
The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
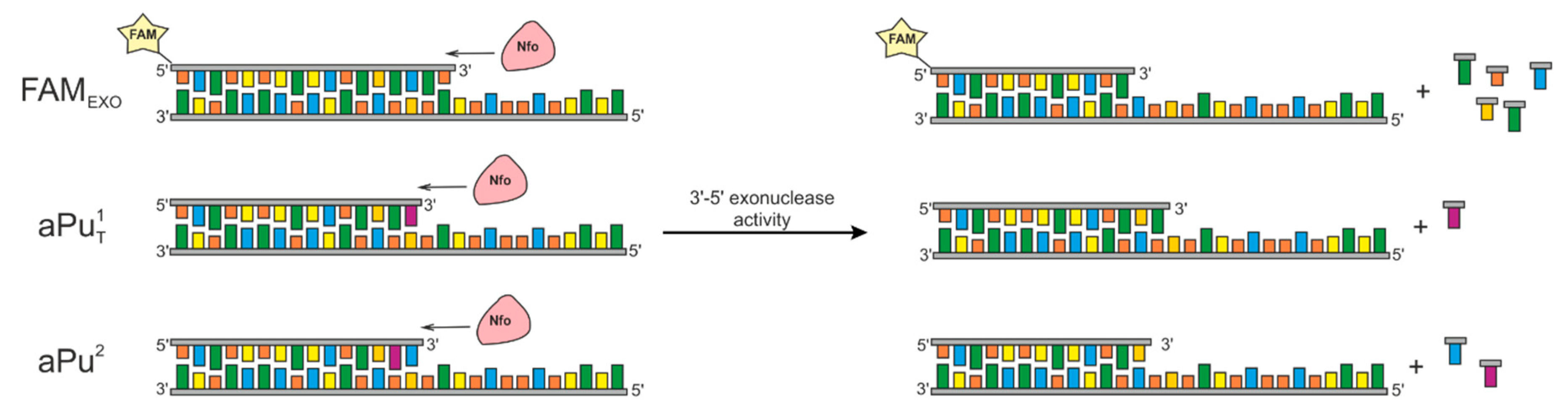
2.1. Model DNA Substrates
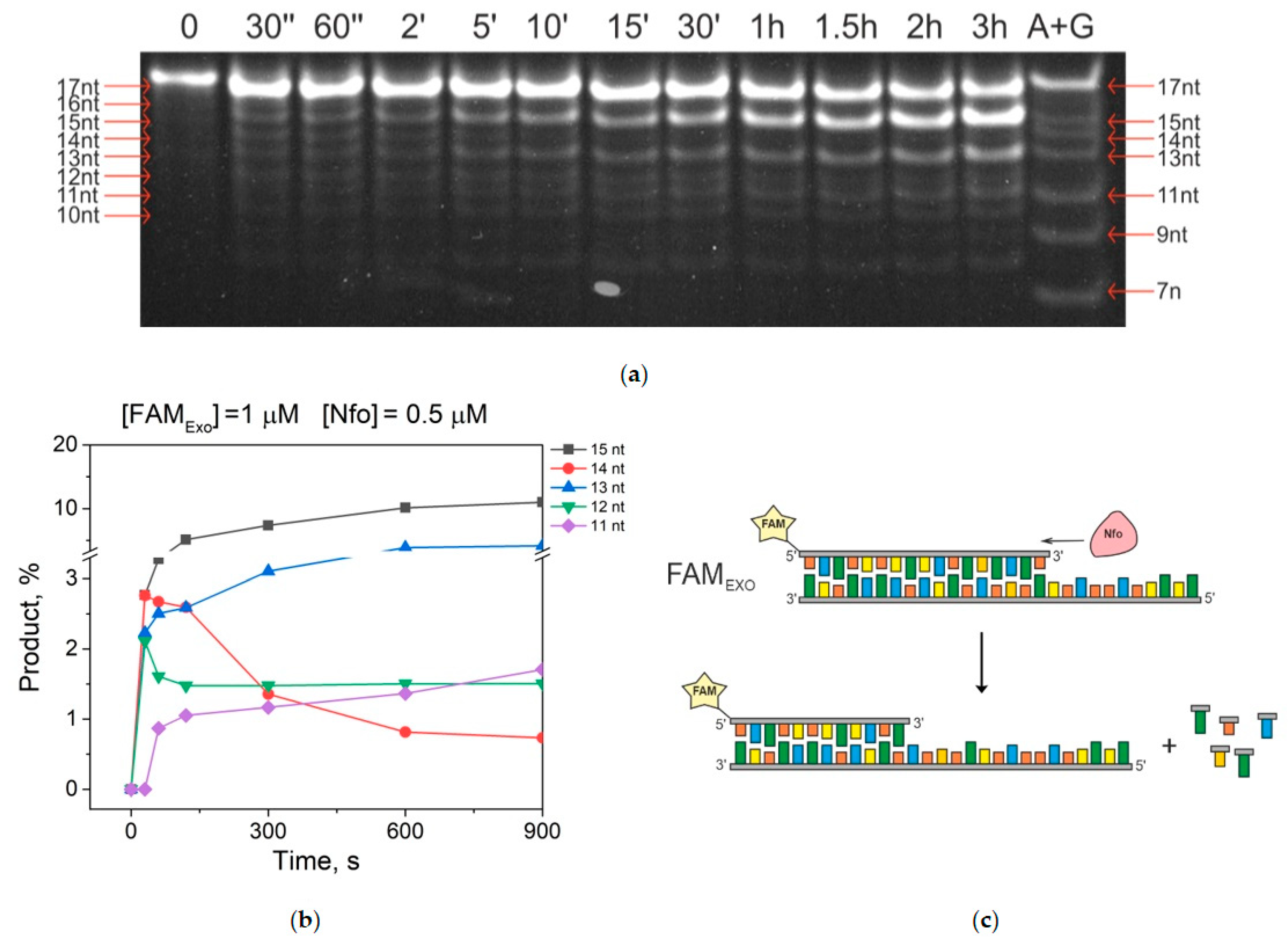
2.2. Accumulation of 3′-5′-Exonucleolytic-Degradation Products
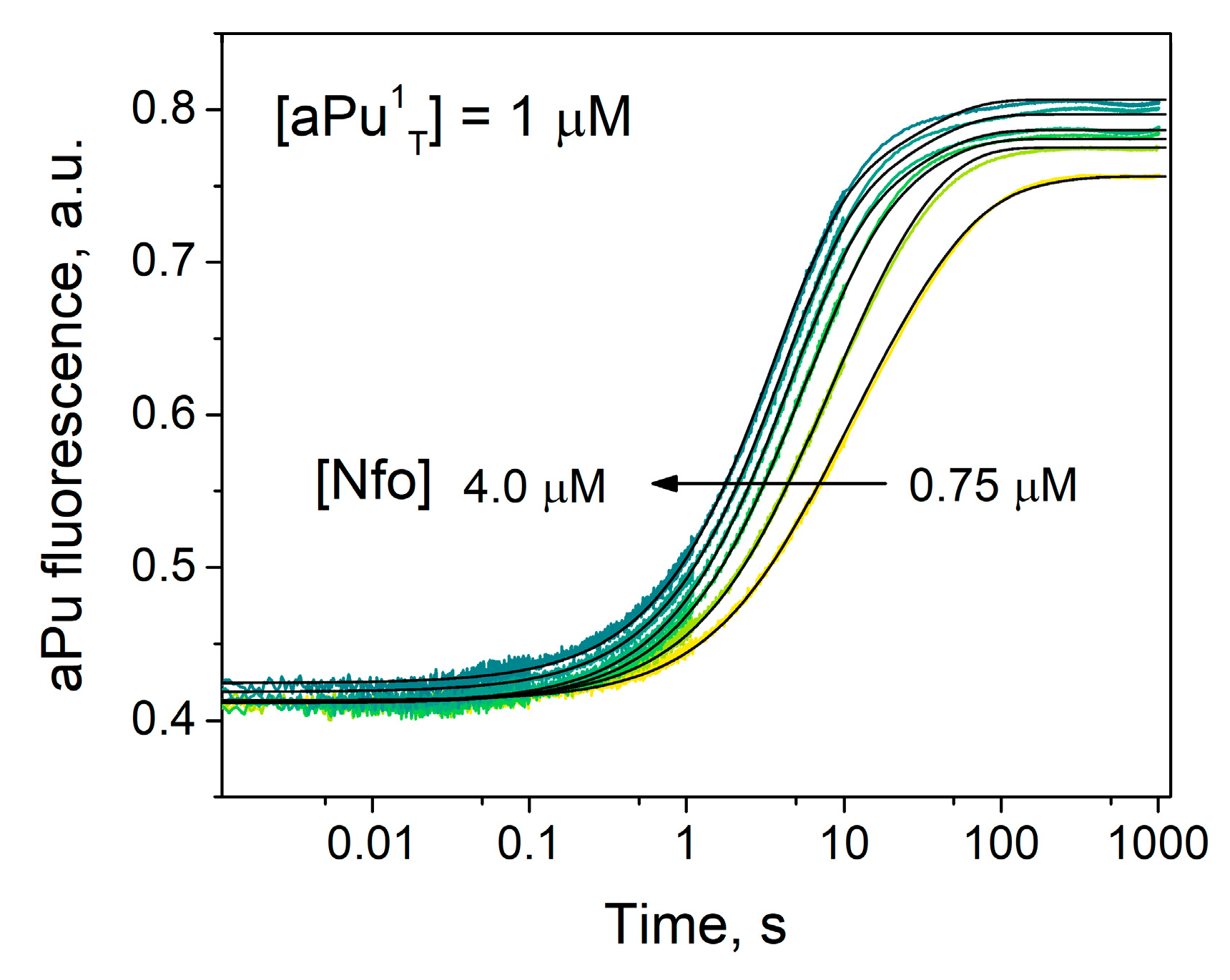
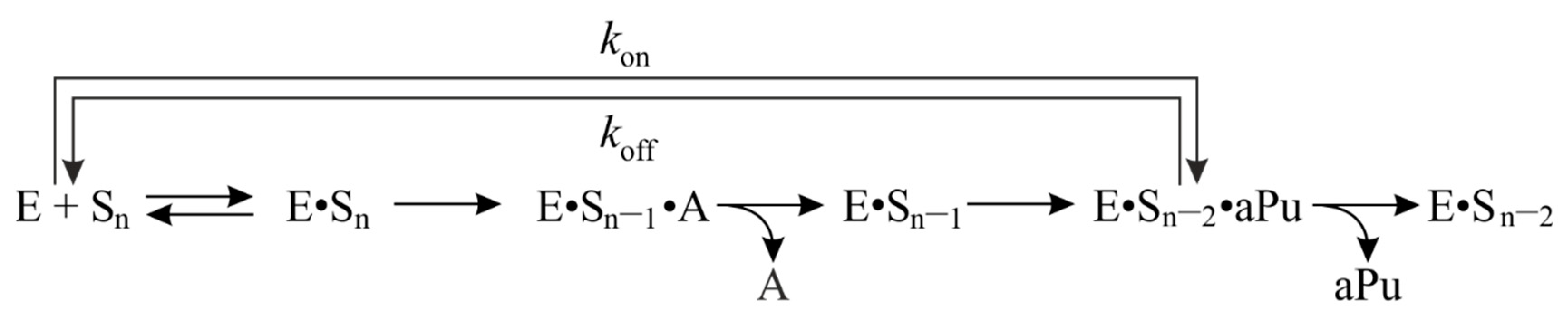
2.3. Interaction with the DNA Substrate Containing aPu at the First Position
2.4. The Influence of Stability of a 3′-Terminal Base Pair on Binding and Cleavage Efficiency
2.5. Interaction with the DNA Substrate Containing aPu at the Second Position
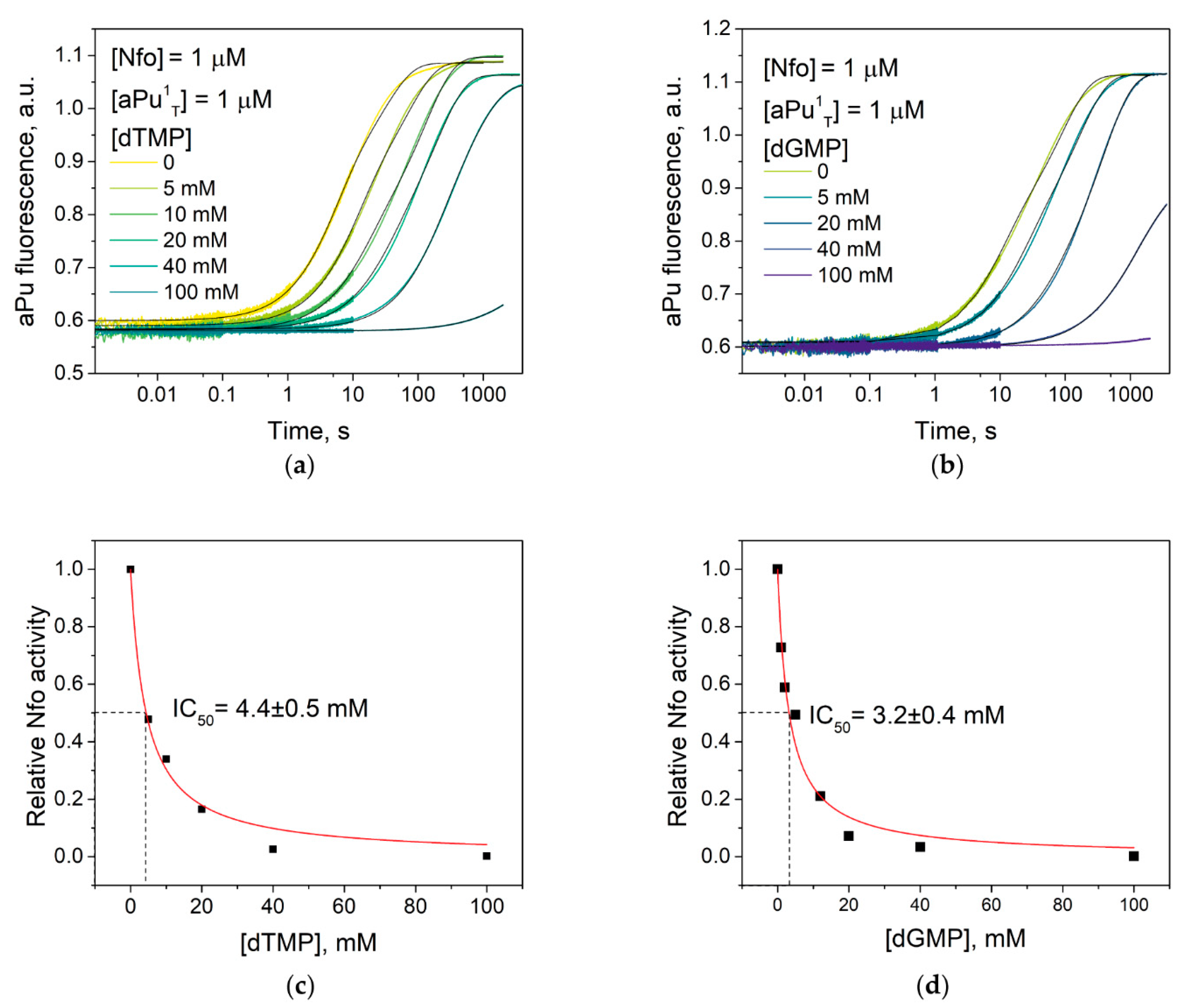
2.6. Determination of the Inhibitory Ability of 2′-Deoxynucleoside Monophosphates
3. Conclusions
4. Materials and Methods
4.1. The Enzyme and Oligonucleotides
4.2. Polyacrylamide Gel (PAAG) Electrophoretic Analysis of the Cleavage of DNA Substrates
4.3. Kinetic Analysis of the Cleavage of DNA Substrates by the Stopped-Flow Method
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wilson III, D.M.; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1261. [Google Scholar] [CrossRef] [PubMed]
- Matta, E.; Aliyaskarova, U.; Kuznetsova, A.A.; Matkarimov, B.T.; Fedorova, O.S.