Phase Behavior of Gradient Copolymer Melts with Different Gradient Strengths Revealed by Mesoscale Simulations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mesoscopic Modelling
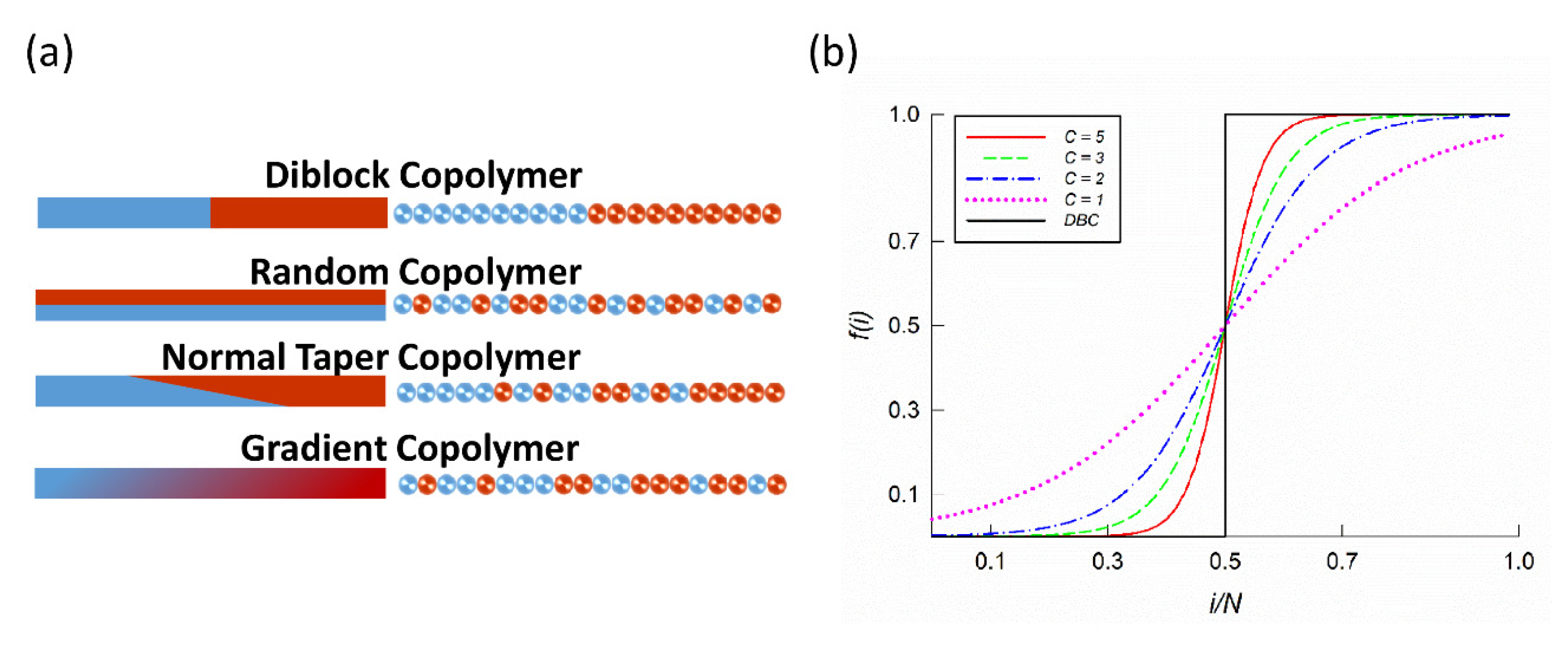
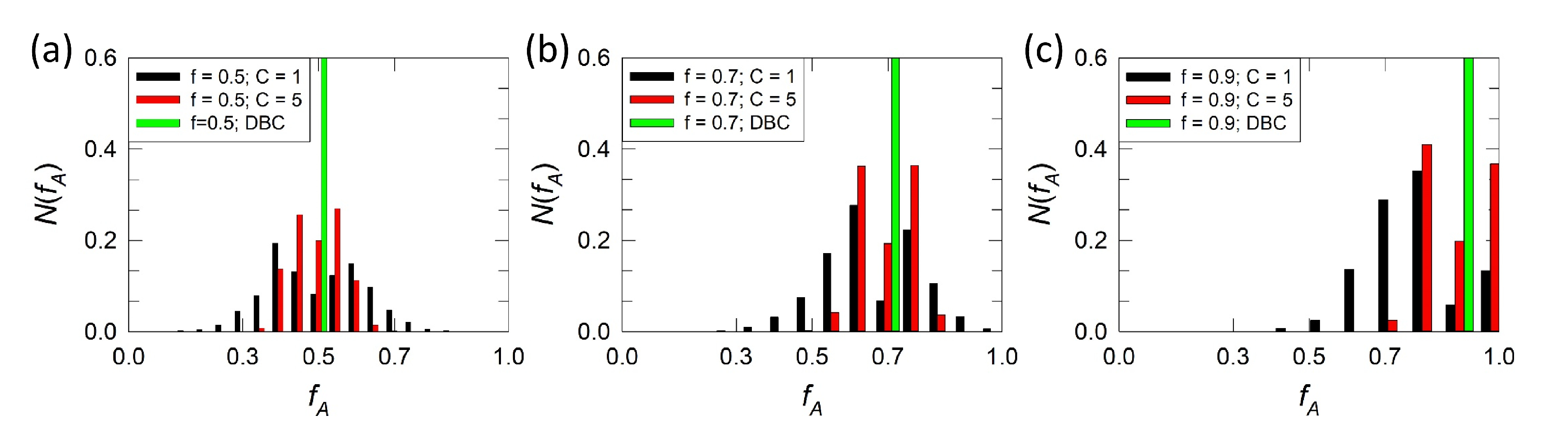
2.1. Gradient Copolymer Melts with Polydisperse Monomer Sequence
2.2. Dissipative Particle Dynamics and Gradient Copolymer Chain Model
2.3. Simulation Details
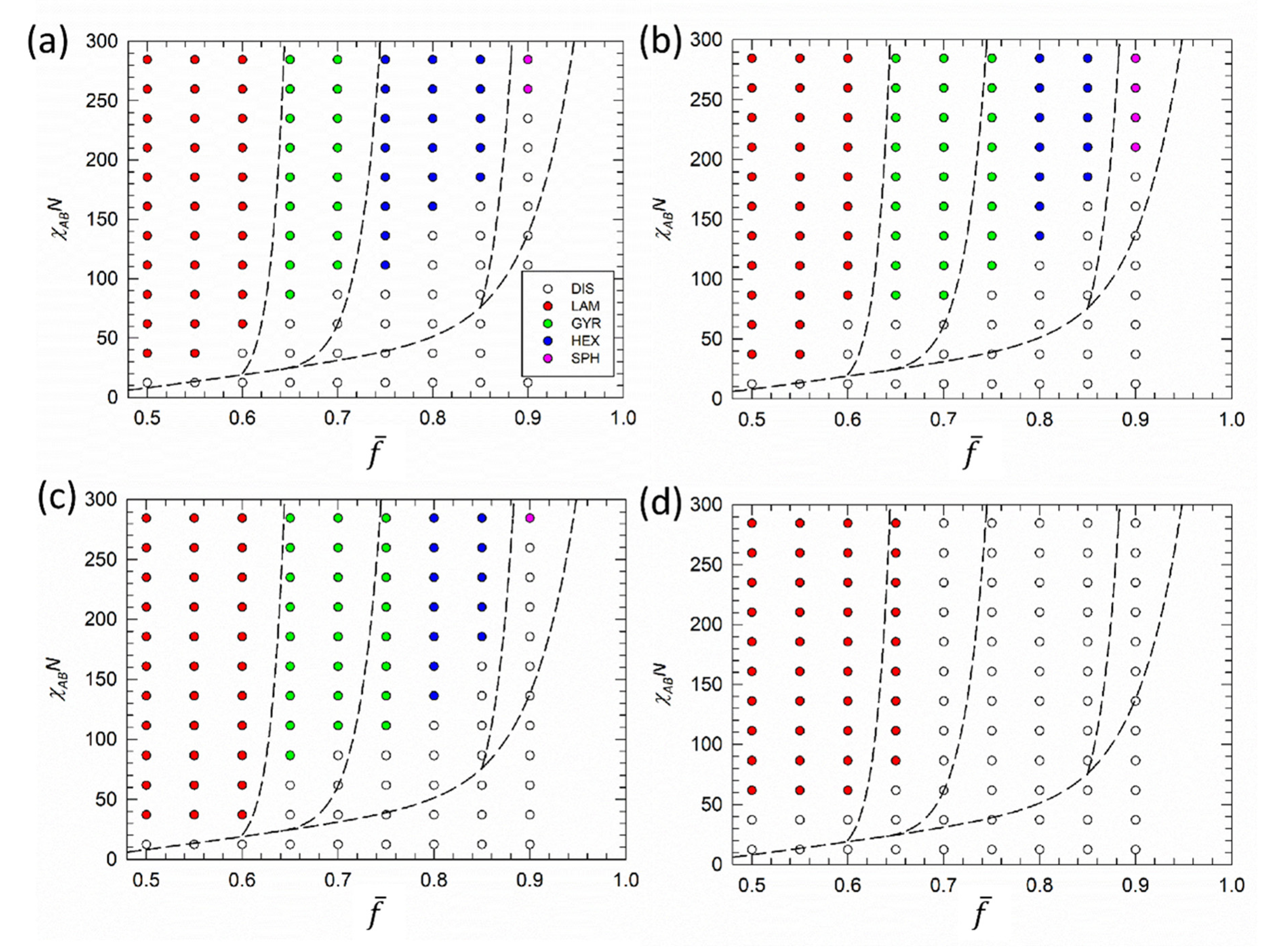
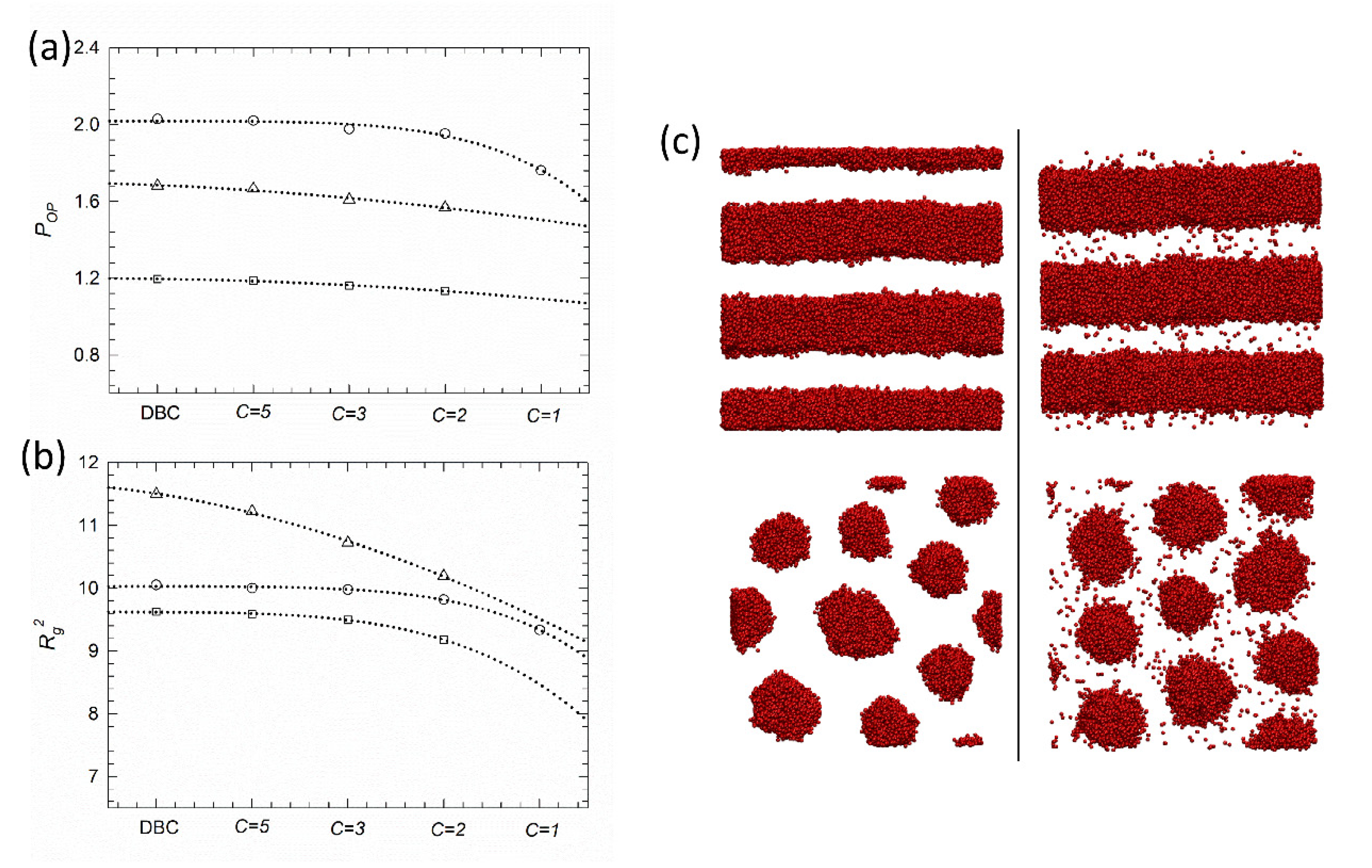
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bates, F.S.; Fredrickson, G.H. Block copolymers—Designer soft materials. Phys. Today 1999, 52, 32–38. [Google Scholar] [CrossRef]
- Feng, H.B.; Lu, X.Y.; Wang, W.Y.; Kang, N.G.; Mays, J.W. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chen, Y.; Zhang, Z.H.; Han, X. Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications. Polymers 2016, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Eggers, S.; Eckert, T.; Abetz, V. Double thermoresponsive block-random copolymers with adjustable phase transition temperatures: From block-like to gradient-like behavior. J. Polym. Sci. Pol. Chem. 2018, 56, 399–411. [Google Scholar] [CrossRef]
- Rabyk, M.; Destephen, A.; Lapp, A.; King, S.; Noirez, L.; Billon, L.; Hruby, M.; Borisov, O.; Stepanek, P.; Deniau, E. Interplay of Thermosensitivity and pH Sensitivity of Amphiphilic Block-Gradient Copolymers of Dimethylaminoethyl Acrylate and Styrene. Macromolecules 2018, 51, 5219–5233. [Google Scholar] [CrossRef]
- Cui, J.; Ma, Z.; Pan, L.; An, C.H.; Liu, J.; Zhou, Y.F.; Li, Y.S. Self-healable gradient copolymers. Mater. Chem. Front. 2019, 3, 464–471. [Google Scholar] [CrossRef]
