


Populations of Tau Conformers Drive Prion-like Strain Effects in Alzheimer’s Disease and Related Dementias


Abstract
1. Phenotypic Spectra of Alzheimer’s Disease (AD) and Frontotemporal Lobar Degeneration (FTLD)
2. Implications of Strain Concept of Human Prion Diversity for Tauopathies
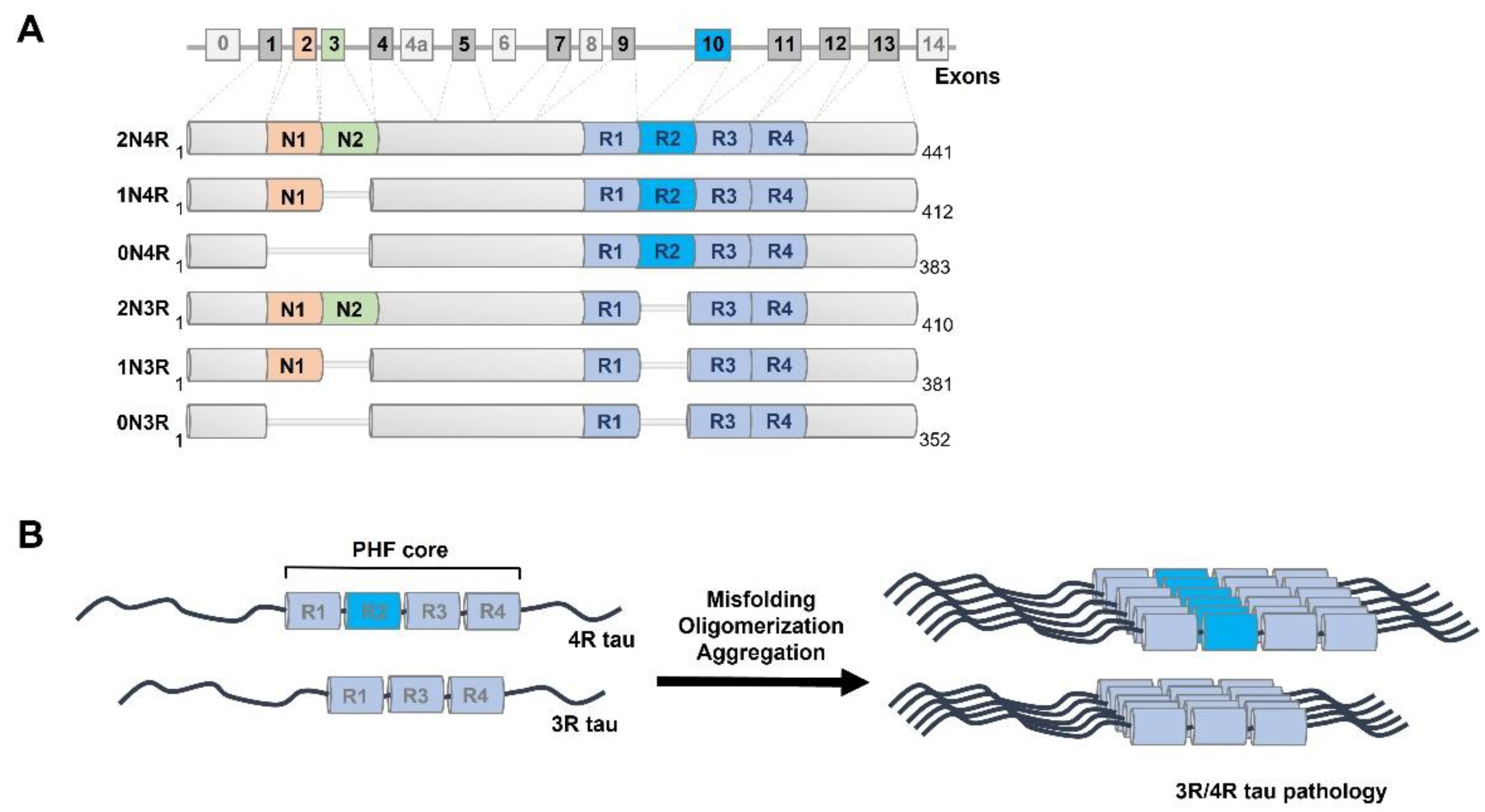
3. Isoforms and Cellular Functions of Normal Human Tau Protein
4. Preferential Misfolding of 4R Tau Isoform in Late Onset AD and FTLD-MAPT-P301L Patients
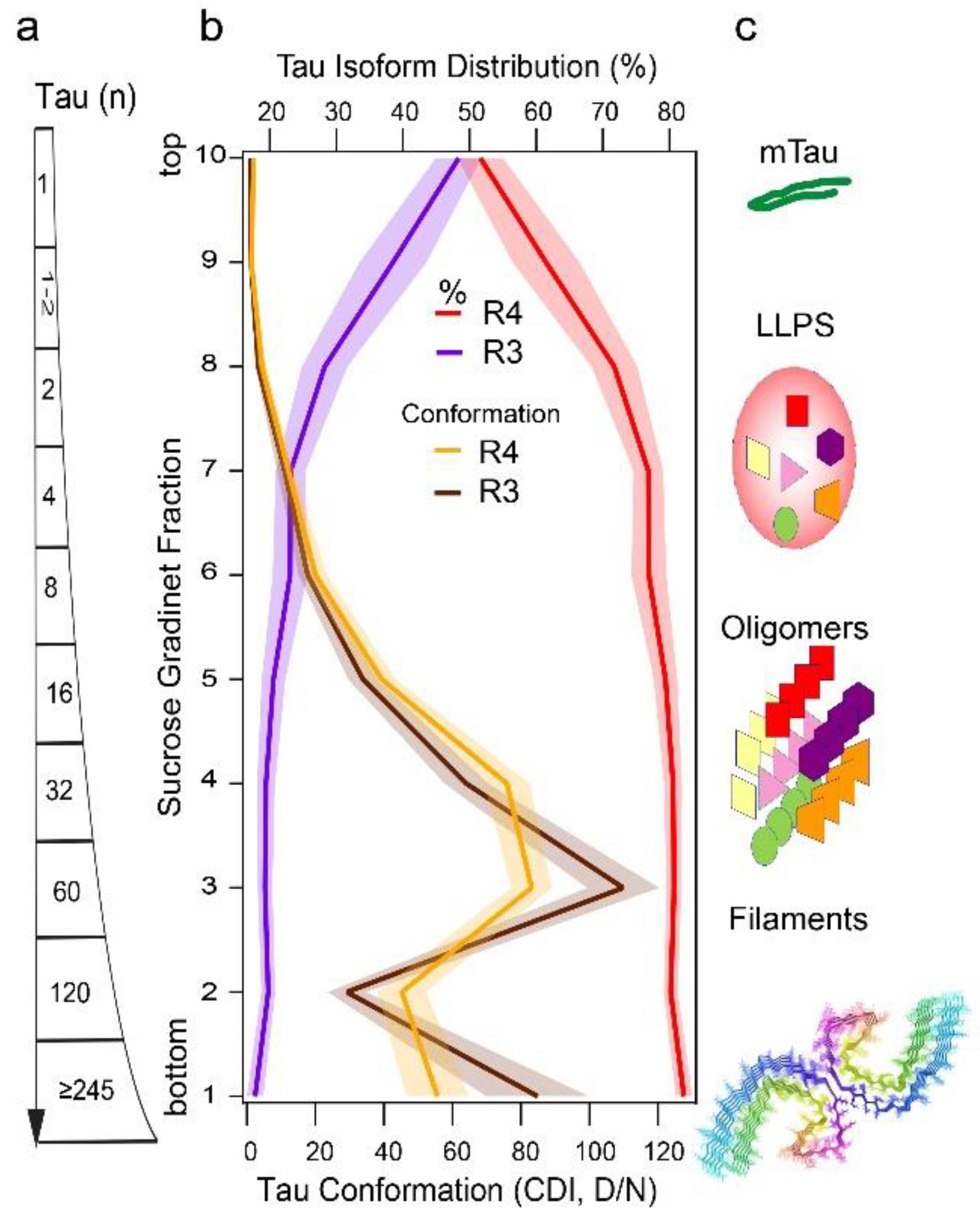
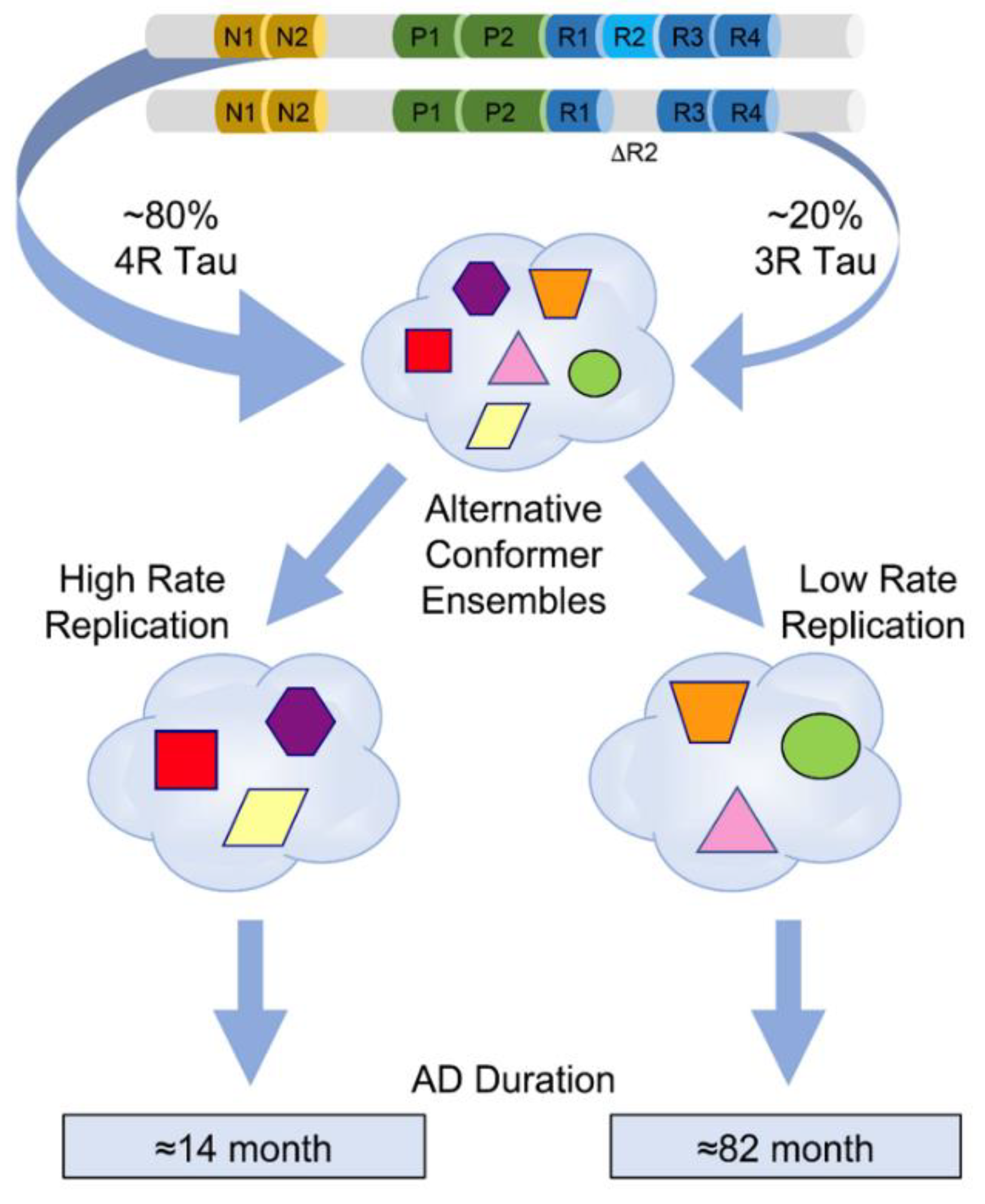
5. Conformational Diversity of Tau Isoforms in Different Phenotypes of AD, FTLD-MAPT-P301L, and TgP301L Model
6. Modelling Replication Mechanism of Tau Conformer Populations In Vitro
7. Strain Effects of Prion-like Tau Conformers in Cell Reporters
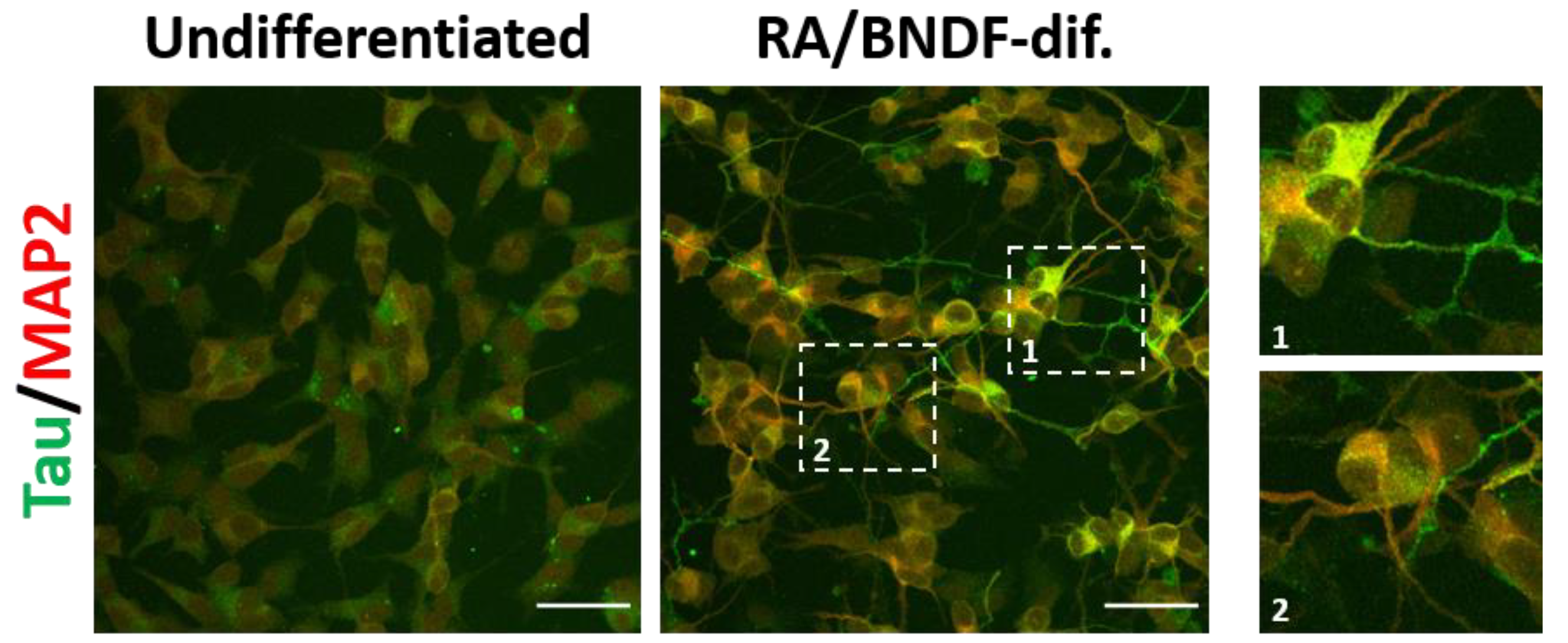
8. Effects of Misfolded Tau in Neuronal Cultures
9. Concluding Remarks and Future Investigations
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CDI | Conformation-Dependent Immunoassay |
| CJD | Creutzfeldt–Jakob disease |
| CSA | Conformational Stability Assay |
| FFI | fatal familial insomnia |
| FTLD | frontotemporal lobar degeneration |
| GSS | Gerstmann–Sträussler–Scheinker syndrome |
| MAPT | microtubule-associated protein tau gene |
| PrP | prion protein |
| PrPC | normal or cellular prion protein |
| PrPSc | pathogenic prion protein |
| PRNP | prion protein gene |
| CJD | sporadic Creutzfeldt–Jakob disease |
| RT QuIC | Real-Time Quaking-Induced Conversion |
| SFI | sporadic fatal insomnia |
| VPSPr | variable protease-sensitive prionopathy |
References
- Nelson, P.T.; Trojanowski, J.Q.; Abner, E.L.; Al-Janabi, O.M.; Jicha, G.A.; Schmitt, F.A.; Smith, C.D.; Fardo, D.W.; Wang, W.X.; Kryscio, R.J.; et al. “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). J. Neuropathol. Exp. Neurol. 2016, 75, 482–498. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef]
- James, B.D.; Wilson, R.S.; Boyle, P.A.; Trojanowski, J.Q.; Bennett, D.A.; Schneider, J.A. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016, 139, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [PubMed]
- Risacher, S.L.; Anderson, W.H.; Charil, A.; Castelluccio, P.F.; Shcherbinin, S.; Saykin, A.J.; Schwarz, A.J.; Weiner, M.W. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 2017, 89, 2176–2186. [Google Scholar] [CrossRef]
- Sepulveda-Falla, D.; Chavez-Gutierrez, L.; Portelius, E.; Vélez, J.I.; Dujardin, S.; Barrera-Ocampo, A.; Dinkel, F.; Hagel, C.; Puig, B.; Mastronardi, C.; et al. A multifactorial model of pathology for age of onset heterogeneity in familial Alzheimer’s disease. Acta Neuropathol. 2021, 141, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef]
- Kim, C.; Haldiman, T.; Kang, S.G.; Hromadkova, L.; Han, Z.Z.; Chen, W.; Lissemore, F.; Lerner, A.; de Silva, R.; Cohen, M.L.; et al. Distinct populations of highly potent TAU seed conformers in rapidly progressing Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabg0253. [Google Scholar] [CrossRef]
- Liu, H.; Kim, C.; Haldiman, T.; Sigurdson, C.J.; Nyström, S.; Nilsson, K.P.R.; Cohen, M.L.; Wisniewski, T.; Hammarström, P.; Safar, J.G. Distinct conformers of amyloid beta accumulate in the neocortex of patients with rapidly progressive Alzheimer’s disease. J. Biol. Chem. 2021, 297, 101267. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Appleby, B.; Safar, J.G. Distinct Prion-Like Strains of Amyloid Beta Implicated in Phenotypic Diversity of Alzheimer Disease. Prion 2016, 10, 9–17. [Google Scholar] [CrossRef]
- Cohen, M.L.; Kim, C.; Haldiman, T.; ElHag, M.; Mehndiratta, P.; Pichet, T.; Lissemore, F.; Shea, M.; Cohen, Y.; Chen, W.; et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-beta. Brain 2015, 138, 1009–1022. [Google Scholar] [CrossRef]
- Schmidt, C.; Wolff, M.; Weitz, M.; Bartlau, T.; Korth, C.; Zerr, I. Rapidly progressive Alzheimer disease. Arch Neurol. 2011, 68, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Astroglia and Tau: New Perspectives. Front. Aging Neurosci. 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Rösler, T.W.; Tayaranian Marvian, A.; Brendel, M.; Nykänen, N.P.; Höllerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Prog. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Fox, E.J.; Bukhari, S.A.; Montine, K.S.; Bendall, S.C.; Montine, T.J. The basis of cellular and regional vulnerability in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004457. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Hasegawa, M.; Ihara, Y. Neuronal and glial tau-positive inclusions in diverse neurologic diseases share common phosphorylation characteristics. Acta Neuropathol. 1994, 88, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef]