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M. Chapter 11. Alternative DNA Repair Pathways to Handle Complex DNA Damage Generated by Oxidative Stress and Anticancer Drugs; The Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Ljungquist, S. A new endonuclease from Escherichia coli acting at apurinic sites in DNA. J. Biol. Chem. 1977, 252, 2808–2814. [Google Scholar] [CrossRef]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic site recognition by two apurinicrapyrimidinic endonuclease families in DNA base excision repair: The 3X ends justify the means, Mutat. Res. DNA Repair 2000, 460, 211–229. [Google Scholar]
- Hosfield, D.J.; Guan, Y.; Haas, B.J.; Cunningham, R.P.; Tainer, J.A. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: Double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell 1999, 98, 397–408. [Google Scholar] [CrossRef]
- Demple, B.; Harrison, L. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 1994, 63, 915–948. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.P.; Saporito, S.M.; Spitzer, S.G.; Weiss, B. Endonuclease IV (nfo) mutant of Escherichia coli. J. Bacteriol. 1986, 168, 1120–1127. [Google Scholar] [CrossRef]
- Cunningham, R.P.; Weiss, B. Endonuclease III (nth) mutants of Escherichia coli. Proc. Natl. Acad. Sci. USA 1985, 82, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Huisman, O. Isolation of a formamidopyrimidine-DNA glycosylase (fpg) mutant of Escherichia coli K12. MGG Mol. Gen. Genet. 