- Zhao, R.B.; Shea, K.J. Gradient Methylidene-Ethylidene Copolymer via C1 Polymerization: An Ersatz Gradient Ethylene-Propylene Copolymer. Acs Macro Lett. 2015, 4, 584–587. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441–1706462. [Google Scholar] [CrossRef]
- Ogura, Y.; Takenaka, M.; Sawamoto, M.; Terashima, T. Fluorous Gradient Copolymers via in-Situ Transesterification of a Perfluoromethacrylate in Tandem Living Radical Polymerization: Precision Synthesis and Physical Properties. Macromolecules 2018, 51, 864–871. [Google Scholar] [CrossRef]
- Posel, Z.; Svoboda, M.; Limpouchova, Z.; Lisal, M.; Prochazka, K. Adsorption of amphiphilic graft copolymers in solvents selective for the grafts on a lyophobic surface: A coarse-grained simulation study. Phys. Chem. Chem. Phys. 2018, 20, 6533–6547. [Google Scholar]
- Wang, W.Y.; Lu, W.; Goodwin, A.; Wang, H.Q.; Yin, P.C.; Kang, N.G.; Hong, K.L.; Mays, J.W. Recent advances in thermoplastic elastomers from living polymerizations: Macromolecular architectures and supramolecular chemistry. Prog. Polym. Sci. 2019, 95, 1–31. [Google Scholar] [CrossRef]
- Sigle, J.L.; Clough, A.; Zhou, J.; White, J.L. Controlling Macroscopic Properties by Tailoring Nanoscopic Interfaces in Tapered Copolymers. Macromolecules 2015, 48, 5714–5722. [Google Scholar] [CrossRef]
- Hodrokoukes, P.; Floudas, G.; Pispas, S.; Hadjichristidis, N. Microphase separation in normal and inverse tapered block copolymers of polystyrene and polyisoprene. 1. Phase state. Macromolecules 2001, 34, 650–657. [Google Scholar] [CrossRef]
- Alam, M.M.; Jack, K.S.; Hill, D.J.T.; Whittaker, A.K.; Peng, H. Gradient copolymers—Preparation, properties and practice. Eur. Polym. J. 2019, 116, 394–414. [Google Scholar] [CrossRef]
