Theory of Flexible Polymer Networks: Elasticity and Heterogeneities


Abstract
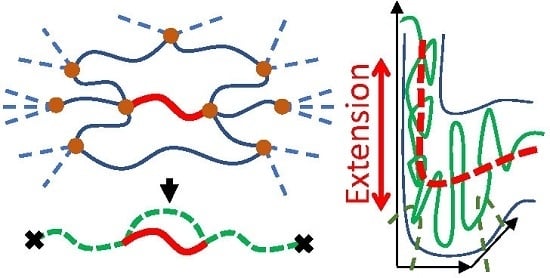
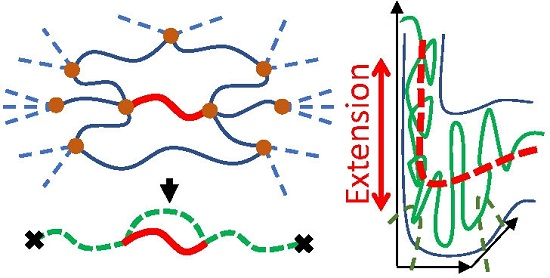{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Phantom Networks
2.1. Replica Method
2.2. Liquid-Solid Order Parameter
2.3. Overlap Parameter
2.4. Combined Chain Model: Monodisperse Networks
2.5. Combined Chain Model: Polydisperse Networks
2.6. Generalized Combined Chain Model
2.7. Finite-Size Loops of Real Networks
3. Entangled Networks
3.1. Physics of Entangled Network Deformation
3.2. Slip-Tube Model
3.3. Crossover from Unentangled to Entangled Regimes
4. Heterogeneities in Polymer Networks
4.1. Theory of Heterogeneities in Polymer Networks
4.2. Scattering Intensity
4.3. Heterogeneities in Charged Gels
4.4. Good Solvent: Scaling Approach
4.5. Amplification of Cross-Linking Density Pattern
5. Discussion
Funding
Conflicts of Interest
Appendix A. Replica Model of the Network
Appendix B. Distribution of Virtual Chains
Appendix C. Free Energy of Entangled Networks
References
- Urayama, K.; Kohjiya, S. Extensive stretch of polysiloxane network chains with random- and super-coiled conformations. Eur. Phys. J. B 1998, 2, 75–78. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Rubinstein, M.; Panyukov, S. Elasticity of Polymer Networks. Macromolecules 2002, 35, 6670–6686. [Google Scholar] [CrossRef]
- Panyukov, S. Loops in Polymer Networks. Macromolecules 2019, 52, 4145–4153. [Google Scholar] [CrossRef]
- Ge, T.; Panyukov, S.; Rubinstein, M. Self-Similar Conformations and Dynamics in Entangled Melts and Solutions of Nonconcatenated Ring Polymers. Macromolecules 2016, 49, 708–722. [Google Scholar] [CrossRef]
- Rubinstein, M.; Panyukov, S. Nonaffine Deformation and Elasticity of Polymer Networks. Macromolecules 1997, 30, 8036–8044. [Google Scholar] [CrossRef]
- Deam, R.T.; Edwards, S.F. The Theory of Rubber Elasticity. Philos. Trans. R. Sot. Lond. Ser. A 1976, 280, 317. [Google Scholar] [CrossRef]
- Panyukov, S.; Rabin, Y. Statistical physics of polymer gels. Phys. Rep. 1996, 269, 1–131. [Google Scholar] [CrossRef]
- Panyukov, S.V.; Rabin, Y. Replica Field Theory Methods in Physics of Polymer Networks. In Theoretical and Mathematical Models in Polymer Research; Modern Methods in Polymer Research and Technology; Grosberg, A.Y., Ed.; Academic Press: Boston, MA, USA, 1989; pp. 83–185. [Google Scholar]
- Panyukov, S.; Rabin, Y.; Fiegel, A. Solid Elasticity and Liquid-Like Behaviour in Randomly Crosslinked Polymer Networks. Europhys. Lett. 1994, 28, 149–154. [Google Scholar] [CrossRef]
- Edwards, S.F. A field theory formulation of polymer networks. J. Phys. 1988, 49, 1673–1682. [Google Scholar] [CrossRef]
- Cai, L.-H.; Panyukov, S.; Rubinstein, M. Hopping Diffusion of Nanoparticles in Polymer Matrices. Macromolecules 2015, 48, 847–862. [Google Scholar] [CrossRef]
- Lang, M.; Kreitmeier, S.; Göritz, D. Trapped Entanglements in Polymer Networks. Rubber Chem. Technol. 2007, 80, 873–894. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK; New York, NY, USA, 2003. [Google Scholar]
- Schieber, J.D.; Horio, K. Fluctuation in entanglement positions via elastic slip-links. J. Chem. Phys. 2010, 132, 074905. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.A. Numerical Estimates of the Topological Effects in the Elasticity of Gaussian Polymer Networks and Their Exact Theoretical Description. Macromolecules 2019, 52, 3244–3251. [Google Scholar] [CrossRef]
- Helfand, E.; Tonelli, A.E. Elastically Ineffective Polymer Chains in Rubbers. Macromolecules 1974, 7, 832–834. [Google Scholar] [CrossRef]
- Tonelli, A.E.; Laboratories, B.; Hill, M. Elastically Ineffective Cross-Links in Rubbers. Macromolecules 1974, 7, 59–63. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, R.; Kawamoto, K.; Olsen, B.D.; Johnson, J.A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264–1268. [Google Scholar] [CrossRef]