- Higuchi, M.; Ishihara, T.; Zhang, B.; Hong, M.; Andreadis, A.; Trojanowski, J.Q.; Lee, V.M.-Y. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron 2002, 35, 433–446. [Google Scholar] [CrossRef]
- Lin, W.-L.; Lewis, J.; Yen, S.-H.; Hutton, M.; Dickson, D.W. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am. J. Pathol. 2003, 162, 213–218. [Google Scholar] [CrossRef]
- Ferrer, I.; Zelaya, M.V.; Aguiló García, M.; Carmona, M.; López-González, I.; Andrés-Benito, P.; Lidón, L.; Gavín, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [Google Scholar] [CrossRef]
- Forrest, S.L.; Kril, J.J.; Kovacs, G.G. Association Between Globular Glial Tauopathies and Frontotemporal Dementia-Expanding the Spectrum of Gliocentric Disorders: A Review. JAMA Neurol. 2021, 78, 1004–1014. [Google Scholar] [CrossRef]
- Hartnell, I.J.; Blum, D.; Nicoll, J.A.; Dorothee, G.; Boche, D. Glial cells and adaptive immunity in frontotemporal dementia with tau pathology. Brain 2021, 144, 724–745. [Google Scholar] [CrossRef]
- Odfalk, K.F.; Bieniek, K.F.; Hopp, S.C. Microglia: Friend and foe in tauopathy. Prog. Neurobiol. 2022, 216, 102306. [Google Scholar] [CrossRef]
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534. [Google Scholar] [CrossRef]
- Wszolek, Z.K.; Tsuboi, Y.; Ghetti, B.; Pickering-Brown, S.; Baba, Y.; Cheshire, W.P. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis. 2006, 1, 30. [Google Scholar] [CrossRef]
- Miyasaka, T.; Morishima-Kawashima, M.; Ravid, R.; Kamphorst, W.; Nagashima, K.; Ihara, Y. Selective deposition of mutant tau in the FTDP-17 brain affected by the P301L mutation. J. Neuropathol. Exp. Neurol. 2001, 60, 872–884. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138 (Suppl. S1), 54–70. [Google Scholar] [CrossRef]
- Josephs, K.A.; Nelson, P.T. Unlocking the mysteries of TDP-43. Neurology 2015, 84, 870–871. [Google Scholar] [CrossRef]
- Borrego-Écija, S.; Morgado, J.; Palencia-Madrid, L.; Grau-Rivera, O.; Reñé, R.; Hernández, I.; Almenar, C.; Balasa, M.; Antonell, A.; Molinuevo, J.L.; et al. Frontotemporal Dementia Caused by the P301L Mutation in the MAPT Gene: Clinicopathological Features of 13 Cases from the Same Geographical Origin in Barcelona, Spain. Dement. Geriatr. Cogn. Disord. 2017, 44, 213–221. [Google Scholar] [CrossRef]
- Stamelou, M.; Respondek, G.; Giagkou, N.; Whitwell, J.L.; Kovacs, G.G.; Höglinger, G.U. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat. Rev. Neurol. 2021, 17, 601–620. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Evolutional aspects of Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S155–S161. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; Holmes, B.B.; Diamond, M.I. Prions and Protein Assemblies that Convey Biological Information in Health and Disease. Neuron 2016, 89, 433–448. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Cali, I.; Cohen, M.L.; Haïk, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.P.; et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Asher, D.M.; Belay, E.; Bigio, E.; Brandner, S.; Brubaker, S.A.; Caughey, B.; Clark, B.; Damon, I.; Diamond, M.; Freund, M.; et al. Risk of Transmissibility From Neurodegenerative Disease-Associated Proteins: Experimental Knowns and Unknowns. J. Neuropathol. Exp. Neurol. 2020, 79, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef]
- Safar, J.G.; Xiao, X.; Kabir, M.E.; Chen, S.; Kim, C.; Haldiman, T.; Cohen, Y.; Chen, W.; Cohen, M.L.; Surewicz, W.K. Structural determinants of phenotypic diversity and replication rate of human prions. PLoS Pathog. 2015, 11, e1004832. [Google Scholar] [CrossRef]
- Safar, J.G. Molecular pathogenesis of sporadic prion diseases in man. Prion 2012, 6, 108–115. [Google Scholar] [CrossRef]
- Puoti, G.; Bizzi, A.; Forloni, G.; Safar, J.G.; Tagliavini, F.; Gambetti, P. Sporadic human prion diseases: Molecular insights and diagnosis. Lancet Neurol. 2012, 11, 618–628. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, I.; Blevins, J.; Castellani, R.; Al-Shekhlee, A.; Yuan, J.; Parchi, P.; Safar, J.; Zou, W.-Q.; Gambetti, P. The Co-Existence of PrPSc Type 1 and 2 in Sporadic Creutzfeldt-Jakob Disease Affects the Phenotype and PrPSc Conformation. In Journal of Neuropathology and Experimental Neurology: 2009; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009; p. 553. [Google Scholar]
- Gibbs, C.J., Jr.; Gajdusek, D.C.; Asher, D.M.; Alpers, M.P.; Beck, E.; Daniel, P.M.; Matthews, W.B. Creutzfeldt-Jakob disease (spongiform encephalopathy): Transmission to the chimpanzee. Science 1968, 161, 388–389. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Gibbs, C.J., Jr.; Alpers, M. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 1966, 209, 794–796. [Google Scholar] [CrossRef]