- Kim, J.; Gray, M.K.; Zhou, H.Y.; Nguyen, S.T.; Torkelson, J.M. Polymer blend compatibilization by gradient copolymer addition during melt processing: Stabilization of dispersed phase to static coarsening. Macromolecules 2005, 38, 1037–1040. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Torkelson, J.M. Achievement of quasi-nano structured polymer blends by solid-state shear pulverization and compatibilization by gradient copolymer addition. Polymer 2006, 47, 6773–6781. [Google Scholar] [CrossRef]
- Mok, M.M.; Kim, J.; Torkelson, J.M. Gradient copolymers with broad glass transition temperature regions: Design of purely interphase compositions for damping applications. J. Polym. Sci. Pol. Phys. 2008, 46, 48–58. [Google Scholar] [CrossRef]
- Aksimentiev, A.; Holyst, R. Phase behavior of gradient copolymers. J. Chem. Phys. 1999, 111, 2329–2339. [Google Scholar] [CrossRef]
- Lefebvre, M.D.; de la Cruz, M.O.; Shull, K.R. Phase segregation in gradient copolymer melts. Macromolecules 2004, 37, 1118–1123. [Google Scholar] [CrossRef]
- Jiang, R.; Jin, Q.H.; Li, B.H.; Ding, D.T.; Wickham, R.A.; Shi, A.C. Phase behavior of gradient copolymers. Macromolecules 2008, 41, 5457–5465. [Google Scholar] [CrossRef]
- Tito, N.B.; Milner, S.T.; Lipson, J.E.G. Self-Assembly of Lamellar Microphases in Linear Gradient Copolymer Melts. Macromolecules 2010, 43, 10612–10620. [Google Scholar] [CrossRef]
- Mok, M.M.; Pujari, S.; Burghardt, W.R.; Dettmer, C.M.; Nguyen, S.T.; Ellison, C.J.; Torkelson, J.M. Microphase separation and shear alignment of gradient copolymers: Melt rheology and small-angle X-ray scattering analysis. Macromolecules 2008, 41, 5818–5829. [Google Scholar] [CrossRef]
