

HCS-Splice: A High-Content Screening Method to Advance the Discovery of RNA Splicing-Modulating Therapeutics

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mutagenesis of Two-Fluorescent Reporter Plasmid Bearing Exon 10
2.2. SH-SY5Y Human Cell Line Culture
2.3. SH-SY5Y: Transfection and Cotransfection
2.4. High Content Imaging Acquisition
2.5. High Content Imaging System Analysis
2.6. Validation of HCS-Splice: RNA Extraction and Semiquantitative RT-PCR Analysis
2.7. Statistical Analysis
- and µ = mean; σ = Standard deviation; SNR: Signal to Noise Ratio.
3. Results
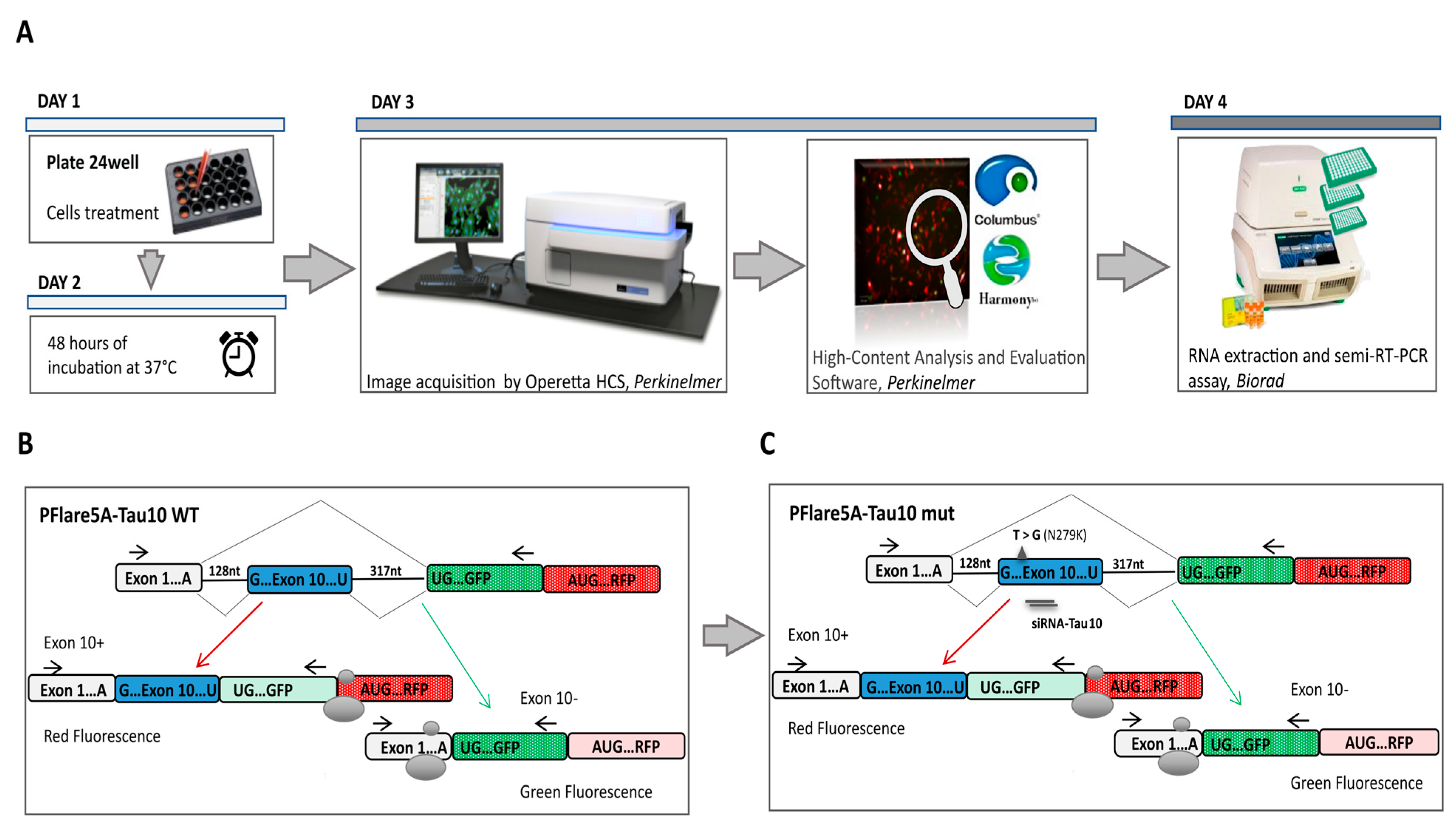
3.1. HCS-Splice Workflow
3.2. Exon 10 Site-Directed Mutagenesis of a Two-Colour (GFP/RFP) Fluorescent Reporter
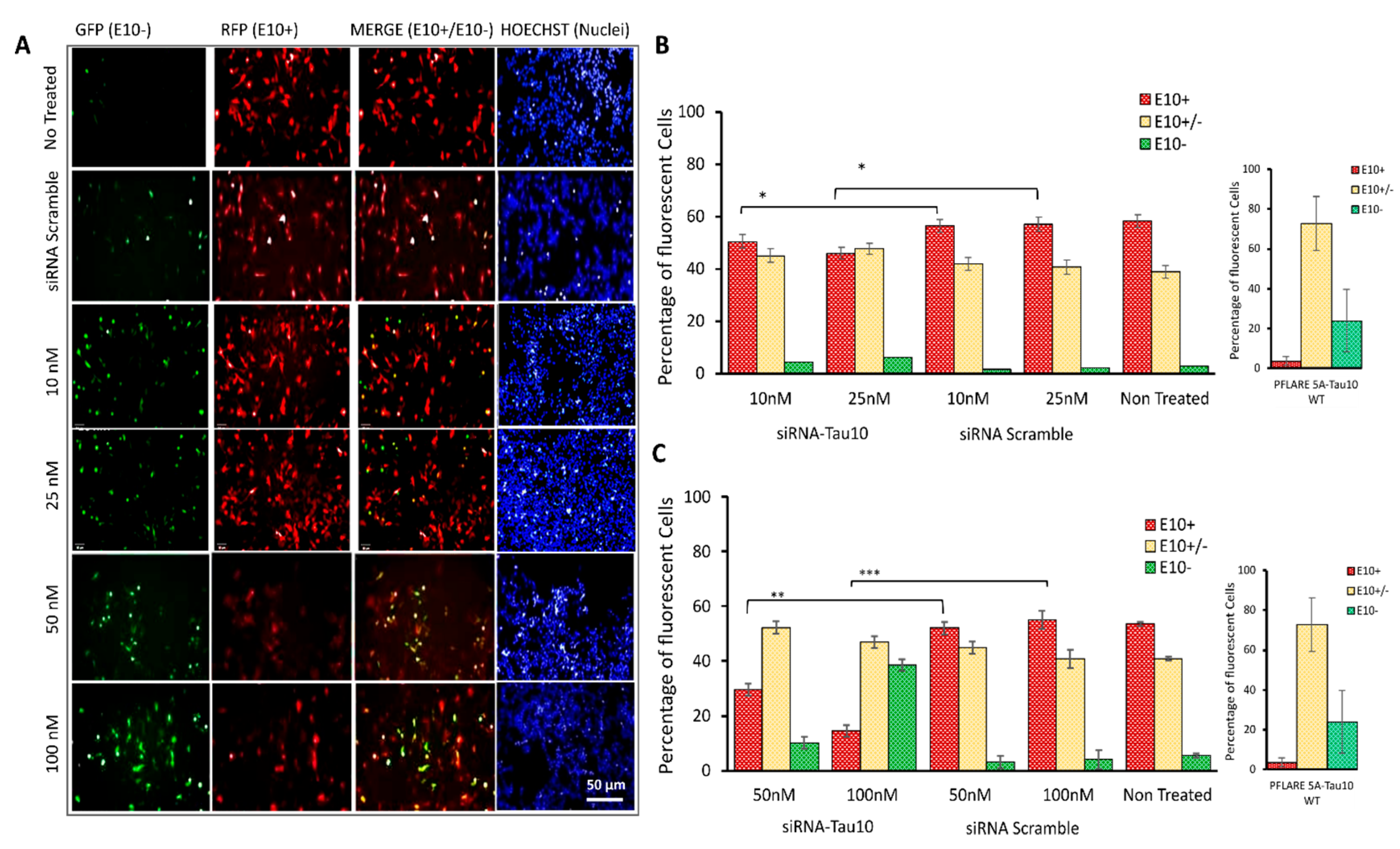
3.3. Development of an Image-Based HCS-Splice Method Using Two-Colour (GFP/RFP) Fluorescent Reporter Plasmids Transfected into SH-SY5Y Cells
3.4. HCS-Splice Data Validated Using Semi-Quantitative RT-PCR
3.5. HCS-Splice Method Validation by Using the siRNA Molecules Modulating the Exon 10 Splicing
3.6. Semiquantitative RT-PCR Analysis to Validate the Effects of the siRNA after HCS-Splice Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep Surveying of Alternative Splicing Complexity in the Human Transcriptome by High-Throughput Sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Bush, S.J.; Tovar-Corona, J.M.; Castillo-Morales, A.; Urrutia, A.O. Correcting for Differential Transcript Coverage Reveals a Strong Relationship between Alternative Splicing and Organism Complexity. Mol. Biol. Evol. 2014, 31, 1402–1413. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative Splicing: Increasing Diversity in the Proteomic World. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the Eukaryotic Proteome by Alternative Splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Wood, M.J.A.; Gait, M.J.; Yin, H. RNA-Targeted Splice-Correction Therapy for Neuromuscular Disease. Brain 2010, 133, 957–972. [Google Scholar] [CrossRef]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. HnRNP Proteins and Splicing Control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.D. Regulation of Splicing by SR Proteins and SR Protein-Specific Kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of Alternative Splicing Regulation: Insights from Molecular and Genomics Approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-MRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Influence of RNA Secondary Structure on the Pre-MRNA Splicing Process. Mol. Cell. Biol. 2004, 24, 10505–10514. [Google Scholar] [CrossRef] [PubMed]