1989, 215, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, J.O.; Wallace, S.S. Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli. Proc. Natl. Acad. Sci. USA 2001, 98, 7426–7430. [Google Scholar] [CrossRef] [PubMed]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, A.A.; Saparbaev, M.K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002, 415, 183–187. [Google Scholar] [CrossRef]
- Kerins, S.M.; Collins, R.; McCarthy, T.V. Characterization of an endonuclease IV 3′-5′ exonuclease activity. J. Biol. Chem. 2003, 278, 3048–3054. [Google Scholar] [CrossRef]
- Golan, G.; Ishchenko, A.A.; Khassenov, B.; Shoham, G.; Saparbaev, M.K. Coupling of the nucleotide incision and 3′-5′ exonuclease activities in Escherichia coli endonuclease IV: Structural and genetic evidences. Mutat Res. 2010, 685, 70–79. [Google Scholar] [CrossRef]
- Senchurova, S.I.; Syryamina, V.N.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. The mechanism of damage recognition by apurinic/apyrimidinic endonuclease Nfo from Escherichia coli. BBA-Gen. Subj. 2022, 1866, 130216. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Senchurova, S.I.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Common Kinetic Mechanism of Abasic Site Recognition by Structurally Different Apurinic/Apyrimidinic Endonucleases. Int. J. Mol. Sci. 2021, 22, 8874. [Google Scholar] [CrossRef]
