
ARH Family of ADP-Ribose-Acceptor Hydrolases

 , ,
, ,
Abstract
1. Dedication to Elaine and Myron Jacobson
1.1. Early Studies on Amino Acid Modification by ADP-Ribose
1.2. Collaborative Studies with the Jacobsons on Acetal Linkages
1.3. Regarding the Path to Dermatology Products
2. Introduction
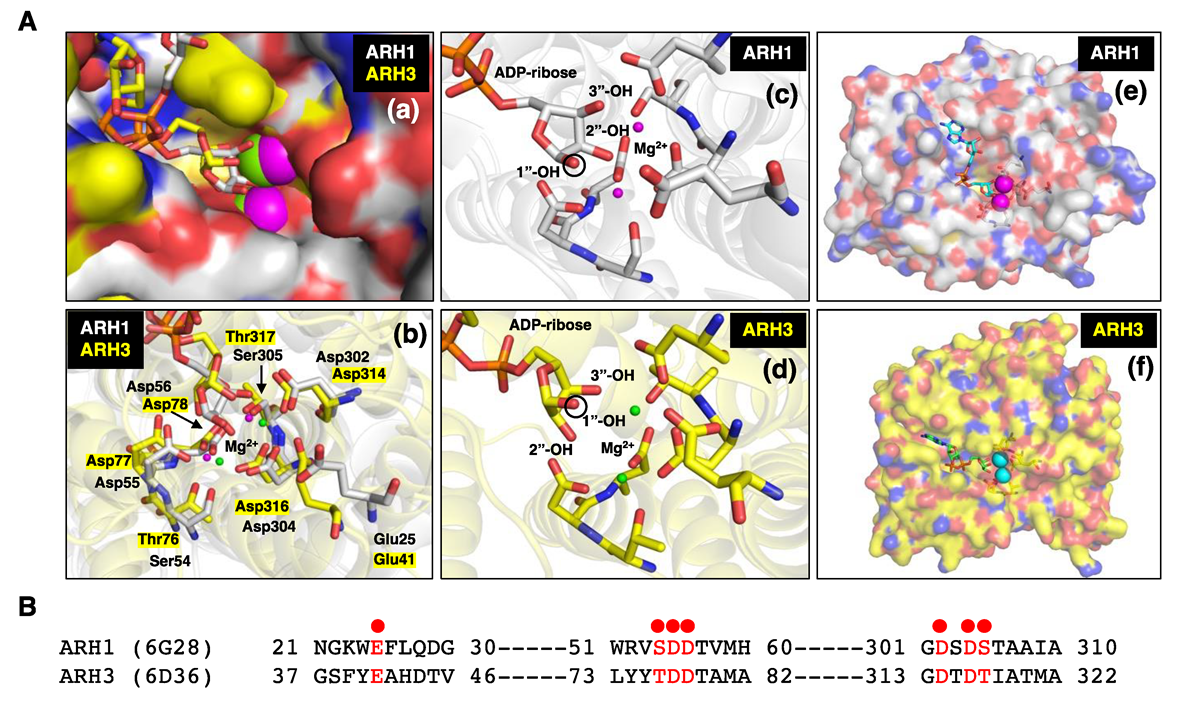
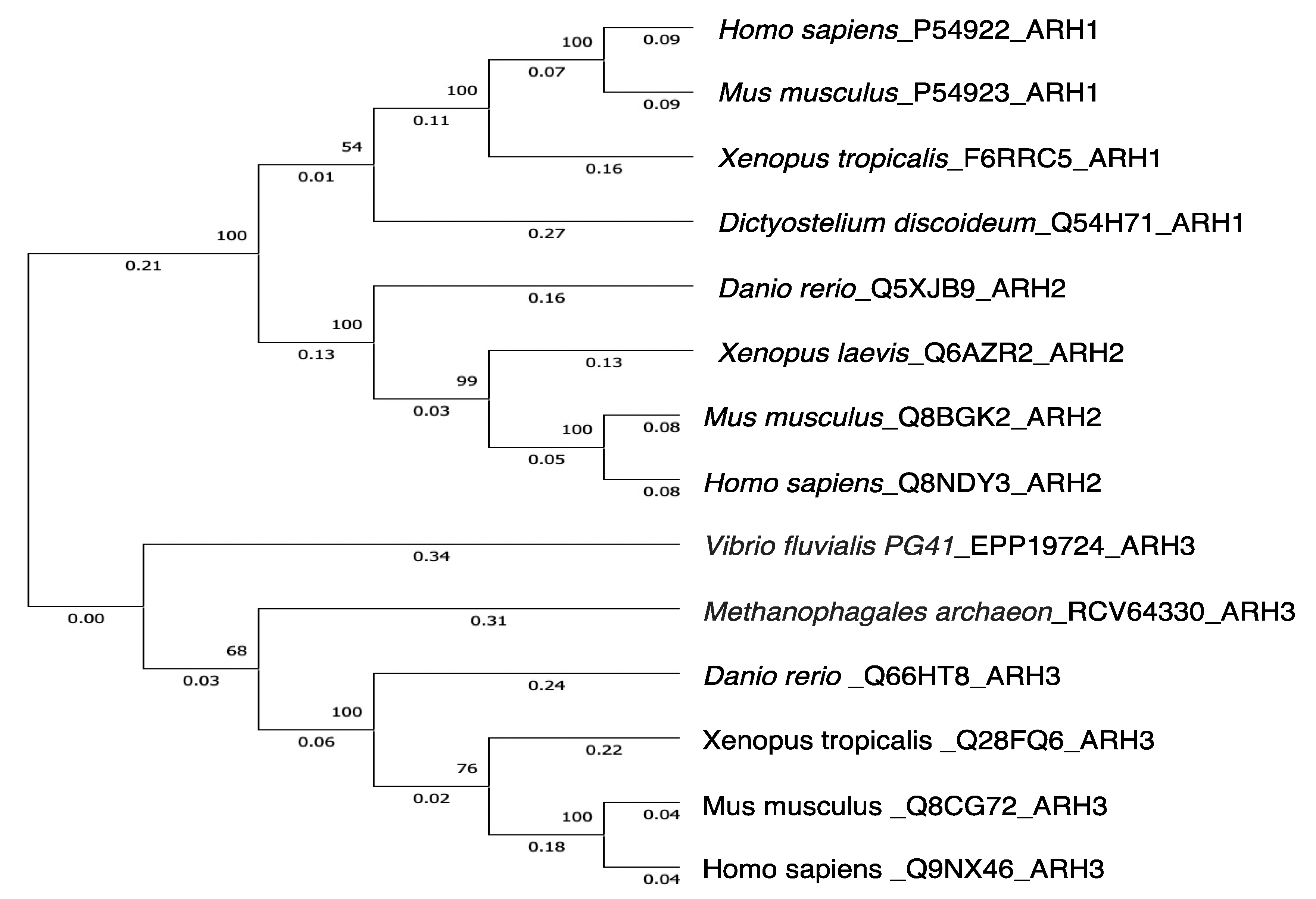
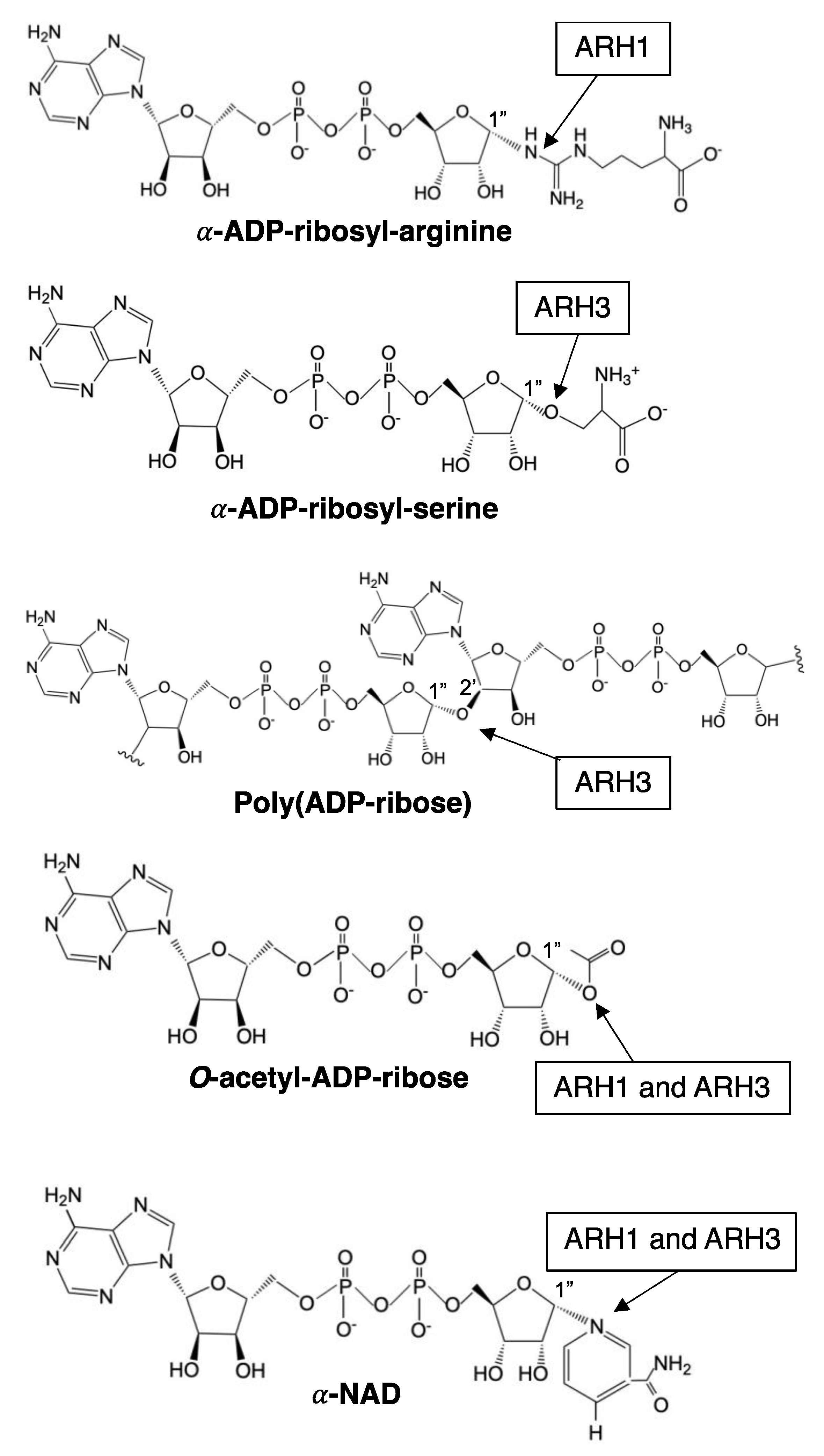
3. ADP-Ribose-Acceptor Hydrolase (ARH) Overview
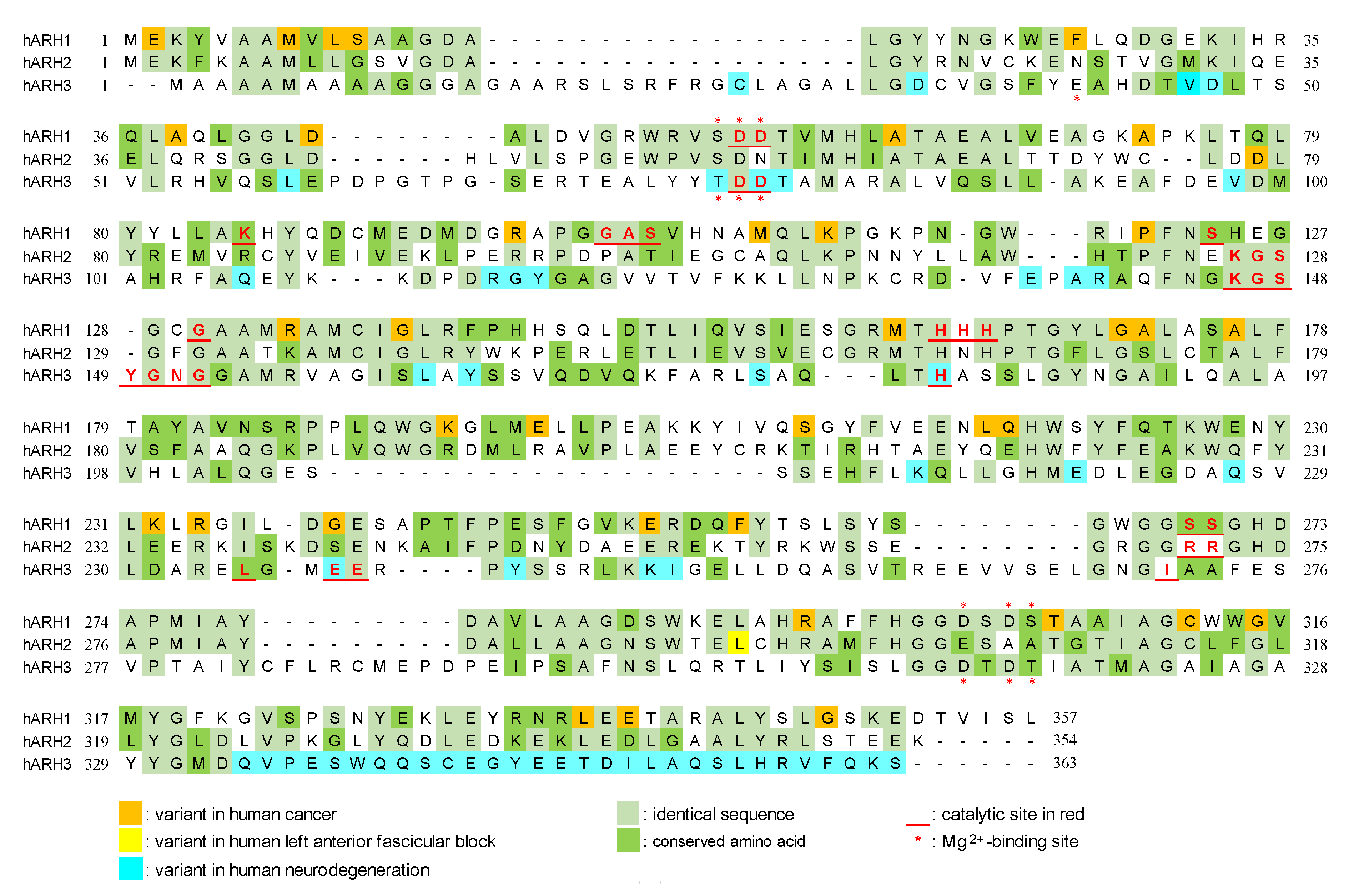
4. ARH1
5. ARH2
6. ARH3
7. Conclusions





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ARH Family | ARH1 | ARH2 | ARH3 | |
|---|---|---|---|---|
| Length (Accession number) | 357 aa (P54922) | 354 aa (Q8NDY3) | 363 aa (Q9NX46) | |
| Identity/similarity of amino acid sequences to ARH1 | 47/68% a | 22/41% a | ||
| Subcellular location | cytoplasm b | cytoplasm c | cytoplasm (65%) mitochondria (25%) nucleus (10%) d | |
| Protein expression | ubiquitous e | muscle f, prostate z, brain g | ubiquitous e | |
| Substrate | α-NAD+ | + h | — | +++ h |
| β-NAD+ | no h | — | no h | |
| OAADPr | + i | — | +++ i | |
| MAR acceptor | ADPr-arginine j | no a | ADPr-serine k | |
| PAR acceptor | no l | no l | yes l | |
| DNA | no m | no m | yes m | |
| RNA | no n | no n | yes n | |
| Physiological function | bacterial infection o tumorigenesis r membrane repair t | cardiac development p | oxidative stress q DNA/RNA repair s | |
| Disease | cholera o lung adenocarcinoma r ovarian cancer u hyperlipoproteinemia x | uveal melanoma v cardiac disease y prostate cancer z | neurodegeneration w | |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Payne, D.M.; Jacobson, E.L.; Moss, J.; Jacobson, M.K. Modification of proteins by mono(ADP-ribosylation) in vivo. Biochemistry 1985, 24, 7540–7549. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.P.; Benjamin, R.C.; Moss, J.; Jacobson, M.K. Identification of enzymatic activities which process protein bound mono(ADP-ribose). Biochem. Biophys. Res. Commun. 1985, 126, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Jacobson, M.K.; Stanley, S.J. Reversibility of arginine-specific mono(ADP-ribosyl)ation: Identification in erythrocytes of an ADP-ribose-L-arginine cleavage enzyme. Proc. Natl. Acad. Sci. USA 1985, 82, 5603–5607. [Google Scholar] [CrossRef] [PubMed]
- Rankin, P.W.; Jacobson, E.L.; Benjamin, R.C.; Moss, J.; Jacobson, M.K. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J. Biol. Chem. 1989, 264, 4312–4317. [Google Scholar] [CrossRef]
- Jacobson, M.K.; Loflin, P.T.; Aboul-Ela, N.; Mingmuang, M.; Moss, J.; Jobson, E.L. Modification of plasma membrane protein cysteine residues by ADP-ribose in vivo. J. Biol. Chem. 1990, 265, 10825–10828. [Google Scholar] [CrossRef]
- Banwell, J.G.; Pierce, N.F.; Mitra, R.C.; Brigham, K.L.; Caranasos, G.J.; Keimowitz, R.I.; Fedson, D.S.; Thomas, J.; Gorbach, S.L.; Sack, R.B.; et al. Intestinal fluid and electrolyte transport in human cholera. J. Clin. Investig. 1970, 49, 183–195. [Google Scholar] [CrossRef]
- Kanungo, S.; Azman, A.S.; Ramamurthy, T.; Deen, J.; Dutta, S. Cholera. Lancet 2022, 399, 1429–1440. [Google Scholar] [CrossRef]
- Vaughan, M.; Pierce, N.F.; Greenough, W.B., 3rd. Stimulation of glycerol production in fat cells by cholera toxin. Nature 1970, 226, 658–659. [Google Scholar] [CrossRef]