- McManus, C.J.; Graveley, B.R. RNA Structure and the Mechanisms of Alternative Splicing. Curr. Opin. Genet. Dev. 2011, 21, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pandya-Jones, A.; Black, D.L. Co-Transcriptional Splicing of Constitutive and Alternative Exons. RNA 2009, 15, 1896–1908. [Google Scholar] [CrossRef]
- Shukla, S.; Oberdoerffer, S. Co-Transcriptional Regulation of Alternative Pre-MRNA Splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 673–683. [Google Scholar] [CrossRef]
- Montes, M.; Sanford, B.L.; Comiskey, D.F.; Chandler, D.S. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. 2019, 35, 68–87. [Google Scholar] [CrossRef]
- Fontana, F.; Siva, K.; Denti, M.A. A Network of RNA and Protein Interactions in Fronto Temporal Dementia. Front. Mol. Neurosci. 2015, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Nikom, D.; Zheng, S. Alternative Splicing in Neurodegenerative Disease and the Promise of RNA Therapies. Nat. Rev. Neurosci. 2023, 24, 457–473. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of Missense and 5′-Splice-Site Mutations in Tau with the Inherited Dementia FTDP-17. Nature 1998, 393, 702–704. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the Tau Gene in Familial Multiple System Tauopathy with Presenile Dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 Update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Kar, A.; Kuo, D.; He, R.; Zhou, J.; Wu, J.Y. Tau Alternative Splicing and Frontotemporal Dementia. Alzheimer Dis. Assoc. Disord. 2005, 19, S29. [Google Scholar] [CrossRef]
- Hefti, M.M.; Farrell, K.; Kim, S.H.; Bowles, K.R.; Fowkes, M.E.; Raj, T.; Crary, J.F. High-Resolution Temporal and Regional Mapping of MAPT Expression and Splicing in Human Brain Development. PLoS ONE 2018, 13, e0195771. [Google Scholar] [CrossRef] [PubMed]
