Poly (1-butene-ran-ethylene) Monomodal Copolymers from Metallocene Catalysts: Structural and Morphological Differences with Increasing Ethylene Content



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Thermal Analysis
2.2. Wide Angle X-ray Diffraction
2.3. Small Angle X-ray Scattering
SAXS Data Analysis
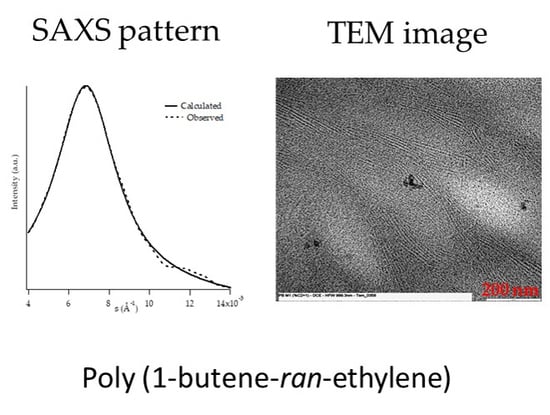
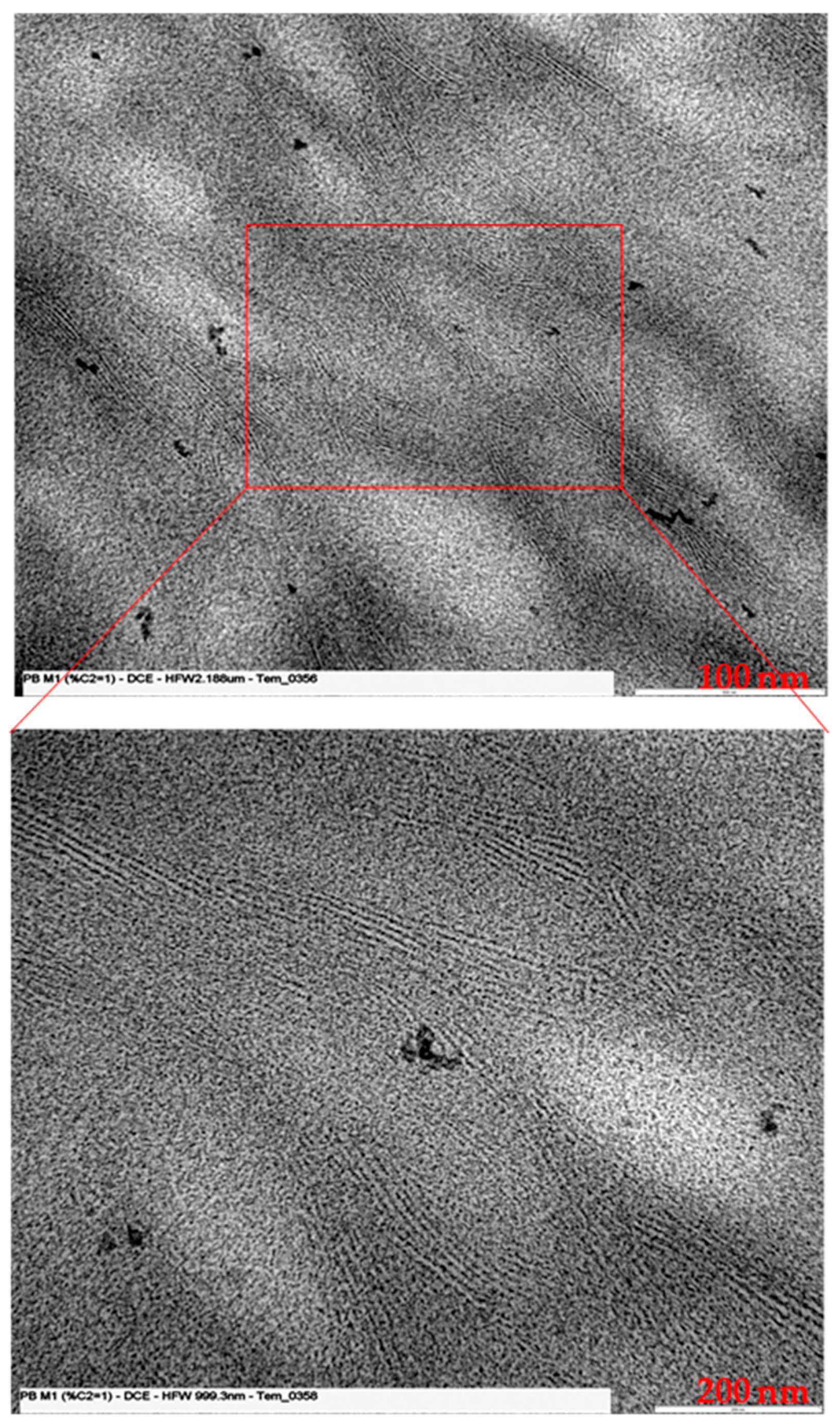
2.4. Transmission Electron Microscopy