- Collier, R.J. Effect of diphtheria toxin on protein synthesis: Inactivation of one of the transfer factors. J. Mol. Biol. 1967, 25, 83–98. [Google Scholar] [CrossRef]
- Honjo, T.; Nishizuka, Y.; Hayaishi, O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J. Biol. Chem. 1968, 243, 3553–3555. [Google Scholar] [CrossRef]
- Gill, D.M.; Pappenheimer, A.M., Jr.; Brown, R.; Kurnick, J.T. Studies on the mode of action of diphtheria toxin. VII. Toxin-stimulated hydrolysis of nicotinamide adenine dinucleotide in mammalian cell extracts. J. Exp. Med. 1969, 129, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Manganiello, V.C.; Vaughan, M. Hydrolysis of nicotinamide adenine dinucleotide by choleragen and its A protomer: Possible role in the activation of adenylate cyclase. Proc. Natl. Acad. Sci. USA 1976, 73, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Kaslow, H.R.; Bourne, H.R. Genetic evidence that cholera toxin substrates are regulatory components of adenylate cyclase. J. Biol. Chem. 1978, 253, 7120–7123. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Meren, R. ADP-ribosylation of membrane proteins catalyzed by cholera toxin: Basis of the activation of adenylate cyclase. Proc. Natl. Acad. Sci. USA 1978, 75, 3050–3054. [Google Scholar] [CrossRef] [PubMed]
- Cassel, D.; Pfeuffer, T. Mechanism of cholera toxin action: Covalent modification of the guanyl nucleotide-binding protein of the adenylate cyclase system. Proc. Natl. Acad. Sci. USA 1978, 75, 2669–2673. [Google Scholar] [CrossRef]
- Moss, J.; Vaughan, M. Mechanism of action of choleragen. Evidence for ADP-ribosyltransferase activity with arginine as an acceptor. J. Biol. Chem. 1977, 252, 2455–2457. [Google Scholar] [CrossRef]
- Moss, J.; Balducci, E.; Cavanaugh, E.; Kim, H.J.; Konczalik, P.; Lesma, E.A.; Okazaki, I.J.; Park, M.; Shoemaker, M.; Stevens, L.A.; et al. Characterization of NAD:arginine ADP-ribosyltransferases. Mol. Cell. Biochem. 1999, 193, 109–113. [Google Scholar] [CrossRef]
- Paone, G.; Stevens, L.A.; Levine, R.L.; Bourgeois, C.; Steagall, W.K.; Gochuico, B.R.; Moss, J. ADP-ribosyltransferase-specific modification of human neutrophil peptide-1. J. Biol. Chem. 2006, 281, 17054–17060. [Google Scholar] [CrossRef]
- Ishiwata-Endo, H.; Kato, J.; Tonouchi, A.; Chung, Y.W.; Sun, J.; Stevens, L.A.; Zhu, J.; Aponte, A.M.; Springer, D.A.; San, H.; et al. Role of a TRIM72 ADP-ribosylation cycle in myocardial injury and membrane repair. JCI Insight 2018, 3, e97898. [Google Scholar] [CrossRef]
- West, R.E., Jr.; Moss, J.; Vaughan, M.; Liu, T.; Liu, T.Y. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J. Biol. Chem. 1985, 260, 14428–14430. [Google Scholar] [CrossRef]
- Itoh, H.; Katada, T.; Ui, M.; Kawasaki, H.; Suzuki, K.; Kaziro, Y. Identification of three pertussis toxin substrates (41, 40 and 39 kDa proteins) in mammalian brain. Comparison of predicted amino acid sequences from G-protein alpha-subunit genes and cDNAs with partial amino acid sequences from purified proteins. FEBS Lett. 1988, 230, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Laurean, D.; Loflin, P.T.; Minter, D.E.; Jacobson, E.L.; Jacobson, M.K. Protein modification by ADP-ribose via acid-labile linkages. J. Biol. Chem. 1995, 270, 7929–7936. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. eLife 2017, 6, e28533. [Google Scholar] [CrossRef] [PubMed]