- Froelich, S.; Basun, H.; Forsell, C.; Lilius, L.; Axelman, K.; Andreadis, A.; Lannfelt, L. Mapping of a disease locus for familial rapidly progressive frontotemporal dementia to chromosome 17q12-21. Am. J. Med. Genet. 1997, 74, 380–385. [Google Scholar] [CrossRef]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.Y.; Bird, T.D.; Schellenberg, G.D. Missense and Silent Tau Gene Mutations Cause Frontotemporal Dementia with Parkinsonism-Chromosome 17 Type, by Affecting Multiple Alternative RNA Splicing Regulatory Elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Cote, J.; Kwon, J.M.; Goate, A.M.; Wu, J.Y. Aberrant Splicing of Tau Pre-MRNA Caused by Intronic Mutations Associated with the Inherited Dementia Frontotemporal Dementia with Parkinsonism Linked to Chromosome 17. Mol. Cell. Biol. 2000, 20, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, L.; Ong, A.A.L.; Shi, J.; Zhong, Z.; Lye, M.L.; Liu, S.; Lisowiec-Wachnicka, J.; Kierzek, R.; Roca, X.; et al. A Disease-Causing Intronic Point Mutation C19G Alters Tau Exon 10 Splicing via RNA Secondary Structure Rearrangement. Biochemistry 2019, 58, 1565–1578. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT Mutations, Tauopathy, and Mechanisms of Neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Levinson, N.; Hinman, R.; Patil, A.; Stephenson, C.R.J.; Werner, S.; Woo, G.H.C.; Xiao, J.; Wipf, P.; Lynch, K.W. Use of Transcriptional Synergy to Augment Sensitivity of a Splicing Reporter Assay. RNA 2006, 12, 925–930. [Google Scholar] [CrossRef]
- Soret, J.; Bakkour, N.; Maire, S.; Durand, S.; Zekri, L.; Gabut, M.; Fic, W.; Divita, G.; Rivalle, C.; Dauzonne, D.; et al. Selective Modification of Alternative Splicing by Indole Derivatives That Target Serine-Arginine-Rich Protein Splicing Factors. Proc. Natl. Acad. Sci. USA 2005, 102, 8764–8769. [Google Scholar] [CrossRef]
- Zhang, M.L.; Lorson, C.L.; Androphy, E.J.; Zhou, J. An in Vivo Reporter System for Measuring Increased Inclusion of Exon 7 in SMN2 MRNA: Potential Therapy of SMA. Gene Ther. 2001, 8, 1532–1538. [Google Scholar] [CrossRef][Green Version]
- Siva, K.; Covello, G.; Denti, M.A. Exon-Skipping Antisense Oligonucleotides to Correct Missplicing in Neurogenetic Diseases. Nucleic Acid Ther. 2014, 24, 69–86. [Google Scholar] [CrossRef]
- Kuroyanagi, H.; Ohno, G.; Sakane, H.; Maruoka, H.; Hagiwara, M. Visualization and Genetic Analysis of Alternative Splicing Regulation In Vivo Using Fluorescence Reporters in Transgenic Caenorhabditis Elegans. Nat. Protoc. 2010, 5, 1495–1517. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Havlioglu, N.; Tarn, W.Y.; Wu, J.Y. RBM4 Interacts with an Intronic Element and Stimulates Tau Exon 10 Inclusion. J. Biol. Chem. 2006, 281, 24479–24488. [Google Scholar] [CrossRef] [PubMed]