- Safar, J.G.; Geschwind, M.D.; Deering, C.; Didorenko, S.; Sattavat, M.; Sanchez, H.; Serban, A.; Vey, M.; Baron, H.; Giles, K. Diagnosis of human prion disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3501–3506. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Giles, K.; Lessard, P.; Letessier, F.; Patel, S.; Serban, A.; DeArmond, S.J.; Prusiner, S.B. Conserved properties of human and bovine prion strains on transmission to guinea pigs. Lab. Investig. 2011, 91, 1326–1336. [Google Scholar] [CrossRef][Green Version]
- Bruce, M.E.; Dickinson, A.G. Biological evidence that the scrapie agent has an independent genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Outram, G.W. Genetic aspects of unconventional virus infections: The basis of the virino hypothesis. In Novel Infectious Agents and the Central Nervous System. Ciba Foundation Symposium 135; Bock, G., Marsh, J., Eds.; John Wiley and Sons: Chichester, UK, 1988; pp. 63–83. [Google Scholar]
- Meyer, N.; Rosenbaum, V.; Schmidt, B.; Gilles, K.; Mirenda, C.; Groth, D.; Prusiner, S.B.; Riesner, D. Search for a putative scrapie genome in purified prion fractions reveals a paucity of nucleic acids. J. Gen. Virol. 1991, 72, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Kellings, K.; Prusiner, S.B.; Riesner, D. Nucleic acids in prion preparations: Unspecific background or essential component? Philos. Trans. R. Soc. Lond. B Biol. Sci. 1994, 343, 425–430. [Google Scholar] [PubMed]
- Kellings, K.; Meyer, N.; Mirenda, C.; Prusiner, S.B.; Riesner, D. Further analysis of nucleic acids in purified scrapie prion preparations by improved return refocussing gel electrophoresis (RRGE). J. Gen. Virol. 1992, 73, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Kellings, K.; Serban, A.; Groth, D.; Cleaver, J.E.; Prusiner, S.B.; Riesner, D. Search for a prion-specific nucleic acid. J. Virol. 2005, 79, 10796–10806. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions (Les Prix Nobel Lecture). In Les Prix Nobel; Frängsmyr, T., Ed.; Almqvist & Wiksell International: Stockholm, Sweden, 1998; pp. 268–323. [Google Scholar]
- Weissmann, C. The state of the prion. Nat. Rev. Microbiol. 2004, 2, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014, 5, 4347. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G. Molecular Mechanisms Encoding Quantitative and Qualitative Traits of Prion Strains. In Prions and Diseases; Zou, W.A.G.P., Ed.; Springer Verlag: New York, NY, USA, 2012; Volume 1. [Google Scholar]
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef]
- Cobb, N.J.; Surewicz, W.K. Prion diseases and their biochemical mechanisms. Biochemistry 2009, 48, 2574–2585. [Google Scholar] [CrossRef]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular and genetic basis of prion diseases. In Principles of Molecular Medicine; Jameson, J.L., Ed.; Humana Press: Totowa, NJ, USA, 1998; pp. 927–939. [Google Scholar]
- Prusiner, S.B.; Legname, G.; DeArmond, S.J.; Cohen, F.E.; Safar, J.; Riesner, D.; Kaneko, K. Some strategies and methods for the study of prions. In Prion Biology and Diseases, 2nd ed.; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004; pp. 857–920. [Google Scholar]
- Kim, C.; Xiao, X.; Chen, S.; Haldiman, T.; Smirnovas, V.; Kofskey, D.; Warren, M.; Surewicz, K.; Maurer, N.R.; Kong, Q.; et al. Artificial strain of human prions created in vitro. Nat. Commun. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Baiardi, S.; Rossi, M.; Mammana, A.; Appleby, B.S.; Barria, M.A.; Calì, I.; Gambetti, P.; Gelpi, E.; Giese, A.; Ghetti, B.; et al. Phenotypic diversity of genetic Creutzfeldt-Jakob disease: A histo-molecular-based classification. Acta Neuropathol. 2021, 142, 707–728. [Google Scholar] [CrossRef]
- Parchi, P.; de Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.J.; Hoftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra329. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Orseth, M.C.; Brandel, J.-P.; Haïk, S.; Laplanche, J.-L.; Zerr, I.; Parchi, P.; Capellari, S.; Safar, J. Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology 2019, 93, e125–e134. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, M.K.; Kim, C.; Haldiman, T.; Kacirova, M.; Wang, B.; Bohon, J.; Chance, M.R.; Kiselar, J.; Safar, J.G. Structurally distinct external solvent-exposed domains drive replication of major human prions. PLoS Pathog. 2021, 17, e1009642. [Google Scholar] [CrossRef]