- Mazouzi, A.; Vigouroux, A.; Aikeshev, B.; Brooks, P.J.; Saparbaev, M.K.; Morera, S.; Ishchenko, A.A. Insight into mechanisms of 3′-5′ exonuclease activity and removal of bulky 8,5′-cyclopurine adducts by apurinic/apyrimidinic endonucleases. Proc. Natl. Acad. Sci. USA 2013, 110, E3071–E3080. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′-5′ Exonuclease Activity of Human AP-Endonuclease APE1. Molecules 2018, 23, 2101. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Khodyreva, S.N.; Lavrik, O.I. Multifunctional human apurinic/apyrimidinic endonuclease 1: The role of additional functions. Mol. Biol. 2007, 41, 450–466. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Lomzov, A.A.; Pyshnyi, D.V.; Khodyreva, S.N.; Lavrik, O.I. Efficiency of exonucleolytic action of apurinic/apyrimidinic endonuclease 1 towards matched and mismatched dNMP at the 3′ terminus of different oligomeric DNA structures correlates with thermal stability of DNA duplexes. Biochim. Biophys. Acta-Proteins Proteom. 2006, 1764, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Jean, J.M.; Hall, K.B. 2-Aminopurine fluorescence quenching and lifetimes: Role of base stacking. Proc. Natl. Acad. Sci. USA 2001, 98, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Kladova, O.A.; Barthes, N.P.F.; Michel, B.Y.; Burger, A.; Fedorova, O.S.; Kuznetsov, N.A. Comparative