- Abplanalp, J.; Leutert, M.; Frugier, E.; Nowak, K.; Feurer, R.; Kato, J.; Kistemaker, H.V.A.; Filippov, D.V.; Moss, J.; Caflisch, A.; et al. Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 2017, 8, 2055. [Google Scholar] [CrossRef]
- Hopp, A.K.; Hottiger, M.O. Uncovering the Invisible: Mono-ADP-ribosylation Moved into the Spotlight. Cells 2021, 10, 680. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Palazzo, L.; Ahel, I. (ADP-ribosyl)hydrolases: Structure, function, and biology. Genes Dev. 2020, 34, 263–284. [Google Scholar] [CrossRef]
- Feijs, K.L.H.; Cooper, C.D.O.; Zaja, R. The Controversial Roles of ADP-Ribosyl Hydrolases MACROD1, MACROD2 and TARG1 in Carcinogenesis. Cancers 2020, 12, 604. [Google Scholar] [CrossRef]
- Rack, J.G.; Perina, D.; Ahel, I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu. Rev. Biochem. 2016, 85, 431–454. [Google Scholar] [CrossRef]
- Plummer, R.; Jones, C.; Middleton, M.; Wilson, R.; Evans, J.; Olsen, A.; Curtin, N.; Boddy, A.; McHugh, P.; Newell, D.; et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 7917–7923. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Longarini, E.J.; Matic, I. The fast-growing business of Serine ADP-ribosylation. DNA Repair 2022, 118, 103382. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.G.; Becker, K.; Huang, H.; Dixon-Salazar, T.; Chai, G.; Salpietro, V.; Al-Gazali, L.; Waisfisz, Q.; Wang, H.; Vaux, K.K.; et al. Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. Am. J. Hum. Genet. 2018, 103, 431–439. [Google Scholar] [CrossRef]
- Mashimo, M.; Bu, X.; Aoyama, K.; Kato, J.; Ishiwata-Endo, H.; Stevens, L.A.; Kasamatsu, A.; Wolfe, L.A.; Toro, C.; Adams, D.; et al. PARP1 inhibition alleviates injury in ARH3-deficient mice and human cells. JCI Insight 2019, 4, e124519. [Google Scholar] [CrossRef] [PubMed]
- Danhauser, K.; Alhaddad, B.; Makowski, C.; Piekutowska-Abramczuk, D.; Syrbe, S.; Gomez-Ospina, N.; Manning, M.A.; Kostera-Pruszczyk, A.; Krahn-Peper, C.; Berutti, R.; et al. Bi-allelic ADPRHL2 Mutations Cause Neurodegeneration with Developmental Delay, Ataxia, and Axonal Neuropathy. Am. J. Hum. Genet. 2018, 103, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Aryan, H.; Razmara, E.; Farhud, D.; Zarif-Yeganeh, M.; Zokaei, S.; Hassani, S.A.; Ashrafi, M.R.; Garshasbi, M.; Tavasoli, A.R. Novel imaging and clinical phenotypes of CONDSIAS disorder caused by a homozygous frameshift variant of ADPRHL2: A case report. BMC Neurol. 2020, 20, 291. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Fatima, S.; Agarwal, A.; Radhakrishnan, D.M.; Garg, A.; Srivastava, A.K. Dystonia and Myelopathy in a Case of Stress-Induced Childhood-Onset Neurodegeneration with Ataxia and Seizures (CONDSIAS). Mov. Disord. Clin. Pract. 2021, 8, 156–158. [Google Scholar] [CrossRef]
- Beijer, D.; Agnew, T.; Rack, J.G.M.; Prokhorova, E.; Deconinck, T.; Ceulemans, B.; Peric, S.; Milic Rasic, V.; De Jonghe, P.; Ahel, I.; et al. Biallelic ADPRHL2 mutations in complex neuropathy affect ADP ribosylation and DNA damage response. Life Sci. Alliance 2021, 4, e202101057. [Google Scholar] [CrossRef]
- Lu, A.; Dong, C.; Chen, B.; Xie, L.; Hu, H. Case Report: Stress-Induced Childhood-Onset Neurodegeneration With Ataxia-Seizures Syndrome Caused by a Novel Compound Heterozygous Mutation in ADPRHL2. Front. Neurol. 2022, 13, 807291. [Google Scholar] [CrossRef]