- Mok, M.M.; Kim, J.; Wong, C.L.H.; Marrou, S.R.; Woo, D.J.; Dettmer, C.M.; Nguyen, S.T.; Ellison, C.J.; Shull, K.R.; Torkelson, J.M. Glass Transition Breadths and Composition Profiles of Weakly, Moderately, and Strongly Segregating Gradient Copolymers: Experimental Results and Calculations from Self-Consistent Mean-Field Theory. Macromolecules 2009, 42, 7863–7876. [Google Scholar] [CrossRef]
- Ganesan, V.; Kumar, N.A.; Pryamitsyn, V. Blockiness and Sequence Polydispersity Effects on the Phase Behavior and Interfacial Properties of Gradient Copolymers. Macromolecules 2012, 45, 6281–6297. [Google Scholar] [CrossRef]
- Jiang, R.; Wang, Z.; Yin, Y.H.; Li, B.H.; Shi, A.C. Effects of compositional polydispersity on gradient copolymer melts. J. Chem. Phys. 2013, 138, 074906. [Google Scholar] [CrossRef] [PubMed]
- Pandav, G.; Pryamitsyn, V.; Gallow, K.C.; Loo, Y.L.; Genzer, J.; Ganesan, V. Phase behavior of gradient copolymer solutions: A Monte Carlo simulation study. Soft Matter 2012, 8, 6471–6482. [Google Scholar] [CrossRef]
- Pakula, T.; Matyjaszewski, K. Copolymers with controlled distribution of comonomers along the chain. 1. Structure, thermodynamics and dynamic properties of gradient copolymers. Computer simulation. Macromol. Theor. Simul. 1996, 5, 987–1006. [Google Scholar] [CrossRef]
- Sun, D.C.; Guo, H.X. Monte Carlo Studies on the Interfacial Properties and Interfacial Structures of Ternary Symmetric Blends with Gradient Copolymers. J. Phys. Chem. B 2012, 116, 9512–9522. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.C.; Guo, H.X. Influence of compositional gradient on the phase behavior of ternary symmetric homopolymer-copolymer blends: A Monte Carlo study. Polymer 2011, 52, 5922–5932. [Google Scholar] [CrossRef]