Analysis of Nucleotide Fluorescent Analogs for Registration of DNA Conformational Changes Induced by Interaction with Formamidopyrimidine-DNA Glycosylase Fpg. Russ. J. Biorgan. Chem. 2019, 45, 591–598. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Vorobjev, Y.N.; Barthes, N.P.F.; Michel, B.Y.; Burger, A.; Fedorova, O.S. New Environment-Sensitive Multichannel DNA Fluorescent Label for Investigation of the Protein-DNA Interactions. PLoS ONE 2014, 9, e100007. [Google Scholar] [CrossRef]
- Law, S.M.; Eritja, R.; Goodman, M.F.; Breslauer, K.J. Spectroscopic and calorimetric characterizations of DNA duplexes containing 2-aminopurine. Biochemistry 1996, 35, 12329–12337. [Google Scholar] [CrossRef]
- Kuzmic, P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of the DNA repair process by endonuclease VIII. Acta Naturae 2019, 11, 29–37. [Google Scholar] [CrossRef]
- Alekseeva, I.V.; Kuznetsova, A.A.; Bakman, A.S.; Fedorova, O.S.; Kuznetsov, N.A. The role of active-site amino acid residues in the cleavage of DNA and RNA substrates by human apurinic/apyrimidinic endonuclease APE1. BBA-Gen. Subj. 2020, 1864, 129718. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rate Constant | aPu1T |
|---|---|
| Kbind, 10−6 M−1 | 0.67 ± 0.02 |
| kdetach, s−1 | 0.54 ± 0.01 |
| krelease, s−1 | 0.039 ± 0.001 |