3. Results and Discussion
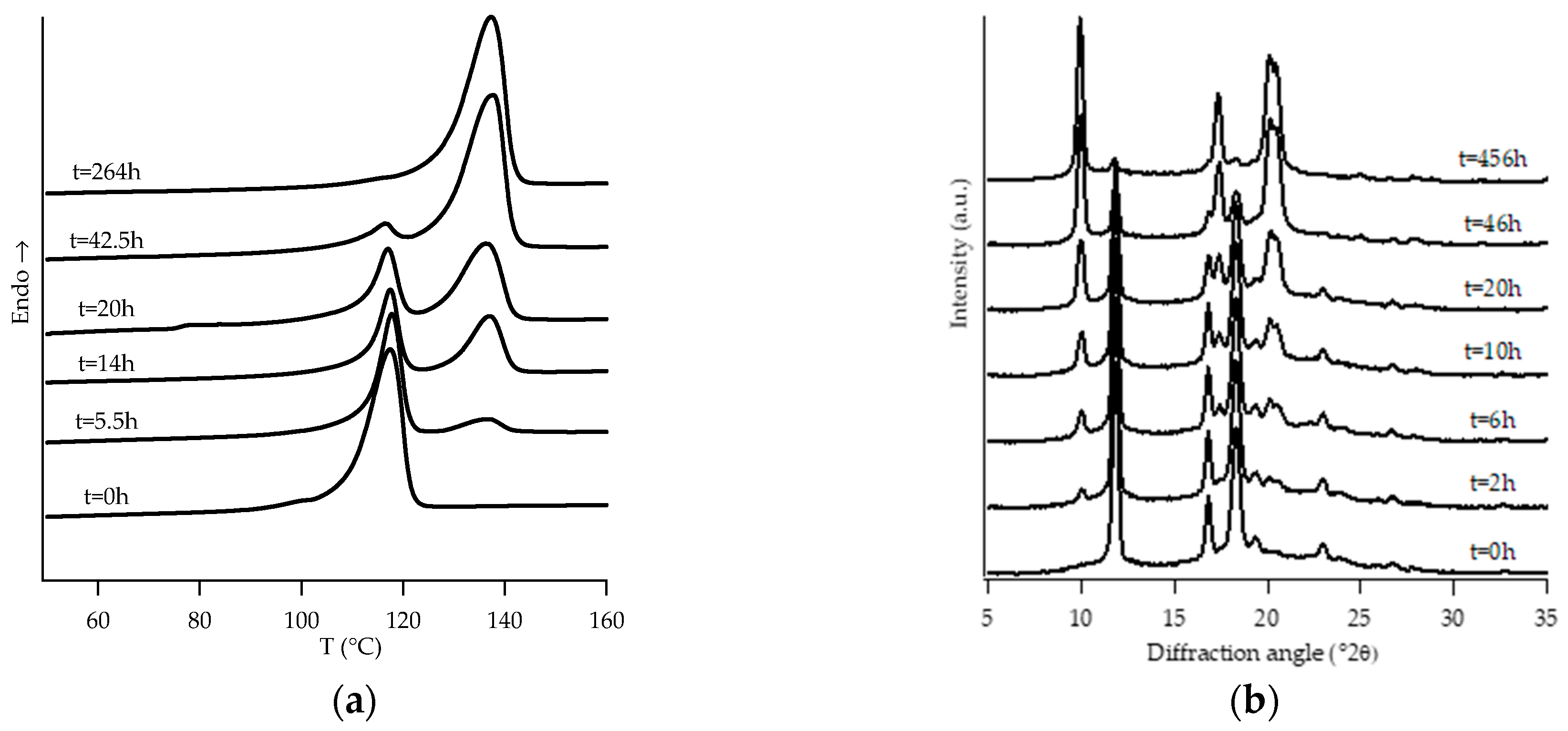
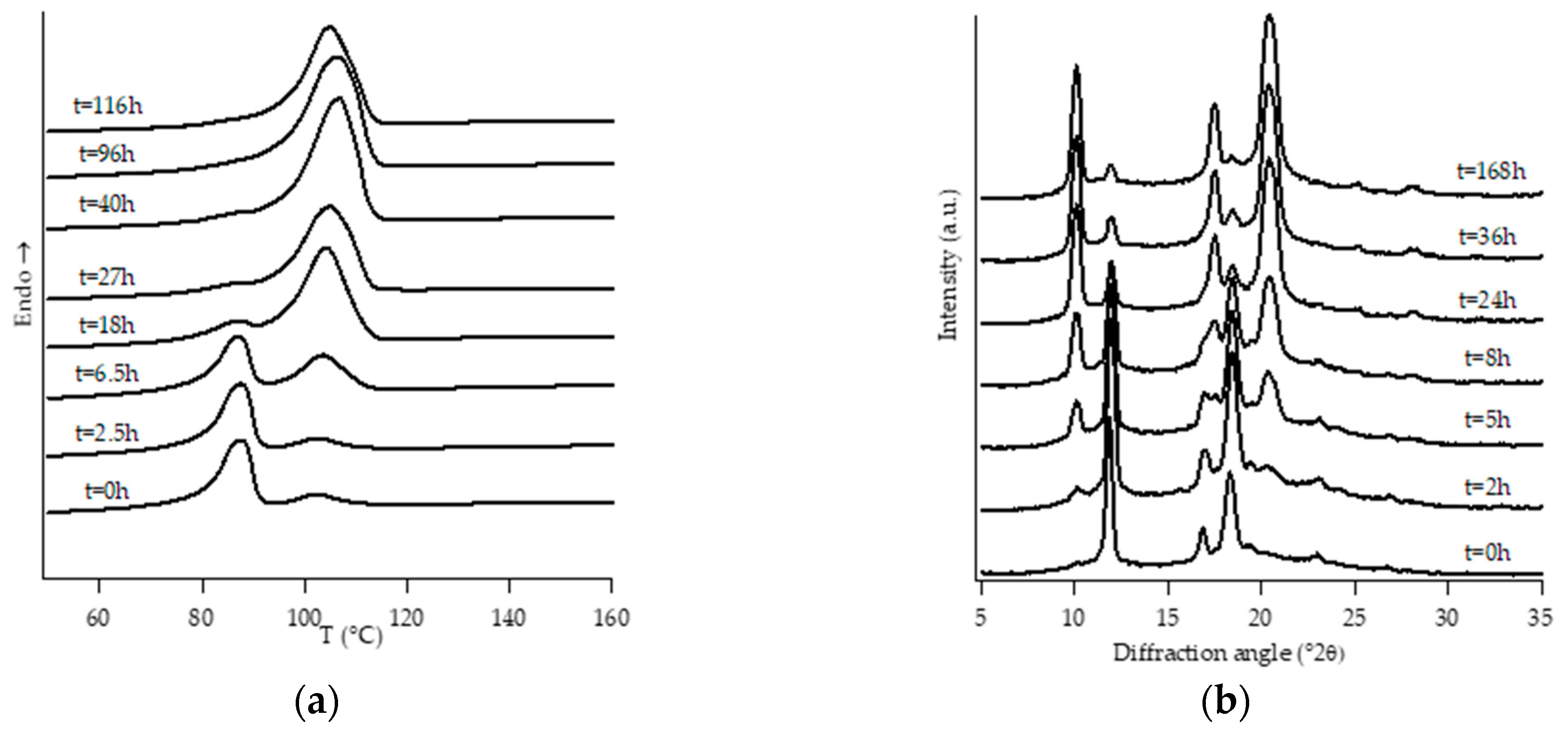