- Collins, S.J.; Sanchez-Juan, P.; Masters, C.L.; Klug, G.M.; van Duijn, C.; Poleggi, A.; Pocchiari, M.; Almonti, S.; Cuadrado-Corrales, N.; de Pedro-Cuesta, J.; et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006, 129, 2278–2287. [Google Scholar] [CrossRef]
- Pocchiari, M.; Puopolo, M.; Croes, E.A.; Budka, H.; Gelpi, E.; Collins, S.; Lewis, V.; Sutcliffe, T.; Guilivi, A.; Delasnerie-Laupretre, N.; et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 2004, 127, 2348–2359. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C. Transgenic mouse models of prion diseases. Methods Mol. Biol. 2008, 459, 249–263. [Google Scholar]
- Bishop, M.T.; Will, R.G.; Manson, J.C. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. USA 2010, 107, 12005–12010. [Google Scholar] [CrossRef]
- Prusiner, S.; Safar, J.; DeArmond, S. Bioassays of prions. Cold Spring Harb. Monogr. Ser. 2004, 41, 143–186. [Google Scholar]
- Wadsworth, J.D.; Collinge, J. Molecular pathology of human prion disease. Acta Neuropathol. 2011, 121, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.D.; Hill, A.F.; Beck, J.A.; Collinge, J. Molecular and clinical classification of human prion disease. Br. Med. Bull. 2003, 66, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Haldiman, T.; Surewicz, K.; Cohen, Y.; Chen, W.; Blevins, J.; Sy, M.S.; Cohen, M.; Kong, Q.; Telling, G.C.; et al. Small Protease Sensitive Oligomers of PrP(Sc) in Distinct Human Prions Determine Conversion Rate of PrP(C). PLoS Pathog. 2012, 8, e1002835. [Google Scholar] [CrossRef]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Haldiman, T.; Cohen, Y.; Chen, W.; Blevins, J.; Sy, M.S.; Cohen, M.; Safar, J.G. Protease-Sensitive Conformers in Broad Spectrum of Distinct PrP Structures in Sporadic Creutzfeldt-Jakob Disease Are Indicator of Progression Rate. PLoS Pathog. 2011, 7, e1002242. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Schmidt, C.; Karch, A.; Artjomova, S.; Hoeschel, M.; Zerr, I. Pre-progression rates in Alzheimer’s disease revisited. J. Alzheimers Dis. 2013, 35, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Haik, S.; Satoh, K.; Rabano, A.; Martinez-Martin, P.; Roeber, S.; Brandel, J.P.; Calero-Lara, M.; de Pedro-Cuesta, J.; Laplanche, J.L.; et al. Rapidly progressive Alzheimer’s disease: A multicenter update. J. Alzheimers Dis. 2012, 30, 751–756. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7, e36584. [Google Scholar] [CrossRef]
- Sharma, A.M.; Thomas, T.L.; Woodard, D.R.; Kashmer, O.M.; Diamond, M.I. Tau monomer encodes strains. Elife 2018, 7, e37813. [Google Scholar] [CrossRef]
- Kaniyappan, S.; Tepper, K.; Biernat, J.; Chandupatla, R.R.; Hübschmann, S.; Irsen, S.; Bicher, S.; Klatt, C.; Mandelkow, E.-M.; Mandelkow, E. FRET-based Tau seeding assay does not represent prion-like templated assembly of Tau filaments. Mol. Neurodegener. 2020, 15, 39. [Google Scholar] [CrossRef]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Muller, R.; Heinrich, M.; Heck, S.; Blohm, D.; Richter-Landsberg, C. Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 1997, 288, 239–249. [Google Scholar] [CrossRef]
- Couchie, D.; Fages, C.; Bridoux, A.M.; Rolland, B.; Tardy, M.; Nunez, J. Microtubule-associated proteins and in vitro astrocyte differentiation. J. Cell Biol. 1985, 101, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Kempf, M.; Clement, A.; Faissner, A.; Lee, G.; Brandt, R. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J. Neurosci. 1996, 16, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kanai, Y.; Cowan, N.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef]
- Feinstein, H.E.; Benbow, S.J.; LaPointe, N.E.; Patel, N.; Ramachandran, S.; Do, T.D.; Gaylord, M.R.; Huskey, N.E.; Dressler, N.; Korff, M. Oligomerization of the microtubule-associated protein tau is mediated by its N-terminal sequences: Implications for normal and pathological tau action. J. Neurochem. 2016, 137, 939–954. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef]
- Lövestam, S.; Koh, F.A.; van Knippenberg, B.; Kotecha, A.; Murzin, A.G.; Goedert, M.; Scheres, S.H. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife 2022, 11, e76494. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Arrasate, M.; Perez, M.; Avila, J. Tau dephosphorylation at tau-1 site correlates with its association to cell membrane. Neurochem. Res. 2000, 25, 43–50. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Perry, G.; Zhu, X.; Moreira, P.I.; Smith, M.A. Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s diseases. Adv. Exp. Med. Biol. 2012, 724, 205–221. [Google Scholar] [PubMed]