- Juarez-Salinas, H.; Sims, J.L.; Jacobson, M.K. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature 1979, 282, 740–741. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Kim, H.; Kim, M.; Williams, J.D.; Coyle, D.L.; Coyle, W.R.; Grove, G.; Rizer, R.L.; Stratton, M.S.; Jacobson, M.K. A topical lipophilic niacin derivative increases NAD, epidermal differentiation and barrier function in photodamaged skin. Exp. Dermatol. 2007, 16, 490–499. [Google Scholar] [CrossRef]
- Jacobson, M.K.; Kim, H.; Coyle, W.R.; Kim, M.; Coyle, D.L.; Rizer, R.L.; Jacobson, E.L. Effect of myristyl nicotinate on retinoic acid therapy for facial photodamage. Exp. Dermatol. 2007, 16, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, Y.; Benavente, C.A.; Meyer, R.G.; Coyle, W.R.; Jacobson, M.K.; Jacobson, E.L. Nicotinic acid receptor abnormalities in human skin cancer: Implications for a role in epidermal differentiation. PLoS ONE 2011, 6, e20487. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.H. Recent advances in identifying the functions of gangliosides. Chem. Phys. Lipids 1986, 42, 137–151. [Google Scholar] [CrossRef]
- Fishman, P.H. Mechanism of Action of Cholera Toxin. In ADP-Ribosylating Toxins and G Proteins: Insights into Signal Transduction; Moss, J., Vaughan, M., Eds.; American Society for Microbiology: Washington, DC, USA, 1990; pp. 127–140. [Google Scholar]
- Schleifer, L.S.; Kahn, R.A.; Hanski, E.; Northup, J.K.; Sternweis, P.C.; Gilman, A.G. Requirements for cholera toxin-dependent ADP-ribosylation of the purified regulatory component of adenylate cyclase. J. Biol. Chem. 1982, 257, 20–23. [Google Scholar] [CrossRef]
- Kahn, R.A.; Gilman, A.G. Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J. Biol. Chem. 1984, 259, 6228–6234. [Google Scholar] [CrossRef]
- Tsai, S.C.; Noda, M.; Adamik, R.; Moss, J.; Vaughan, M. Enhancement of choleragen ADP-ribosyltransferase activities by guanyl nucleotides and a 19-kDa membrane protein. Proc. Natl. Acad. Sci. USA 1987, 84, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K. ADP-Ribosylating Toxins and G Proteins: Insights into Signal Transduction: Mono-ADP-Ribosyltransferases and ADP-Ribosylarginine Hydrolases: A Mono-ADP-Ribosylation Cycle in Animal Cells; Moss, J., Vaughan, M., Eds.; American Society for Microbiology: Washington, DC, USA, 1990; pp. 493–510. [Google Scholar]
- Glowacki, G.; Braren, R.; Firner, K.; Nissen, M.; Kuhl, M.; Reche, P.; Bazan, F.; Cetkovic-Cvrlje, M.; Leiter, E.; Haag, F.; et al. The family of toxin-related ecto-ADP-ribosyltransferases in humans and the mouse. Protein Sci. 2002, 11, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Martello, R.; Leutert, M.; Jungmichel, S.; Bilan, V.; Larsen, S.C.; Young, C.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016, 7, 12917. [Google Scholar] [CrossRef]
- Zolkiewska, A.; Nightingale, M.S.; Moss, J. Molecular characterization of NAD:arginine ADP-ribosyltransferase from rabbit skeletal muscle. Proc. Natl. Acad. Sci. USA 1992, 89, 11352–11356. [Google Scholar] [CrossRef]
- Moss, J.; Stevens, L.A.; Cavanaugh, E.; Okazaki, I.J.; Bortell, R.; Kanaitsuka, T.; Mordes, J.P.; Greiner, D.L.; Rossini, A.A. Characterization of mouse Rt6.1 NAD:arginine ADP-ribosyltransferase. J. Biol. Chem. 1997, 272, 4342–4346. [Google Scholar] [CrossRef]