- Ferri, L.; Covello, G.; Caciotti, A.; Guerrini, R.; Denti, M.A.; Morrone, A. Double-Target Antisense U1snRNAs Correct Mis-Splicing Due to c.639+861C>T and c.639+919G>A GLA Deep Intronic Mutations. Mol. Ther. Nucleic Acids 2016, 5, e380. [Google Scholar] [CrossRef]
- Covello, G.; Ibrahim, G.H.; Bacchi, N.; Casarosa, S.; Denti, M.A. Exon Skipping Through Chimeric Antisense >U1 SnRNAs to Correct Retinitis Pigmentosa GTPase-Regulator (RPGR) Splice Defect. Nucleic Acid Ther. 2022, 32, 333–349. [Google Scholar] [CrossRef]
- Newman, E.A.; Muh, S.J.; Hovhannisyan, R.H.; Warzecha, C.C.; Jones, R.B.; Mckeehan, W.L.; Carstens, R.P. Identification of RNA-Binding Proteins That Regulate FGFR2 Splicing through the Use of Sensitive and Specific Dual Color Fluorescence Minigene Assays. RNA 2006, 12, 1129–1141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orengo, J.P.; Bundman, D.; Cooper, T.A. A Bichromatic Fluorescent Reporter for Cell-Based Screens of Alternative Splicing. Nucleic Acids Res. 2006, 34, e148. [Google Scholar] [CrossRef]
- Stoilov, P.; Lin, C.-H.; Damoiseaux, R.; Nikolic, J.; Black, D.L. A High-Throughput Screening Strategy Identifies Cardiotonic Steroids as Alternative Splicing Modulators. Proc. Natl. Acad. Sci. USA 2008, 105, 11218–11223. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.G.; Black, D.L. Two-Color Fluorescent Reporter for Alternative Pre-MRNA Splicing. U.S. Patent 2010/0233685 A1, 16 September 2010. [Google Scholar]
- Zheng, S.; Damoiseaux, R.; Chen, L.; Black, D.L. A Broadly Applicable High-Throughput Screening Strategy Identifies New Regulators of Dlg4 (Psd-95) Alternative Splicing. Genome Res. 2013, 23, 998–1007. [Google Scholar] [CrossRef]
- Zheng, S. A Cell-Based High-Throughput Method for Identifying Modulators of Alternative Splicing. Methods Mol. Biol. 2017, 1648, 221–233. [Google Scholar] [CrossRef]
- Delisle, M.B.; Murrell, J.R.; Richardson, R.; Trofatter, J.A.; Rascol, O.; Soulages, X.; Mohr, M.; Calvas, P.; Ghetti, B. A Mutation at Codon 279 (N279K) in Exon 10 of the Tau Gene Causes a Tauopathy with Dementia and Supranuclear Palsy. Acta Neuropathol. 1999, 98, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Microtubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 643115. [Google Scholar] [CrossRef] [PubMed]