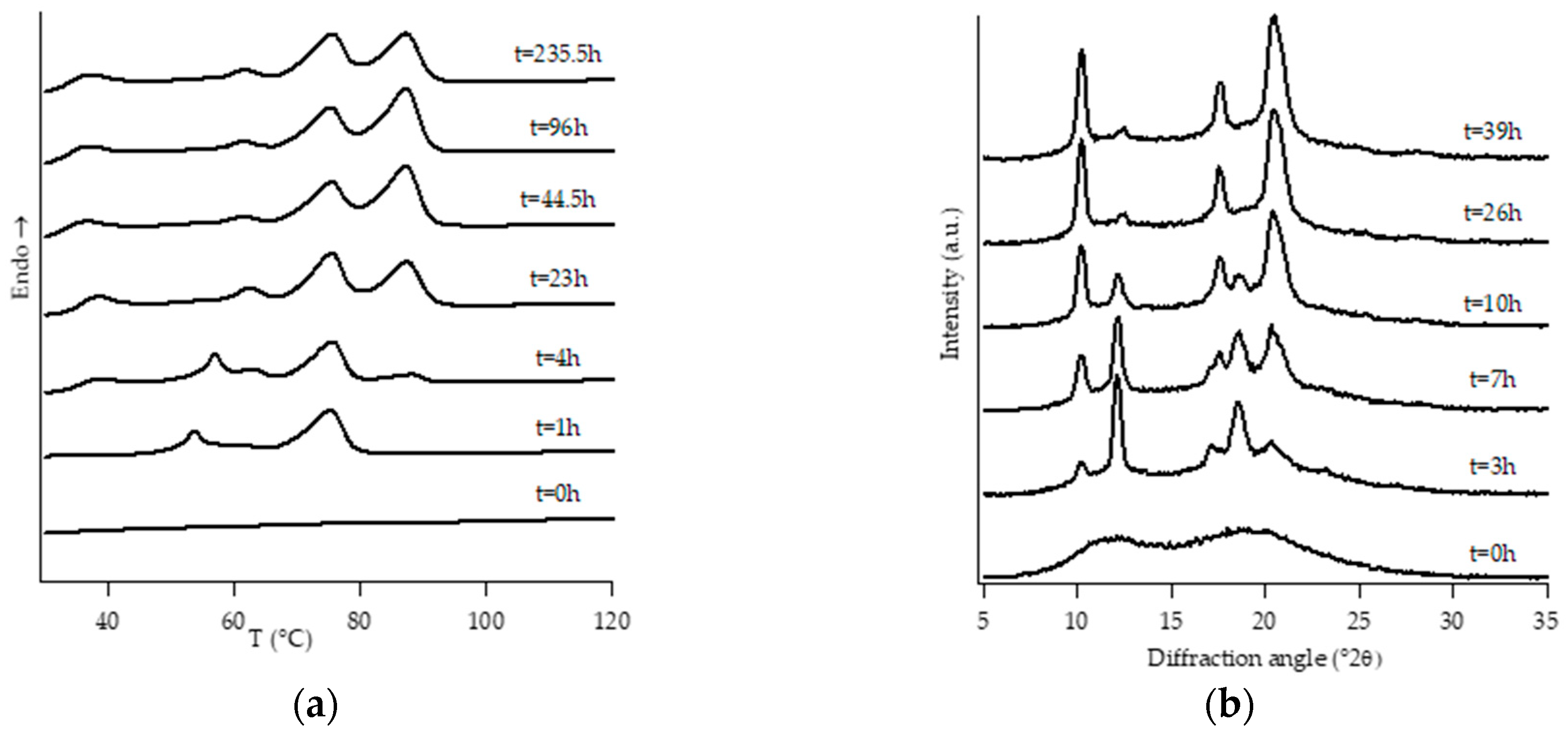
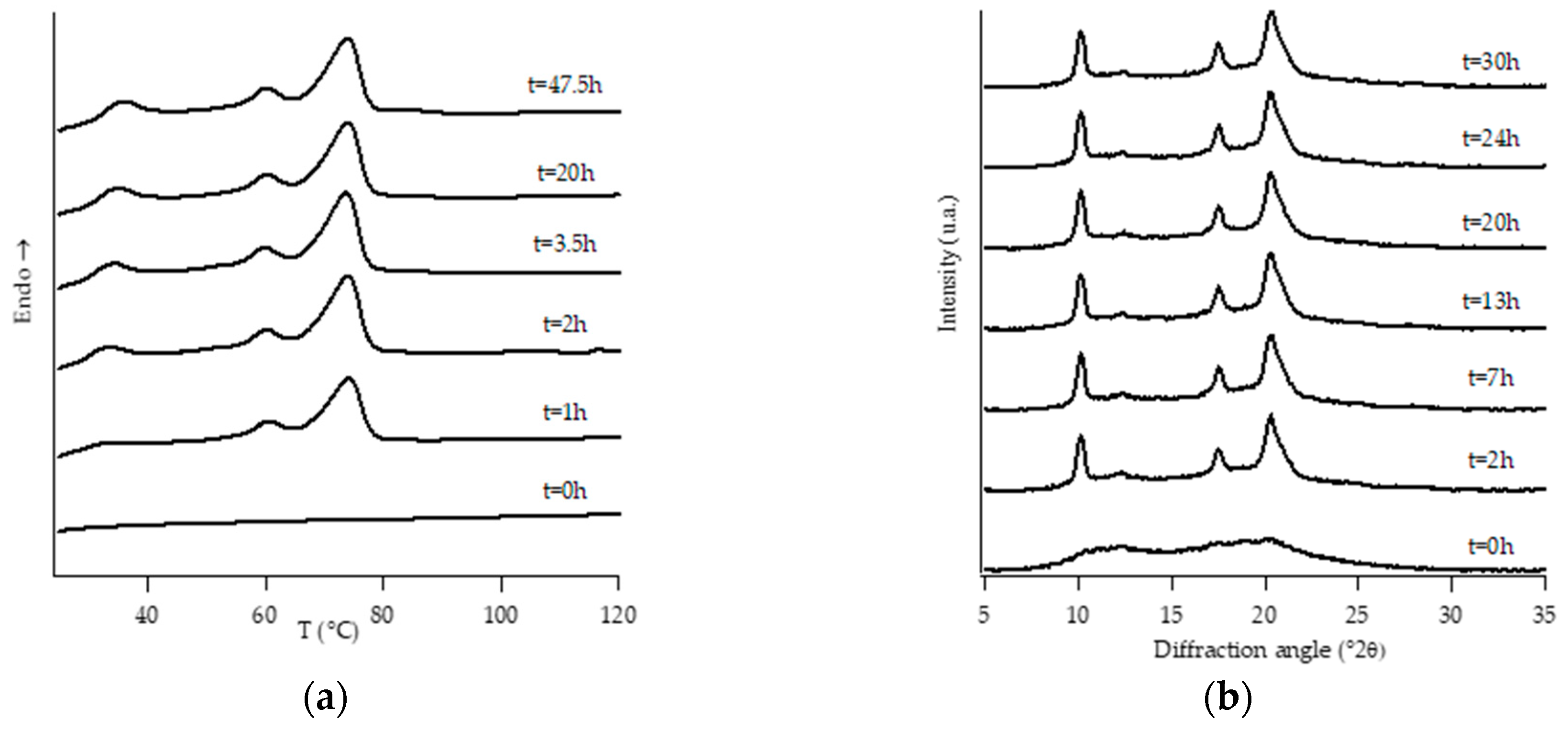
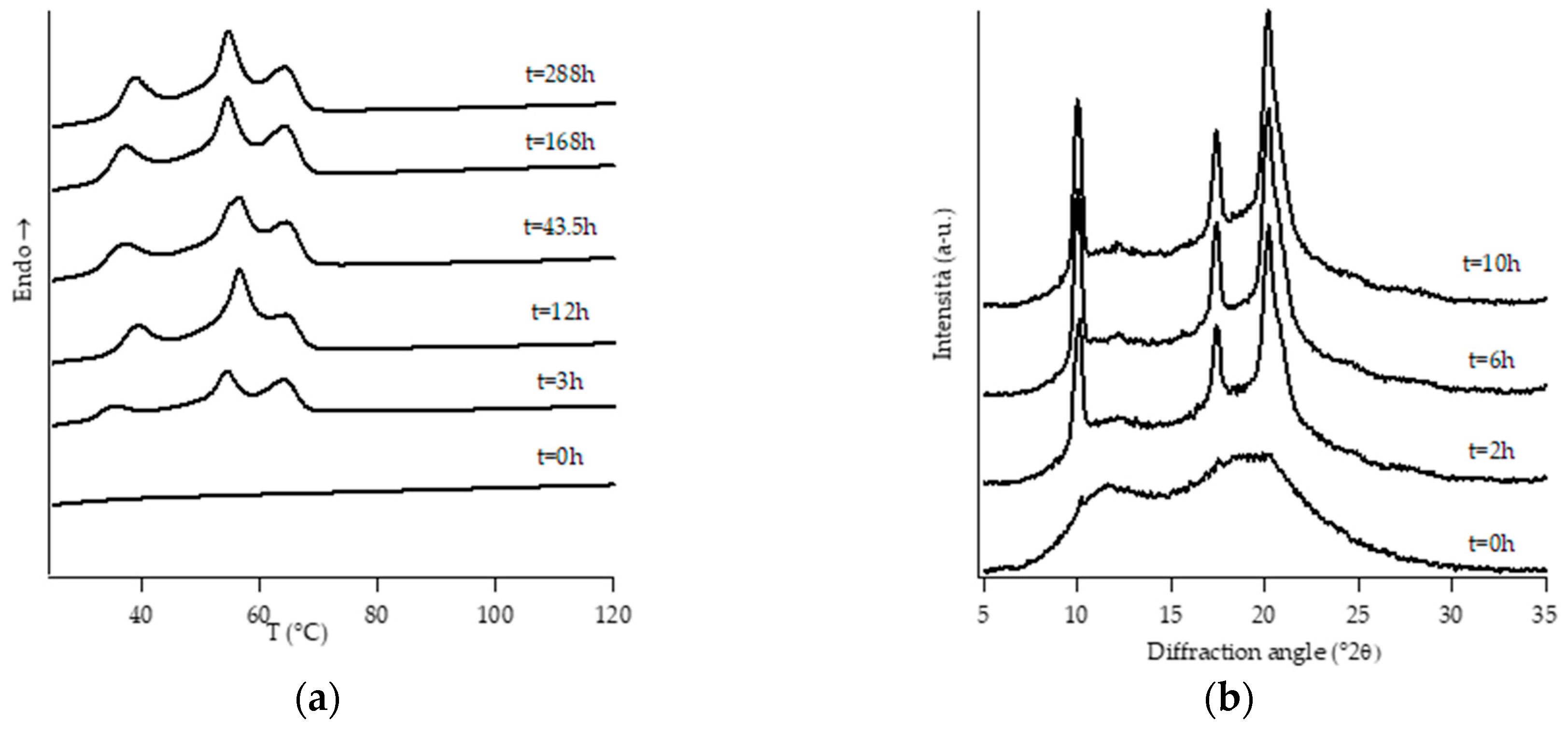