- Maehama, T.; Hoshino, S.; Katada, T. Increase in ADP-ribosyltransferase activity of rat T lymphocyte alloantigen RT6.1 by a single amino acid mutation. FEBS Lett. 1996, 388, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Haag, F.; Koch-Nolte, F.; Kuhl, M.; Lorenzen, S.; Thiele, H.G. Premature stop codons inactivate the RT6 genes of the human and chimpanzee species. J. Mol. Biol. 1994, 243, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Karsten, S.; Schroder, J.; da Silva, C.; Kahlke, D.; Thiele, H.G.; Koch-Noite, F.; Haag, F. Expression and comparative analysis of recombinant rat and mouse RT6 T cell mono(ADP-ribosyl)transferases in E. coli. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 1997; Volume 419, pp. 175–180. [Google Scholar] [CrossRef]
- Okazaki, I.J.; Kim, H.J.; Moss, J. Cloning and characterization of a novel membrane-associated lymphocyte NAD:arginine ADP-ribosyltransferase. J. Biol. Chem. 1996, 271, 22052–22057. [Google Scholar] [CrossRef] [PubMed]
- Weng, B.; Thompson, W.C.; Kim, H.J.; Levine, R.L.; Moss, J. Modification of the ADP-ribosyltransferase and NAD glycohydrolase activities of a mammalian transferase (ADP-ribosyltransferase 5) by auto-ADP-ribosylation. J. Biol. Chem. 1999, 274, 31797–31803. [Google Scholar] [CrossRef] [PubMed]
- Leutert, M.; Menzel, S.; Braren, R.; Rissiek, B.; Hopp, A.K.; Nowak, K.; Bisceglie, L.; Gehrig, P.; Li, H.; Zolkiewska, A.; et al. Proteomic Characterization of the Heart and Skeletal Muscle Reveals Widespread Arginine ADP-Ribosylation by the ARTC1 Ectoenzyme. Cell Rep. 2018, 24, 1916–1929.e1915. [Google Scholar] [CrossRef]
- Oka, S.; Kato, J.; Moss, J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2006, 281, 705–713. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Ding, M.; Yu, Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 2013, 10, 981–984. [Google Scholar] [CrossRef]
- Ogata, N.; Ueda, K.; Hayaishi, O. ADP-ribosylation of histone H2B. Identification of glutamic acid residue 2 as the modification site. J. Biol. Chem. 1980, 255, 7610–7615. [Google Scholar] [CrossRef]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef]
- Tao, Z.; Gao, P.; Liu, H.W. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: Analysis and implications. J. Am. Chem. Soc. 2009, 131, 14258–14260. [Google Scholar] [CrossRef]
- Du, J.; Jiang, H.; Lin, H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 2009, 48, 2878–2890. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Jacobson, M.K.; Bernofsky, C. Evidence against the natural occurrence of alpha-nicotinamide adenine dinucleotide in Azotobacter vinelandii. J. Biol. Chem. 1973, 248, 7891–7897. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J.; Kaplan, N.O. The alpha beta epimerization of reduced nicotinamide adenine dinucleotide. Arch. Biochem. Biophys. 1975, 166, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Kasamatsu, A.; Oka, S.; Moss, J. The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O-acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proc. Natl. Acad. Sci. USA 2006, 103, 16687–16691. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Stanley, S.J.; Oppenheimer, N.J. Substrate specificity and partial purification of a stereospecific NAD- and guanidine-dependent ADP-ribosyltransferase from avian erythrocytes. J. Biol. Chem. 1979, 254, 8891–8894. [Google Scholar] [CrossRef]
- Moss, J.; Stanley, S.J.; Nightingale, M.S.; Murtagh, J.J., Jr.; Monaco, L.; Mishima, K.; Chen, H.C.; Williamson, K.C.; Tsai, S.C. Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J. Biol. Chem. 1992, 267, 10481–10488. [Google Scholar] [CrossRef]