| Rate Constant | aPu2 |
|---|---|
| kon, M−1 s−1 | 3600 ± 400 |
| koff, s−1 | 0.020 ± 0.001 |
| K, 10−6 M−1 | 0.18 ± 0.02 |
| Name | Sequence | Structure |
|---|---|---|
| aPu1N N = A, C, G, T | 5′-CAGCTCTGTACGTG(aPu)-3′ 3′-GTCGAGACATGCAC N CGTCACCACTGTG-5′ |  |
| aPu2 | 5′-CAGCTCTGTACG(aPu)A-3′ 3′-GTCGAGACATGCA C TCGTCACCACTGTG-5′ |  |
| FAMExo | 5′-FAM-CAGCTCTGTACGTGAGC-3′ 3′-GTCGAGACATGCACTCGTCACCACTGTG-5′ |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senchurova, S.I.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli. Cells 2022, 11, 2998. https://doi.org/10.3390/cells11192998
Senchurova SI, Kuznetsova AA, Ishchenko AA, Saparbaev M, Fedorova OS, Kuznetsov NA. The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli. Cells. 2022; 11(19):2998. https://doi.org/10.3390/cells11192998
Chicago/Turabian StyleSenchurova, Svetlana I., Aleksandra A. Kuznetsova, Alexander A. Ishchenko, Murat Saparbaev, Olga S. Fedorova, and Nikita A. Kuznetsov. 2022. "The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli" Cells 11, no. 19: 2998. https://doi.org/10.3390/cells11192998
APA StyleSenchurova, S. I., Kuznetsova, A. A., Ishchenko, A. A., Saparbaev, M., Fedorova, O. S., & Kuznetsov, N. A. (2022). The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli. Cells, 11(19), 2998. https://doi.org/10.3390/cells11192998