- Fredrickson, G.H.; Milner, S.T.; Leibler, L. Multicritical Phenomena and Microphase Ordering in Random Block Copolymer Melts. Macromolecules 1992, 25, 6341–6354. [Google Scholar] [CrossRef]
- Skvor, J.; Posel, Z. Simulation Aspects of Lamellar Morphology: Incommensurability Effect. Macromol. Theor. Simul. 2015, 24, 141–151. [Google Scholar] [CrossRef]
- Posel, Z.; Rousseau, B.; Lisal, M. Scaling behaviour of different polymer models in dissipative particle dynamics of unentangled melts. Mol. Simulat. 2014, 40, 1274–1289. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kroger, M.; Clarke, N. Modeling of Entangled Polymer Diffusion in Melts and Nanocomposites: A Review. Polymers 2019, 11, 876. [Google Scholar] [CrossRef]
- Posel, Z.; Posocco, P. Tuning the Properties of Nanogel Surfaces by Grafting Charged Alkylamine Brushes. Nanomaterials 2019, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Guskova, O.A.; Seidel, C. Mesoscopic Simulations of Morphological Transitions of Stimuli-Responsive Diblock Copolymer Brushes. Macromolecules 2011, 44, 671–682. [Google Scholar] [CrossRef]
- Posocco, P.; Hassan, Y.M.; Barandiaran, I.; Kortaberria, G.; Pricl, S.; Fermeglia, M. Combined Mesoscale/Experimental Study of Selective Placement of Magnetic Nanoparticles in Diblock Copolymer Films via Solvent Vapor Annealing. J. Phys. Chem. C 2016, 120, 7403–7411. [Google Scholar] [CrossRef]
- Posel, Z.; Posocco, P.; Fermeglia, M.; Lisal, M.; Pricl, S. Modeling hierarchically structured nanoparticle/diblock copolymer systems. Soft Matter 2013, 9, 2936–2946. [Google Scholar] [CrossRef]
- Posocco, P.; Posel, Z.; Fermeglia, M.; Lisal, M.; Pricl, S. A molecular simulation approach to the prediction of the morphology of self-assembled nanoparticles in diblock copolymers. J. Mater. Chem. 2010, 20, 10511–10520. [Google Scholar] [CrossRef]