- Moss, J.; Oppenheimer, N.J.; West, R.E., Jr.; Stanley, S.J. Amino acid specific ADP-ribosylation: Substrate specificity of an ADP-ribosylarginine hydrolase from turkey erythrocytes. Biochemistry 1986, 25, 5408–5414. [Google Scholar] [CrossRef]
- Takada, T.; Okazaki, I.J.; Moss, J. ADP-ribosylarginine hydrolases. Mol. Cell. Biochem. 1994, 138, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J. Structural determination and stereospecificity of the choleragen-catalyzed reaction of NAD+ with guanidines. J. Biol. Chem. 1978, 253, 4907–4910. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Kato, J.; Kasamatsu, A.; Oda, H.; Lee, D.Y.; Moss, J. The ARH and Macrodomain Families of alpha-ADP-ribose-acceptor Hydrolases Catalyze alpha-NAD(+) Hydrolysis. ACS Chem. Biol. 2019, 14, 2576–2584. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Vekhter, D.; Heath, J.; Zhu, J.; Barbieri, J.T.; Moss, J. Mutations of the functional ARH1 allele in tumors from ARH1 heterozygous mice and cells affect ARH1 catalytic activity, cell proliferation and tumorigenesis. Oncogenesis 2015, 4, e151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, J.; Zhu, J.; Liu, C.; Stylianou, M.; Hoffmann, V.; Lizak, M.J.; Glasgow, C.G.; Moss, J. ADP-ribosylarginine hydrolase regulates cell proliferation and tumorigenesis. Cancer Res. 2011, 71, 5327–5335. [Google Scholar] [CrossRef]
- Kato, J.; Zhu, J.; Liu, C.; Moss, J. Enhanced sensitivity to cholera toxin in ADP-ribosylarginine hydrolase-deficient mice. Mol. Cell. Biol. 2007, 27, 5534–5543. [Google Scholar] [CrossRef]
- Watanabe, K.; Kato, J.; Zhu, J.; Oda, H.; Ishiwata-Endo, H.; Moss, J. Enhanced sensitivity to cholera toxin in female ADP-ribosylarginine hydrolase (ARH1)-deficient mice. PLoS ONE 2018, 13, e0207693. [Google Scholar] [CrossRef]
- Stevens, L.A.; Levine, R.L.; Gochuico, B.R.; Moss, J. ADP-ribosylation of human defensin HNP-1 results in the replacement of the modified arginine with the noncoded amino acid ornithine. Proc. Natl. Acad. Sci. USA 2009, 106, 19796–19800. [Google Scholar] [CrossRef]
- Kasamatsu, A.; Nakao, M.; Smith, B.C.; Comstock, L.R.; Ono, T.; Kato, J.; Denu, J.M.; Moss, J. Hydrolysis of O-acetyl-ADP-ribose isomers by ADP-ribosylhydrolase 3. J. Biol. Chem. 2011, 286, 21110–21117. [Google Scholar] [CrossRef]
- Smith, S.J.; Towers, N.; Saldanha, J.W.; Shang, C.A.; Mahmood, S.R.; Taylor, W.R.; Mohun, T.J. The cardiac-restricted protein ADP-ribosylhydrolase-like 1 is essential for heart chamber outgrowth and acts on muscle actin filament assembly. Dev. Biol. 2016, 416, 373–388. [Google Scholar] [CrossRef]
- Norland, K.; Sveinbjornsson, G.; Thorolfsdottir, R.B.; Davidsson, O.B.; Tragante, V.; Rajamani, S.; Helgadottir, A.; Gretarsdottir, S.; van Setten, J.; Asselbergs, F.W.; et al. Sequence variants with large effects on cardiac electrophysiology and disease. Nat. Commun. 2019, 10, 4803. [Google Scholar] [CrossRef] [PubMed]