- Lin, T.-S.; Wang, R.; Johnson, J.A.; Olsen, B.D. Topological Structure of Networks Formed from Symmetric Four-Arm Precursors. Macromolecules 2018, 51, 1224–1231. [Google Scholar] [CrossRef]
- Lin, T.-S.; Wang, R.; Johnson, J.A.; Olsen, B.D. Revisiting the Elasticity Theory for Real Gaussian Phantom Networks. Macromolecules 2019, 52, 1685–1694. [Google Scholar] [CrossRef]
- Lang, M. Elasticity of Phantom Model Networks with Cyclic Defects. ACS Macro Lett. 2018, 7, 536–539. [Google Scholar] [CrossRef]
- Lange, F.; Schwenke, K.; Kurakazu, M.; Akagi, Y.; Chung, U.I.; Lang, M.; Sommer, J.-U.; Sakai, T.; Saalächter, K. Connectivity and structural defects in model hydrogels: A combined proton NMR and Monte Carlo simulation study. Macromolecules 2011, 44, 9666–9674. [Google Scholar] [CrossRef]
- Zhou, H.; Woo, J.; Cok, A.M.; Wang, M.; Olsen, B.D.; Johnson, J.A. Counting primary loops in polymer gels. PNAS 2012, 109, 19119–19124. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Alexander-Katz, A.; Johnson, J.A.; Olsen, B.D. Universal Cyclic Topology in Polymer Networks. Phys. Rev. Lett. 2016, 116, 188302. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Miller, T. Analysis of the Gel Point of Polymer Model Networks by Computer Simulations. Macromolecules 2020, 53, 498–512. [Google Scholar] [CrossRef]
- Panagiotou, E.; Kruger, M.; Millett, K.C. Writhe and mutual entanglement combine to give the entanglement length. Phys. Rev. E 2013, 88, 062604. [Google Scholar] [CrossRef]
- Panyukov, S.V. Topology fluctuations in polymer networks. Sov. Phys. JETP 1989, 69, 342–353. [Google Scholar]
- Panyukov, S.V. Topological interactions in the statistical theory of polymers. Sov. Phys. JETP 1988, 67, 2274–2284. [Google Scholar]
- Gumbrell, S.M.; Mullins, L.; Rivlin, R.S. Departures of the elastic behaviour of rubbers in simple extension from the kinetic theory. Rubber Chem. Technol. 1955, 28, 24–35. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Seiffert, S. Nanostructural heterogeneity in polymer networks and gels. Polym. Chem. 2015, 6, 5515–5528. [Google Scholar] [CrossRef]
- Seiffert, S. Origin of Nanostructural Inhomogeneity in Polymer-Network Gels. Polym. Chem. 2016, 7, 36–43. [Google Scholar] [CrossRef]
- Vilgis, T.A.; Heinrich, G. The Essential Role of Network Topology in Rubber Elasticity. Angew. Makromol. Chem. 1992, 202, 243–259. [Google Scholar] [CrossRef]
- Vilgis, T.A.; Sommer, J.-U.; Heinrich, G. Swelling and fractal heterogeneities in networks. Macromol. Symp. 1995, 93, 205–212. [Google Scholar] [CrossRef]
- Svaneborg, C.; Grest, G.S.; Everaers, R. Disorder effects on the strain response of model polymer networks. Polymer 2005, 46, 4283–4295. [Google Scholar] [CrossRef]
- Panyukov, S.V. Inhomogeneities as consequences of a stretching of polymer networks. JETP Lett. 1993, 58, 118–122. [Google Scholar]
- Panyukov, S.; Rabin, Y. Polymer Gels: Frozen Inhomogeneities and Density Fluctuations. Macromolecules 1996, 29, 7960–7975. [Google Scholar] [CrossRef]
- Panyukov, S.V. Theory of heterogeneities in polymer networks. Polym. Sci. Ser. A 2016, 58, 582–594. [Google Scholar] [CrossRef]
- Lifshitz, I.M.; Grosberg, A.Y.; Khokhlov, A.R. Some problems of the statistical physics of polymer chains with volume interaction. Rev. Mod. Phys. 1978, 50, 683. [Google Scholar] [CrossRef]
- de Gennes, P.G. Effect of cross-links on a mixture of polymers. J. Phys. Lett. 1979, 40, 69–72. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Orkisz, M.; Sun, S.-T.; Li, Y.; Tanaka, T. Origin of Structural Inhomogeneities in Polymer Gels. Macromolecules 1994, 27, 6791–6796. [Google Scholar] [CrossRef]
- Kizilay, M.Y.; Okay, O. Effect of Initial Monomer Concentration on Spatial Inhomogeneity in Poly(acrylamide) Gels. Macromolecules 2003, 36, 6856–6862. [Google Scholar] [CrossRef]
- Kizilay, M.Y.; Okay, O. Effect of swelling on spatial inhomogeneity in poly(acrylamide) gels formed at various monomer concentrations. Polymer 2004, 45, 2567–2576. [Google Scholar] [CrossRef]