- Posel, Z.; Posocco, P.; Lisal, M.; Fermeglia, M.; Pricl, S. Highly grafted polystyrene/polyvinylpyridine polymer gold nanoparticles in a good solvent: Effects of chain length and composition. Soft Matter 2016, 12, 3600–3611. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Kroger, M. Modeling of Polymer Structure and Conformations in Polymer Nanocomposites from Atomistic to Mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Posel, Z.; Svoboda, M.; Colina, C.M.; Lisal, M. Flow and aggregation of rod-like proteins in slit and cylindrical pores coated with polymer brushes: An insight from dissipative particle dynamics. Soft Matter 2017, 13, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Espanol, P.; Warren, P.B. Perspective: Dissipative particle dynamics. J. Chem. Phys. 2017, 146, 150901. [Google Scholar] [CrossRef]
- Groot, R.D.; Madden, T.J. Dynamic simulation of diblock copolymer microphase separation. J. Chem. Phys. 1998, 108, 8713–8724. [Google Scholar] [CrossRef]
- Martinez-Veracoechea, F.J.; Escobedo, F.A. Simulation of the gyroid phase in off-lattice models of pure diblock copolymer melts. J. Chem. Phys. 2006, 125, 104907. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Plimpton, S.J. Accelerating dissipative particle dynamics simulations for soft matter systems. Comp. Mater. Sci 2015, 100, 173–180. [Google Scholar] [CrossRef]