- Pourhaghighi, R.; Ash, P.E.A.; Phanse, S.; Goebels, F.; Hu, L.Z.M.; Chen, S.; Zhang, Y.; Wierbowski, S.D.; Boudeau, S.; Moutaoufik, M.T.; et al. BraInMap Elucidates the Macromolecular Connectivity Landscape of Mammalian Brain. Cell Syst. 2020, 10, 333–350.e314. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Tang, J.; Han, Y.; Wang, D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp. Eye Res. 2018, 166, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Z.; Bavarva, J.; Kuhns, K.J.; Guo, J.; Ledet, E.M.; Qian, C.; Lin, Y.; Fang, Z.; Zabaleta, J.; et al. A Recurrent ADPRHL1 Germline Mutation Activates PARP1 and Confers Prostate Cancer Risk in African American Families. Mol. Cancer Res. 2022, 21, 874. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Crawford, K.; Bonfiglio, J.J.; Mikoc, A.; Matic, I.; Ahel, I. Specificity of reversible ADP-ribosylation and regulation of cellular processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef]
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e936. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Buch-Larsen, S.C.; Prokhorova, E.; Elsborg, J.D.; Rebak, A.; Zhu, K.; Ahel, D.; Lukas, C.; Ahel, I.; Nielsen, M.L. The regulatory landscape of the human HPF1- and ARH3-dependent ADP-ribosylome. Nat. Commun. 2021, 12, 5893. [Google Scholar] [CrossRef]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef]
- Mashimo, M.; Kato, J.; Moss, J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 18964–18969. [Google Scholar] [CrossRef]
- Cohen, M.S.; Chang, P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, K.; Nemoto, Y.; Ueda, K.; Hayaishi, O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J. Biol. Chem. 1986, 261, 14902–14911. [Google Scholar] [CrossRef]
- Niere, M.; Mashimo, M.; Agledal, L.; Dolle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J. Biol. Chem. 2012, 287, 16088–16102. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, M.; Morozumi, A.; Nobeyama, A.; Kanzaki, M.; Negi, S.; Kato, J.; Moss, J.; Nomura, A.; Fujii, T. Poly(ADP-ribose) Polymerase 1 Mediates Rab5 Inactivation after DNA Damage. Int. J. Mol. Sci. 2022, 23, 7827. [Google Scholar] [CrossRef] [PubMed]
- Rack, J.G.M.; Ariza, A.; Drown, B.S.; Henfrey, C.; Bartlett, E.; Shirai, T.; Hergenrother, P.J.; Ahel, I. (ADP-ribosyl)hydrolases: Structural Basis for Differential Substrate Recognition and Inhibition. Cell Chem. Biol. 2018, 25, 1533–1546.e1512. [Google Scholar] [CrossRef] [PubMed]
- Pourfarjam, Y.; Ventura, J.; Kurinov, I.; Cho, A.; Moss, J.; Kim, I.K. Structure of human ADP-ribosyl-acceptor hydrolase 3 bound to ADP-ribose reveals a conformational switch that enables specific substrate recognition. J. Biol. Chem. 2018, 293, 12350–12359. [Google Scholar] [CrossRef] [PubMed]
- Pourfarjam, Y.; Ma, Z.; Kurinov, I.; Moss, J.; Kim, I.K. Structural and biochemical analysis of human ADP-ribosyl-acceptor hydrolase 3 reveals the basis of metal selectivity and different roles for the two magnesium ions. J. Biol. Chem. 2021, 296, 100692. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Kernstock, S.; Koch-Nolte, F.; Mueller-Dieckmann, J.; Weiss, M.S.; Mueller-Dieckmann, C. Cloning, expression, purification and crystallization as well as X-ray fluorescence and preliminary X-ray diffraction analyses of human ADP-ribosylhydrolase 1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 529–532. [Google Scholar] [CrossRef]