- Lu, J.; Li, T.; He, R.; Bartlett, P.F.; Götz, J. Visualizing the microtubule-associated protein tau in the nucleus. Sci. China Life Sci. 2014, 57, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.L.; Tsai, M.-Y.; Aloe, S.; Bechberger, K.; König, S.; Morfini, G.; Brady, S.T. Defined tau phosphospecies differentially inhibit fast axonal transport through activation of two independent signaling pathways. Front. Mol. Neurosci. 2021, 13, 610037. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef]
- Kang, S.-G.; Eskandari-Sedighi, G.; Hromadkova, L.; Safar, J.G.; Westaway, D. Cellular biology of tau diversity and pathogenic conformers. Front. Neurol. 2020, 11, 590199. [Google Scholar] [CrossRef]
- Biundo, F.; Del Prete, D.; Zhang, H.; Arancio, O.; D’Adamio, L. A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 2018, 8, 3184. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from τ deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [CrossRef]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [CrossRef]
- Cantarero, L.A.; Butler, J.E.; Osborne, J.W. The binding characteristics of proteins for polystyrene and their significance in solid-phase immunoassays. Anal. Biochem. 1980, 105, 375–382. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Ordered Assembly of Tau Protein and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1184, 3–21. [Google Scholar] [PubMed]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.-Y.; Bird, T.D.; Schellenberg, G.D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Gamblin, T.C. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry 2012, 51, 8597–8607. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Probst, A.; Götz, J.; Wiederhold, K.; Tolnay, M.; Mistl, C.; Jaton, A.; Hong, M.; Ishihara, T.; Lee, V.-Y.; Trojanowski, J. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; Barmettler, R.; Nitsch, R.M. Tau filament formation in transgenic mice expressing P301L tau. J. Biol. Chem. 2001, 276, 529–534. [Google Scholar]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Tanemura, K.; Murayama, M.; Akagi, T.; Hashikawa, T.; Tominaga, T.; Ichikawa, M.; Yamaguchi, H.; Takashima, A. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J. Neurosci. 2002, 22, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tatebayashi, Y.; Miyasaka, T.; Chui, D.-H.; Akagi, T.; Mishima, K.-i.; Iwasaki, K.; Fujiwara, M.; Tanemura, K.; Murayama, M.; Ishiguro, K. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA 2002, 99, 13896–13901. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Paitel, E.; Kawarabayashi, T.; Ikeda, M.; Chishti, M.A.; Janus, C.; Matsubara, E.; Sasaki, A.; Kawarai, T.; Phinney, A.L. Cortical neuronal and glial pathology in TgTauP301L transgenic mice: Neuronal degeneration, memory disturbance, and phenotypic variation. Am. J. Pathol. 2006, 169, 1365–1375. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Banks, R.A.; Kim, B.; Xu, H.; Changolkar, L.; Leight, S.N.; Riddle, D.M.; Li, C.; Gathagan, R.J.; Brown, H.J. GFP-mutant human tau transgenic mice develop Tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J. Neurosci. 2017, 37, 11485–11494. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013, 587, 717–723. [Google Scholar] [CrossRef]
- Eskandari-Sedighi, G.; Daude, N.; Gapeshina, H.; Sanders, D.W.; Kamali-Jamil, R.; Yang, J.; Shi, B.; Wille, H.; Ghetti, B.; Diamond, M.I. The CNS in inbred transgenic models of 4-repeat Tauopathy develops consistent tau seeding capacity yet focal and diverse patterns of protein deposition. Mol. Neurodegener. 2017, 12, 72. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.-W.; Bajaj, L.; Bader, D.A.; Lee, V.M.; Trojanowski, J.Q.; Liu, Z. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Kenyon, T.K.; Trojanowski, J.Q. Transgenic animal models of tauopathies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1739, 251–259. [Google Scholar] [CrossRef]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nuñez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 2022, 23, 5404. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.Z.; Kang, S.-G.; Arce, L.; Westaway, D. Prion-like strain effects in tauopathies. Cell Tissue Res. 2022, 1–21, online ahead of print. [Google Scholar] [CrossRef]
- Houben, S.; Homa, M.; Yilmaz, Z.; Leroy, K.; Brion, J.-P.; Ando, K. Tau pathology and adult hippocampal neurogenesis: What tau mouse models tell us? Front. Neurol. 2021, 12, 610330. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Schöll, M.; Vogels, T. Tau Seeding Mouse Models with Patient Brain-Derived Aggregates. Int. J. Mol. Sci. 2021, 22, 6132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H.W. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef]