- Beranek, P.; Posel, Z. Phase Behavior of Semiflexible-Flexible Diblock Copolymer Melt: Insight from Mesoscale Modeling. J. Nanosci. Nanotechno. 2016, 16, 7832–7835. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Kudryavtsev, Y.V.; Chertovich, A.V. Phase diagrams of block copolymer melts by dissipative particle dynamics simulations. J. Chem. Phys. 2013, 139, 224901. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Sides, S.W.; Hall, L.M. Phase Behavior of Tapered Diblock Copolymers from Self-Consistent Field Theory. Acs Macro Lett. 2013, 2, 1105–1109. [Google Scholar] [CrossRef]
- Brown, J.R.; Seo, Y.; Maula, T.A.D.; Hall, L.M. Fluids density functional theory and initializing molecular dynamics simulations of block copolymers. J. Chem. Phys. 2016, 144, 124904. [Google Scholar] [CrossRef]
- Brown, J.R.; Seo, Y.M.; Sides, S.W.; Hall, L.M. Unique Phase Behavior of Inverse Tapered Block Copolymers: Self Consistent Field Theory and Molecular Dynamics Simulations. Macromolecules 2017, 50, 5619–5626. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beránek, P.; Posocco, P.; Posel, Z. Phase Behavior of Gradient Copolymer Melts with Different Gradient Strengths Revealed by Mesoscale Simulations. Polymers 2020, 12, 2462. https://doi.org/10.3390/polym12112462
Beránek P, Posocco P, Posel Z. Phase Behavior of Gradient Copolymer Melts with Different Gradient Strengths Revealed by Mesoscale Simulations. Polymers. 2020; 12(11):2462. https://doi.org/10.3390/polym12112462
Chicago/Turabian StyleBeránek, Pavel, Paola Posocco, and Zbyšek Posel. 2020. "Phase Behavior of Gradient Copolymer Melts with Different Gradient Strengths Revealed by Mesoscale Simulations" Polymers 12, no. 11: 2462. https://doi.org/10.3390/polym12112462
APA StyleBeránek, P., Posocco, P., & Posel, Z. (2020). Phase Behavior of Gradient Copolymer Melts with Different Gradient Strengths Revealed by Mesoscale Simulations. Polymers, 12(11), 2462. https://doi.org/10.3390/polym12112462