utility of circulating tumor cells in metastatic cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef]
- Aya-Bonilla, C.A.; Morici, M.; Hong, X.; McEvoy, A.C.; Sullivan, R.J.; Freeman, J.; Calapre, L.; Khattak, M.A.; Meniawy, T.; Millward, M.; et al. Detection and prognostic role of heterogeneous populations of melanoma circulating tumour cells. Br. J. Cancer 2020, 122, 1059–1067. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chang, S.C.; Lam, S.; Irene Ramos, R.; Tran, K.; Ohe, S.; Salomon, M.P.; Bhagat, A.A.S.; Teck Lim, C.; Fischer, T.D.; et al. Prospective Molecular Profiling of Circulating Tumor Cells from Patients with Melanoma Receiving Combinatorial Immunotherapy. Clin. Chem. 2020, 66, 169–177. [Google Scholar] [CrossRef]
- Khattak, M.A.; Reid, A.; Freeman, J.; Pereira, M.; McEvoy, A.; Lo, J.; Frank, M.H.; Meniawy, T.; Didan, A.; Spencer, I.; et al. PD-L1 Expression on Circulating Tumor Cells May Be Predictive of Response to Pembrolizumab in Advanced Melanoma: Results from a Pilot Study. Oncologist 2020, 25, e520–e527. [Google Scholar] [CrossRef]
- Ring, A.; Nguyen-Sträuli, B.D.; Wicki, A.; Aceto, N. Biology, vulnerabilities and clinical applications of circulating tumour cells. Nat. Rev. Cancer 2023, 23, 95–111. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; Van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef]
- Asleh, K.; Dery, V.; Taylor, C.; Davey, M.; Djeungoue-Petga, M.-A.; Ouellette, R.J. Extracellular vesicle-based liquid biopsy biomarkers and their application in precision immuno-oncology. Biomark. Res. 2023, 11, 99. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef]
- Taylor, J.; Azimi, I.; Monteith, G.; Bebawy, M. Ca2+ mediates extracellular vesicle biogenesis through alternate pathways in malignancy. J. Extracell. Vesicles 2020, 9, 1734326. [Google Scholar] [CrossRef]
- Yeung, C.-Y.C.; Dondelinger, F.; Schoof, E.M.; Georg, B.; Lu, Y.; Zheng, Z.; Zhang, J.; Hannibal, J.; Fahrenkrug, J.; Kjaer, M. Circadian regulation of protein cargo in extracellular vesicles. Sci. Adv. 2022, 8, eabc9061. [Google Scholar] [CrossRef]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.-P.; Low, L.-H.; Silke, J.; Tan, S.-S. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef]
- Ruivo, C.F.; Adem, B.; Silva, M.; Melo, S.A. The biology of cancer exosomes: Insights and new perspectives. Cancer Res. 2017, 77, 6480–6488. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, H.; Natalia, A.; Lim, C.Z.; Ho, N.R.; Ong, C.-A.J.; Teo, M.C.; So, J.B.; Shao, H. Exosome-templated nanoplasmonics for multiparametric molecular profiling. Sci. Adv. 2020, 6, eaba2556. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, X.; Gardashova, G.; Yang, Y.; Zhang, Y.; Xu, L.; Zeng, Y. Molecular and functional extracellular vesicle analysis using nanopatterned microchips monitors tumor progression and metastasis. Sci. Transl. Med. 2020, 12, eaaz2878. [Google Scholar] [CrossRef]
- McKiernan, J.; Donovan, M.J.; O’Neill, V.; Bentink, S.; Noerholm, M.; Belzer, S.; Skog, J.; Kattan, M.W.; Partin, A.; Andriole, G. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016, 2, 882–889. [Google Scholar] [CrossRef]
- Xu, K.; Jin, Y.; Li, Y.; Huang, Y.; Zhao, R. Recent progress of exosome isolation and peptide recognition-guided strategies for exosome research. Front. Chem. 2022, 10, 844124. [Google Scholar] [CrossRef]