- Stopschinski, B.E.; Del Tredici, K.; Estill-Terpack, S.-J.; Ghebremdehin, E.; Yu, F.F.; Braak, H.; Diamond, M.I. Anatomic survey of seeding in Alzheimer’s disease brains reveals unexpected patterns. Acta Neuropathol. Commun. 2021, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Han, Z.Z.; Daude, N.; McNamara, E.; Wohlgemuth, S.; Molina-Porcel, L.; Safar, J.G.; Mok, S.A.; Westaway, D. Pathologic tau conformer ensembles induce dynamic, liquid-liquid phase separation events at the nuclear envelope. BMC Biol. 2021, 19, 199. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Moore, R.A.; Sim, V.L.; Hughson, A.G.; Dorward, D.W.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 2007, 4, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Vaquer-Alicea, J.; Diamond, M.I.; Joachimiak, L.A. Tau strains shape disease. Acta Neuropathol. 2021, 142, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Daude, N.; Kim, C.; Kang, S.G.; Eskandari-Sedighi, G.; Haldiman, T.; Yang, J.; Fleck, S.C.; Gomez-Cardona, E.; Han, Z.Z.; Borrego-Ecija, S.; et al. Diverse, evolving conformer populations drive distinct phenotypes in frontotemporal lobar degeneration caused by the same MAPT-P301L mutation. Acta Neuropathol. 2020, 139, 1045–1070. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chance, M.R. Protein footprinting comes of age: Mass spectrometry for biophysical structure assessment. Mol. Cell. Proteom. 2017, 16, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713.e1613. [Google Scholar] [CrossRef] [PubMed]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.-M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.H.; Ajit, A.; Tabassum, Z.; Patel, N.; Tian, X.; Chen, Y.; Prevatte, A.W.; Ling, K.; Rigo, F.; Meeker, R.B.; et al. Tau seeds are subject to aberrant modifications resulting in distinct signatures. Cell Rep. 2021, 35, 109037. [Google Scholar] [CrossRef]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L.; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Chen, D.; Mullapudi, V.; Terpack, S.J.; White, C.L., III; Joachimiak, L.A.; Diamond, M.I. Seed-competent tau monomer initiates pathology in a tauopathy mouse model. J. Biol. Chem. 2022, 298, 102163. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, F.; Xiao, X.; Kim, C.; Bohon, J.; Kiselar, J.; Safar, J.G.; Ma, J.; Surewicz, W.K. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J. Biol. Chem. 2018, 293, 18494–18503. [Google Scholar] [CrossRef]
- Kraus, A.; Saijo, E.; Metrick, M.A., 2nd; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2019, 137, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Orru, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.J.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated alpha-synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry 2008, 47, 10526–10539. [Google Scholar] [CrossRef] [PubMed]
- Daebel, V.; Chinnathambi, S.; Biernat, J.; Schwalbe, M.; Habenstein, B.; Loquet, A.; Akoury, E.; Tepper, K.; Muller, H.; Baldus, M.; et al. beta-Sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc. 2012, 134, 13982–13989. [Google Scholar] [CrossRef]
- Saijo, E.; Metrick, M.A., 2nd; Koga, S.; Parchi, P.; Litvan, I.; Spina, S.; Boxer, A.; Rojas, J.C.; Galasko, D.; Kraus, A.; et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020, 139, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Metrick, M.A., 2nd; Ferreira, N.D.C.; Saijo, E.; Kraus, A.; Newell, K.; Zanusso, G.; Vendruscolo, M.; Ghetti, B.; Caughey, B. A single ultrasensitive assay for detection and discrimination of tau aggregates of Alzheimer and Pick diseases. Acta Neuropathol. Commun. 2020, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sahara, N. Characteristics of tau oligomers. Front. Neurol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Michaels, T.C.T.; Šarić, A.; Habchi, J.; Chia, S.; Meisl, G.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2018, 69, 273–298. [Google Scholar] [CrossRef]
- Noble, G.P.; Wang, D.W.; Walsh, D.J.; Barone, J.R.; Miller, M.B.; Nishina, K.A.; Li, S.; Supattapone, S. A Structural and Functional Comparison Between Infectious and Non-Infectious Autocatalytic Recombinant PrP Conformers. PLoS Pathog. 2015, 11, e1005017. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef]
- Arrasate, M.; Perez, M.; Armas-Portela, R.; Avila, J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999, 446, 199–202. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999, 450, 306–311. [Google Scholar] [CrossRef]
- Von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Holmes, B.B.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 2012, 287, 19440–19451. [Google Scholar] [CrossRef]