- Bastide, J.; Leibler, L.; Prost, J. Scattering by deformed swollen gels: butterfly isointensity patterns. Macromolecules 1990, 23, 1821–1825. [Google Scholar] [CrossRef]
- Onuki, A. Scattering from deformed swollen gels with heterogeneities. J. Phys. II 1992, 2, 45–61. [Google Scholar] [CrossRef][Green Version]
- Panyukov, S.V. Microscopic theory of anisotropic scattering by deformed polymer networks. Sov. Phys. JETP 1992, 75, 347–352. [Google Scholar]
- Koizumi, S.; Monkenbusch, M.; Richter, D.; Schwahn, D.; Farago, B. Concentration fluctuations in polymer gel investigated by neutron scattering: Static inhomogeneity in swollen gel. J. Chem. 2004, 121, 12721. [Google Scholar] [CrossRef] [PubMed]
- Norisuye, T.; Masui, N.; Kida, Y.; Ikuta, D.; Kokufuta, E.; Ito, K.; Panyukov, S.; Shibayama, M. Small angle neutron scattering studies on structural inhomogeneities in polymer gels: Irradiation cross-linked gels vs chemically cross-linked gels. Polymer 2002, 43, 5289–5297. [Google Scholar] [CrossRef]
- Mendes, E.; Schosseler, F.; Isel, F.; Boué, F.; Bastide, J.; Candau, S.J. A SANS Study of Uniaxially Elongated Polyelectrolyte Gels. Europhys. Lett. (EPL) 1995, 32, 273–278. [Google Scholar] [CrossRef]
- Shibayama, M.; Kawakubo, K.; Ikkai, F.; Imai, M. Small-Angle Neutron Scattering Study on Charged Gels in Deformed State. Macromolecules 1998, 31, 2586–2592. [Google Scholar] [CrossRef]
- Rabin, Y.; Panyukov, S. Scattering Profiles of Charged Gels: Frozen Inhomogeneities, Thermal Fluctuations, and Microphase Separation. Macromolecules 1997, 30, 301–312. [Google Scholar] [CrossRef]
- Shibayama, M.; Kawakubo, K.; Norisuye, T. Comparison of the Experimental and Theoretical Structure Factors of Temperature Sensitive Polymer Gels. Macromolecules 1998, 31, 1608–1614. [Google Scholar] [CrossRef]
- Shibayama, M.; Ikkai, F.; Shiwa, Y.; Rabin, Y. Effect of Degree of Cross-linking on Spatial Inhomogeneity in Charged Gels. I. Theoretical Predictions and Light Scattering Study. J. Chem. Phys. 1997, 107, 5227–5235. [Google Scholar] [CrossRef]
- Ikkai, F.; Shibayama, M.; Han, C.C. Effect of Degree of Cross-Linking on Spatial Inhomogeneity in Charged Gels. 2. Small-Angle Neutron Scattering Study. Macromolecules 1998, 31, 3275–3281. [Google Scholar] [CrossRef]
- Panyukov, S.V. Scaling theory of high elasticity. Sov. Phys. JETP 1990, 71, 372–379. [Google Scholar]
- de Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Panyukov, S.; Rabin, Y. Cross-Linking Patterns and Their Images in Swollen and Deformed Gels. Macromolecules 2015, 48, 7378–7381. [Google Scholar] [CrossRef]
- Bastide, J.; Leibler, L. Large-scale heterogeneities in randomly cross-linked networks. Macromolecules 1988, 21, 2647–2649. [Google Scholar] [CrossRef]
- Wang, S.; Panyukov, S.; Rubinstein, M.; Craig, S.L. Quantitative Adjustment to the Molecular Energy Parameter in the Lake–Thomas Theory of Polymer Fracture Energy. Macromolecules 2019, 52, 2772–2777. [Google Scholar] [CrossRef]
- Panyukov, S.; Rubinstein, M. Stress-Induced Ordering in Microphase-Separated Multicomponent Networks. Macromolecules 1996, 29, 8220–8230. [Google Scholar] [CrossRef]




© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panyukov, S. Theory of Flexible Polymer Networks: Elasticity and Heterogeneities. Polymers 2020, 12, 767. https://doi.org/10.3390/polym12040767
Panyukov S. Theory of Flexible Polymer Networks: Elasticity and Heterogeneities. Polymers. 2020; 12(4):767. https://doi.org/10.3390/polym12040767
Chicago/Turabian StylePanyukov, Sergey. 2020. "Theory of Flexible Polymer Networks: Elasticity and Heterogeneities" Polymers 12, no. 4: 767. https://doi.org/10.3390/polym12040767
APA StylePanyukov, S. (2020). Theory of Flexible Polymer Networks: Elasticity and Heterogeneities. Polymers, 12(4), 767. https://doi.org/10.3390/polym12040767