- Choi, D.-Y.; Park, J.-N.; Paek, S.-H.; Choi, S.-C.; Paek, S.-H. Detecting early-stage malignant melanoma using a calcium switch-enriched exosome subpopulation containing tumor markers as a sample. Biosens. Bioelectron. 2022, 198, 113828. [Google Scholar] [CrossRef]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; Gonzalez, A. Study of circulating microRNA-125b levels in serum exosomes in advanced melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Wang, L.; Li, M.; Shen, M.; Zhou, Z.; Zhu, S.; Li, K.; Fang, Z.; Yan, B. The plasma exosomal miR-1180-3p serves as a novel potential diagnostic marker for cutaneous melanoma. Cancer Cell Int. 2021, 21, 487. [Google Scholar] [CrossRef]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta 2016, 454, 28–32. [Google Scholar] [CrossRef]
- Pessuti, C.L.; Costa, D.F.; Ribeiro, K.S.; Abdouh, M.; Tsering, T.; Nascimento, H.; Commodaro, A.G.; Marcos, A.A.A.; Torrecilhas, A.C.; Belfort, R.N. Characterization of extracellular vesicles isolated from different liquid biopsies of uveal melanoma patients. J. Circ. Biomark. 2022, 11, 36. [Google Scholar] [CrossRef]
- Wróblewska, J.P.; Lach, M.S.; Rucinski, M.; Piotrowski, I.; Galus, L.; Suchorska, W.M.; Kreis, S.; Marszałek, A. MiRNAs from serum-derived extracellular vesicles as biomarkers for uveal melanoma progression. Front. Cell Dev. Biol. 2022, 10, 1008901. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, X.; Cong, Z.; Teng, S. Identification of 5 microRNA biomarkers associated with the prognosis of uveal melanoma. Medicine 2022, 101, e30366. [Google Scholar] [CrossRef]
- Wang, J.; Wuethrich, A.; Sina, A.A.I.; Lane, R.E.; Lin, L.L.; Wang, Y.; Cebon, J.; Behren, A.; Trau, M. Tracking extracellular vesicle phenotypic changes enables treatment monitoring in melanoma. Sci. Adv. 2020, 6, eaax3223. [Google Scholar] [CrossRef]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.-P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles 2020, 9, 1710899. [Google Scholar] [CrossRef]
- Shi, A.; Kasumova, G.G.; Michaud, W.A.; Cintolo-Gonzalez, J.; Díaz-Martínez, M.; Ohmura, J.; Mehta, A.; Chien, I.; Frederick, D.T.; Cohen, S. Plasma-derived extracellular vesicle analysis and deconvolution enable prediction and tracking of melanoma checkpoint blockade outcome. Sci. Adv. 2020, 6, eabb3461. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Varrone, F.; Caputo, E. The miRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef]
- Kanemaru, H.; Fukushima, S.; Yamashita, J.; Honda, N.; Oyama, R.; Kakimoto, A.; Masuguchi, S.; Ishihara, T.; Inoue, Y.; Jinnin, M.; et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J. Dermatol. Sci. 2011, 61, 187–193. [Google Scholar] [CrossRef]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, J. Non-coding RNAs in melanoma: Biological functions and potential clinical applications. Mol. Ther. Oncolytics 2021, 22, 219–231. [Google Scholar] [CrossRef]
- Liu, T.; Shen, S.K.; Xiong, J.G.; Xu, Y.; Zhang, H.Q.; Liu, H.J.; Lu, Z.G. Clinical significance of long noncoding RNA SPRY4-IT1 in melanoma patients. FEBS Open Bio. 2016, 6, 147–154. [Google Scholar] [CrossRef]
- Khaitan, D.; Dinger, M.E.; Mazar, J.; Crawford, J.; Smith, M.A.; Mattick, J.S.; Perera, R.J. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011, 71, 3852–3862. [Google Scholar] [CrossRef]
- Ju, H.; Zhang, L.; Mao, L.; Liu, S.; Xia, W.; Hu, J.; Ruan, M.; Ren, G. Altered expression pattern of circular RNAs in metastatic oral mucosal melanoma. Am. J. Cancer Res. 2018, 8, 1788–1800. [Google Scholar]