- Dravid, A.; Raos, B.; Svirskis, D.; O’Carroll, S.J. Optimised Techniques for High-Throughput Screening of Differentiated SH-SY5Y Cells and Application for Neurite Outgrowth Assays. Sci. Rep. 2021, 11, 23935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Wszolek, Z.K.; Pfeiffer, R.F.; Bhatt, M.H.; Schelper, R.L.; Cordes, M.; Snow, B.J.; Rodnitzky, R.L.; Wolters, E.C.; Arwert, F.; Calne, D.B. Rapidly Progressive Autosomal Dominant Padansonism and Dementia with Palhdo-Ponto-Nigral Degeneration. Ann. Neurol. 1992, 32, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Cheshire, W.P.; Tsuboi, Y.; Wszolek, Z.K. Physiologic Assessment of Autonomic Dysfunction in Pallidopontonigral Degeneration with N279K Mutation in the Tau Gene on Chromosome 17. Auton. Neurosci. Basic Clin. 2002, 102, 71–77. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, Q.; Zou, T. Alternative Splicing of Exon 10 in the Tau Gene as a Target for Treatment of Tauopathies. BMC Neurosci. 2008, 9, S10. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Tau Mutations in Frontotemporal Dementia FTDP-17 and Their Relevance for Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 110–121. [Google Scholar] [CrossRef]
- Covello, G.; Siva, K.; Denti, M.A. Rna Interference Mediated Therapy for Neurodegenerative Diseases. WO2016151523A1, 29 September 2016. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Covello, G.; Siva, K.; Adami, V.; Denti, M.A. HCS-Splice: A High-Content Screening Method to Advance the Discovery of RNA Splicing-Modulating Therapeutics. Cells 2023, 12, 1959. https://doi.org/10.3390/cells12151959
Covello G, Siva K, Adami V, Denti MA. HCS-Splice: A High-Content Screening Method to Advance the Discovery of RNA Splicing-Modulating Therapeutics. Cells. 2023; 12(15):1959. https://doi.org/10.3390/cells12151959
Chicago/Turabian StyleCovello, Giuseppina, Kavitha Siva, Valentina Adami, and Michela Alessandra Denti. 2023. "HCS-Splice: A High-Content Screening Method to Advance the Discovery of RNA Splicing-Modulating Therapeutics" Cells 12, no. 15: 1959. https://doi.org/10.3390/cells12151959
APA StyleCovello, G., Siva, K., Adami, V., & Denti, M. A. (2023). HCS-Splice: A High-Content Screening Method to Advance the Discovery of RNA Splicing-Modulating Therapeutics. Cells, 12(15), 1959. https://doi.org/10.3390/cells12151959