- Mueller-Dieckmann, C.; Kernstock, S.; Lisurek, M.; von Kries, J.P.; Haag, F.; Weiss, M.S.; Koch-Nolte, F. The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc. Natl. Acad. Sci. USA 2006, 103, 15026–15031. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, Z.; Xie, R.; Ma, Y.; Liu, X.; Yu, X. Structure-function analyses reveal the mechanism of the ARH3-dependent hydrolysis of ADP-ribosylation. J. Biol. Chem. 2018, 293, 14470–14480. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Liu, Q.; Zorzini, V.; Voorneveld, J.; Ariza, A.; Honarmand Ebrahimi, K.; Reber, J.M.; Krassnig, S.C.; Ahel, D.; van der Marel, G.A.; et al. Mechanistic insights into the three steps of poly(ADP-ribosylation) reversal. Nat. Commun. 2021, 12, 4581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Koch-Nolte, F.; Kernstock, S.; Mueller-Dieckmann, C.; Weiss, M.S.; Haag, F. Mammalian ADP-ribosyltransferases and ADP-ribosylhydrolases. Front. Biosci. 2008, 13, 6716–6729. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Munnur, D.; Ahel, I. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 2017, 284, 4002–4016. [Google Scholar] [CrossRef]
- Weixler, L.; Feijs, K.L.H.; Zaja, R. ADP-ribosylation of RNA in mammalian cells is mediated by TRPT1 and multiple PARPs. Nucleic Acids Res. 2022, 50, 9426–9441. [Google Scholar] [CrossRef]
- Smith, S.J.; Towers, N.; Demetriou, K.; Mohun, T.J. Defective heart chamber growth and myofibrillogenesis after knockout of adprhl1 gene function by targeted disruption of the ancestral catalytic active site. PLoS ONE 2020, 15, e0235433. [Google Scholar] [CrossRef]
- Prokhorova, E.; Agnew, T.; Wondisford, A.R.; Tellier, M.; Kaminski, N.; Beijer, D.; Holder, J.; Groslambert, J.; Suskiewicz, M.J.; Zhu, K.; et al. Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Mol. Cell 2021, 81, 2640–2655.e8. [Google Scholar] [CrossRef]
- Bu, X.; Kato, J.; Hong, J.A.; Merino, M.J.; Schrump, D.S.; Lund, F.E.; Moss, J. CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 2018, 39, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Prokhorova, E.; Krejcikova, K.; Cihlarova, Z.; Kalasova, I.; Kubovciak, J.; Sachova, J.; Hailstone, R.; Brazina, J.; Ghosh, S.; et al. Pathogenic ARH3 mutations result in ADP-ribose chromatin scars during DNA strand break repair. Nat. Commun. 2020, 11, 3391. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, J.; Pang, B.; Li, C.; Zhao, J.; Shen, K. EZH2-induced H3K27me3 is associated with epigenetic repression of the ARHI tumor-suppressor gene in ovarian cancer. Cell Biochem. Biophys. 2015, 71, 105–112. [Google Scholar] [CrossRef]
- Eden, E.R.; Patel, D.D.; Sun, X.M.; Burden, J.J.; Themis, M.; Edwards, M.; Lee, P.; Neuwirth, C.; Naoumova, R.P.; Soutar, A.K. Restoration of LDL receptor function in cells from patients with autosomal recessive hypercholesterolemia by retroviral expression of ARH1. J. Clin. Investig. 2002, 110, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishiwata-Endo, H.; Kato, J.; Yamashita, S.; Chea, C.; Koike, K.; Lee, D.-Y.; Moss, J. ARH Family of ADP-Ribose-Acceptor Hydrolases. Cells 2022, 11, 3853. https://doi.org/10.3390/cells11233853
Ishiwata-Endo H, Kato J, Yamashita S, Chea C, Koike K, Lee D-Y, Moss J. ARH Family of ADP-Ribose-Acceptor Hydrolases. Cells. 2022; 11(23):3853. https://doi.org/10.3390/cells11233853
Chicago/Turabian StyleIshiwata-Endo, Hiroko, Jiro Kato, Sachiko Yamashita, Chanbora Chea, Kazushige Koike, Duck-Yeon Lee, and Joel Moss. 2022. "ARH Family of ADP-Ribose-Acceptor Hydrolases" Cells 11, no. 23: 3853. https://doi.org/10.3390/cells11233853
APA StyleIshiwata-Endo, H., Kato, J., Yamashita, S., Chea, C., Koike, K., Lee, D.-Y., & Moss, J. (2022). ARH Family of ADP-Ribose-Acceptor Hydrolases. Cells, 11(23), 3853. https://doi.org/10.3390/cells11233853