- Bian, D.; Wu, Y.; Song, G. Novel circular RNA, hsa_circ_0025039 promotes cell growth, invasion and glucose metabolism in malignant melanoma via the miR-198/CDK4 axis. Biomed. Pharmacother. 2018, 108, 165–176. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef]
- Xavier, J.B.; Young, V.B.; Skufca, J.; Ginty, F.; Testerman, T.; Pearson, A.T.; Macklin, P.; Mitchell, A.; Shmulevich, I.; Xie, L.; et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer 2020, 6, 192–204. [Google Scholar] [CrossRef]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef]
- Witt, R.G.; Cass, S.H.; Tran, T.; Damania, A.; Nelson, E.E.; Sirmans, E.; Burton, E.M.; Chelvanambi, M.; Johnson, S.; Tawbi, H.A.; et al. Gut Microbiome in Patients with Early-Stage and Late-Stage Melanoma. JAMA Dermatol. 2023, 159, 1076–1084. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Fidelle, M.; Iebba, V.; Alla, L.; Pasolli, E.; Segata, N.; Desnoyer, A.; Pietrantonio, F.; Ferrere, G.; et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur. Urol. 2020, 78, 195–206. [Google Scholar] [CrossRef]
- Jiang, S.; Geng, S.; Chen, Q.; Zhang, C.; Cheng, M.; Yu, Y.; Zhang, S.; Shi, N.; Dong, M. Effects of Concomitant Antibiotics Use on Immune Checkpoint Inhibitor Efficacy in Cancer Patients. Front. Oncol. 2022, 12, 823705. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Yuan, M.; Tao, M.; Kong, C.; Li, H.; Tong, J.; Zhu, H.; Yan, X. Antibiotic administration shortly before or after immunotherapy initiation is correlated with poor prognosis in solid cancer patients: An up-to-date systematic review and meta-analysis. Int. Immunopharmacol. 2020, 88, 106876. [Google Scholar] [CrossRef]
- Hopkins, A.M.; Kichenadasse, G.; Karapetis, C.S.; Rowland, A.; Sorich, M.J. Concomitant Antibiotic Use and Survival in Urothelial Carcinoma Treated with Atezolizumab. Eur. Urol. 2020, 78, 540–543. [Google Scholar] [CrossRef]
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812. [Google Scholar] [CrossRef]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8(+) T cell immunity. Cell Metab. 2021, 33, 988–1000.e7. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopes, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Fluckiger, A.; Daillère, R.; Sassi, M.; Sixt, B.S.; Liu, P.; Loos, F.; Richard, C.; Rabu, C.; Alou, M.T.; Goubet, A.-G.; et al. Cross-reactivity between tumor MHC class I–restricted antigens and an enterococcal bacteriophage. Science 2020, 369, 936–942. [Google Scholar] [CrossRef]
- Bessell, C.A.; Isser, A.; Havel, J.J.; Lee, S.; Bell, D.R.; Hickey, J.W.; Chaisawangwong, W.; Glick Bieler, J.; Srivastava, R.; Kuo, F.; et al. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight 2020, 5, 135597. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef] [PubMed]
- Ragone, C.; Manolio, C.; Mauriello, A.; Cavalluzzo, B.; Buonaguro, F.M.; Tornesello, M.L.; Tagliamonte, M.; Buonaguro, L. Molecular mimicry between tumor associated antigens and microbiota-derived epitopes. J. Transl. Med. 2022, 20, 316. [Google Scholar] [CrossRef]
- Lee, K.A.; Thomas, A.M.; Bolte, L.A.; Björk, J.R.; de Ruijter, L.K.; Armanini, F.; Asnicar, F.; Blanco-Miguez, A.; Board, R.; Calbet-Llopart, N.; et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat. Med. 2022, 28, 535–544. [Google Scholar] [CrossRef]
- Cresci, G.A.; Bawden, E. Gut Microbiome: What We Do and Don’t Know. Nutr. Clin. Pract. 2015, 30, 734–746. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).