- Falcon, B.; Cavallini, A.; Angers, R.; Glover, S.; Murray, T.K.; Barnham, L.; Jackson, S.; O’Neill, M.J.; Isaacs, A.M.; Hutton, M.L.; et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J. Biol. Chem. 2015, 290, 1049–1065. [Google Scholar] [CrossRef]
- Stancu, I.C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 4579. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells: Insights into Therapies For Tauopathies. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, H.; Hasegawa, M.; Tamaoka, A. Fibrillogenic nuclei composed of P301L mutant tau induce elongation of P301L tau but not wild-type tau. J. Biol. Chem. 2007, 282, 20309–20318. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Kamphorst, W.; Heutink, P.; van Swieten, J.C. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am. J. Pathol. 1998, 153, 1359–1363. [Google Scholar] [CrossRef]
- Matsumoto, G.; Matsumoto, K.; Kimura, T.; Suhara, T.; Higuchi, M.; Sahara, N.; Mori, N. Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy. Int. J. Mol. Sci. 2018, 19, 1497. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, B.; Li, G.; Yin, H.; Kuret, J. Tau aggregation and toxicity in a cell culture model of tauopathy. J. Biol. Chem. 2007, 282, 16454–16464. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Stohr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-G.; Han, Z.Z.; Daude, N.; McNamara, E.; Wohlgemuth, S.; Safar, J.G.; Mok, S.-A.; Westaway, D. Tau conformers in FTLD-MAPT undergo liquid-liquid phase separation and perturb the nuclear envelope. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, L.; Shi, R.; Gu, J.; Tung, Y.C.; Zhou, Y.; Zhou, D.; Wu, R.; Chu, D.; Jin, N.; Deng, K.; et al. Alzheimer’s disease brain contains tau fractions with differential prion-like activities. Acta Neuropathol. Commun. 2021, 9, 28. [Google Scholar] [CrossRef]
- Löffler, T.; Flunkert, S.; Taub, N.; Schofield, E.L.; Ward, M.A.; Windisch, M.; Hutter-Paier, B. Stable mutated tau441 transfected SH-SY5Y cells as screening tool for Alzheimer’s disease drug candidates. J. Mol. Neurosci. 2012, 47, 192–203. [Google Scholar] [CrossRef][Green Version]
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.-J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Saragoni, L.; Hernández, P.; Maccioni, R.B. Differential association of tau with subsets of microtubules containing posttranslationally-modified tubulin variants in neuroblastoma cells. Neurochem. Res. 2000, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hou, L.X.E.; Aktiv, A.; Dahlström, A. Studies of the central nervous system-derived CAD cell line, a suitable model for intraneuronal transport studies? J. Neurosci. Res. 2007, 85, 2601–2609. [Google Scholar] [CrossRef]
- Hromadkova, L.; Bezdekova, D.; Pala, J.; Schedin-Weiss, S.; Tjernberg, L.O.; Hoschl, C.; Ovsepian, S.V. Brain-derived neurotrophic factor (BDNF) promotes molecular polarization and differentiation of immature neuroblastoma cells into definitive neurons. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118737. [Google Scholar] [CrossRef]
- Nonaka, T.; Watanabe, S.T.; Iwatsubo, T.; Hasegawa, M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: Cellular models of neurodegenerative diseases. J. Biol. Chem. 2010, 285, 34885–34898. [Google Scholar] [CrossRef]
- Chastagner, P.; Loria, F.; Vargas, J.Y.; Tois, J.; Diamond, I.M.; Okafo, G.; Brou, C.; Zurzolo, C. Fate and propagation of endogenously formed Tau aggregates in neuronal cells. EMBO Mol. Med. 2020, 12, e12025. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Cena, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Bell, M.; Zempel, H. SH-SY5Y-derived neurons: A human neuronal model system for investigating TAU sorting and neuronal subtype-specific TAU vulnerability. Rev. Neurosci. 2022, 33, 1–15. [Google Scholar] [CrossRef]
- Pollack, S.J.; Trigg, J.; Khanom, T.; Biasetti, L.; Marshall, K.E.; Al-Hilaly, Y.K.; Rickard, J.E.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Paired helical filament-forming region of tau (297–391) influences endogenous tau protein and accumulates in acidic compartments in human neuronal cells. J. Mol. Biol. 2020, 432, 4891–4907. [Google Scholar] [CrossRef]
- Shamir, D.B.; Deng, Y.; Wu, Q.; Modak, S.; Congdon, E.E.; Sigurdsson, E.M. Dynamics of internalization and intracellular interaction of tau antibodies and human pathological tau protein in a human neuron-like model. Front. Neurol. 2020, 11, 602292. [Google Scholar] [CrossRef]
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell Neurosci. 1997, 9, 220–234. [Google Scholar] [CrossRef]
- Woerman, A.L.; Aoyagi, A.; Patel, S.; Kazmi, S.A.; Lobach, I.; Grinberg, L.T.; McKee, A.C.; Seeley, W.W.; Olson, S.H.; Prusiner, S.B. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc. Natl. Acad. Sci. USA 2016, 113, E8187–E8196. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; McBride, J.D.; Xu, H.; Changolkar, L.; Kim, S.-J.; Zhang, B.; Narasimhan, S.; Gibbons, G.S.; Guo, J.L.; Kozak, M.; et al. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; O’Reilly, M.; Gibbons, G.S.; Changolkar, L.; McBride, J.D.; Riddle, D.M.; Zhang, B.; Stieber, A.; Nirschl, J.; Kim, S.-J. In vitro amplification of pathogenic tau conserves disease-specific bioactive characteristics. Acta Neuropathol. 2021, 141, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.-H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-Derived Neural Models for Studying Alzheimer’s Disease: From Neural Stem Cells to Cerebral Organoids. Stem Cell Rev. Rep. 2022, 18, 792–820. [Google Scholar] [CrossRef]
- Iovino, M.; Agathou, S.; González-Rueda, A.; Del Castillo Velasco-Herrera, M.; Borroni, B.; Alberici, A.; Lynch, T.; O’Dowd, S.; Geti, I.; Gaffney, D. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015, 138, 3345–3359. [Google Scholar] [CrossRef]
- Wray, S. Modeling tau pathology in human stem cell derived neurons. Brain Pathol. 2017, 27, 525–529. [Google Scholar] [CrossRef]
- Miguel, L.; Rovelet-Lecrux, A.; Feyeux, M.; Frebourg, T.; Nassoy, P.; Campion, D.; Lecourtois, M. Detection of all adult Tau isoforms in a 3D culture model of iPSC-derived neurons. Stem Cell Res. 2019, 40, 101541. [Google Scholar] [CrossRef]
- Verheyen, A.; Diels, A.; Dijkmans, J.; Oyelami, T.; Meneghello, G.; Mertens, L.; Versweyveld, S.; Borgers, M.; Buist, A.; Peeters, P. Using human iPSC-derived neurons to model TAU aggregation. PLoS ONE 2015, 10, e0146127. [Google Scholar] [CrossRef]
- Medda, X.; Mertens, L.; Versweyveld, S.; Diels, A.; Barnham, L.; Bretteville, A.; Buist, A.; Verheyen, A.; Royaux, I.; Ebneth, A. Development of a scalable, high-throughput-compatible assay to detect tau aggregates using iPSC-derived cortical neurons maintained in a three-dimensional culture format. J. Biomol. Screen. 2016, 21, 804–815. [Google Scholar] [CrossRef]
- Manos, J.D.; Preiss, C.N.; Venkat, N.; Tamm, J.; Reinhardt, P.; Kwon, T.; Wu, J.; Winter, A.D.; Jahn, T.R.; Yanamandra, K. Uncovering specificity of endogenous TAU aggregation in a human iPSC-neuron TAU seeding model. Iscience 2022, 25, 103658. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef]
- Aoyagi, A.; Condello, C.; Stöhr, J.; Yue, W.; Rivera, B.M.; Lee, J.C.; Woerman, A.L.; Halliday, G.; van Duinen, S.; Ingelsson, M.; et al. Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 2019, 11, eaat8462. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification Criteria of Human Prion Strains | References |
|---|---|
| Species of human prion strain are determined by the amino acid sequence and polymorphism of the misfolded conformer (PrPSc) of normal human prion protein (PrPC) coded by prion gene (PRNP) | [47,48,74,75] |
| Clinical characteristics of the disease in affected humans | [45,47,48,76] |
| Disease progression rates | [45,48,64,77,78] |
| Incubation times in Tg mice expressing homologous human prion protein or its chimera | [52,79,80,81] |
| Unique neuropathological phenotypes and anatomical distributions of pathogenic PrPSc in the brain | [45,70,82] |
| Distinct N-linked glycosylation profiles of human PrPSc | [83] |
| Differential susceptibility of different human prion strains to proteases | [73,84,85,86]; |
| Unique structural organizations of pathogenic prion protein (PrPSc) | [70,76,84,85,86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hromadkova, L.; Siddiqi, M.K.; Liu, H.; Safar, J.G. Populations of Tau Conformers Drive Prion-like Strain Effects in Alzheimer’s Disease and Related Dementias. Cells 2022, 11, 2997. https://doi.org/10.3390/cells11192997
Hromadkova L, Siddiqi MK, Liu H, Safar JG. Populations of Tau Conformers Drive Prion-like Strain Effects in Alzheimer’s Disease and Related Dementias. Cells. 2022; 11(19):2997. https://doi.org/10.3390/cells11192997
Chicago/Turabian StyleHromadkova, Lenka, Mohammad Khursheed Siddiqi, He Liu, and Jiri G. Safar. 2022. "Populations of Tau Conformers Drive Prion-like Strain Effects in Alzheimer’s Disease and Related Dementias" Cells 11, no. 19: 2997. https://doi.org/10.3390/cells11192997
APA StyleHromadkova, L., Siddiqi, M. K., Liu, H., & Safar, J. G. (2022). Populations of Tau Conformers Drive Prion-like Strain Effects in Alzheimer’s Disease and Related Dementias. Cells, 11(19), 2997. https://doi.org/10.3390/cells11192997