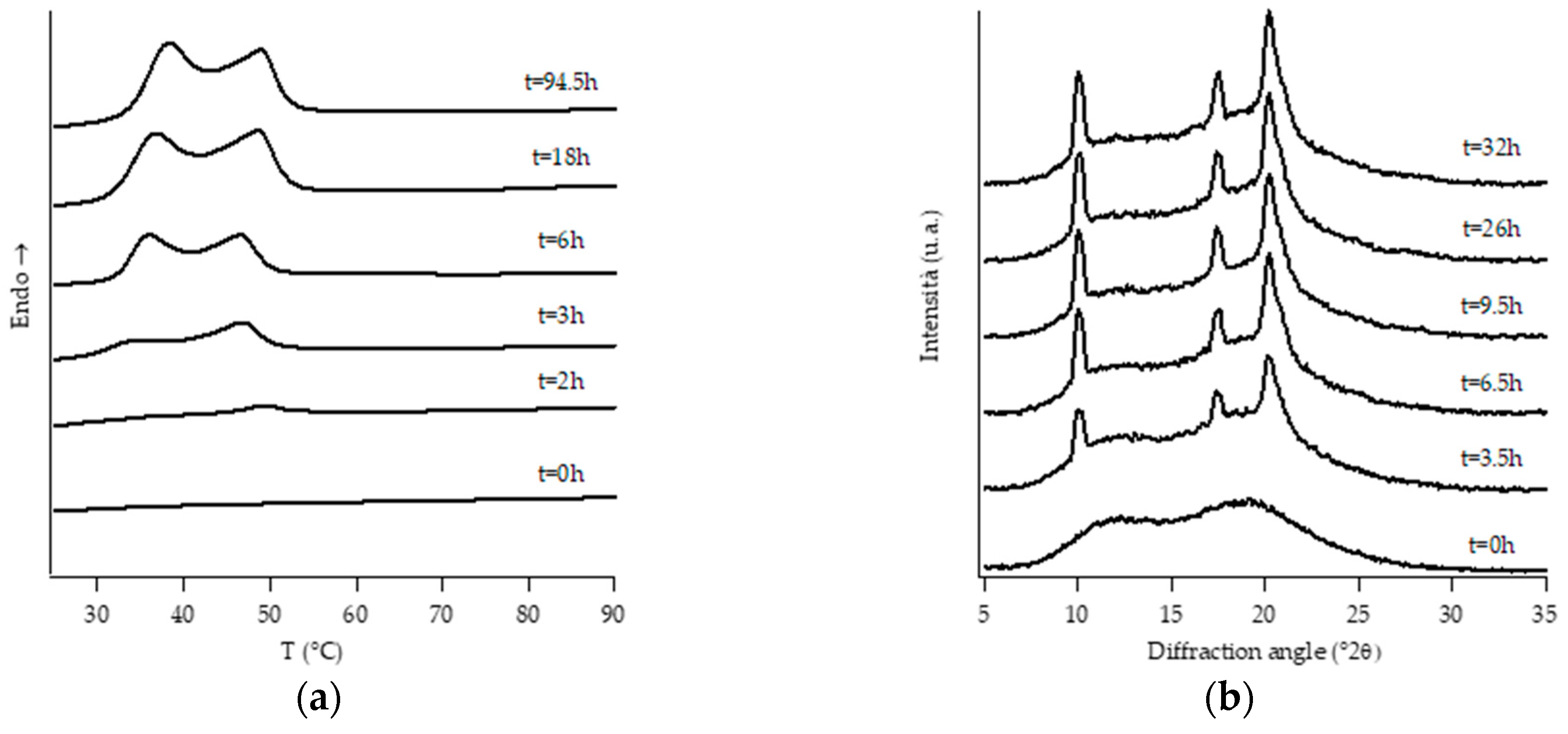
3.1. Structural Analysis: DTA and WAXD Measurements
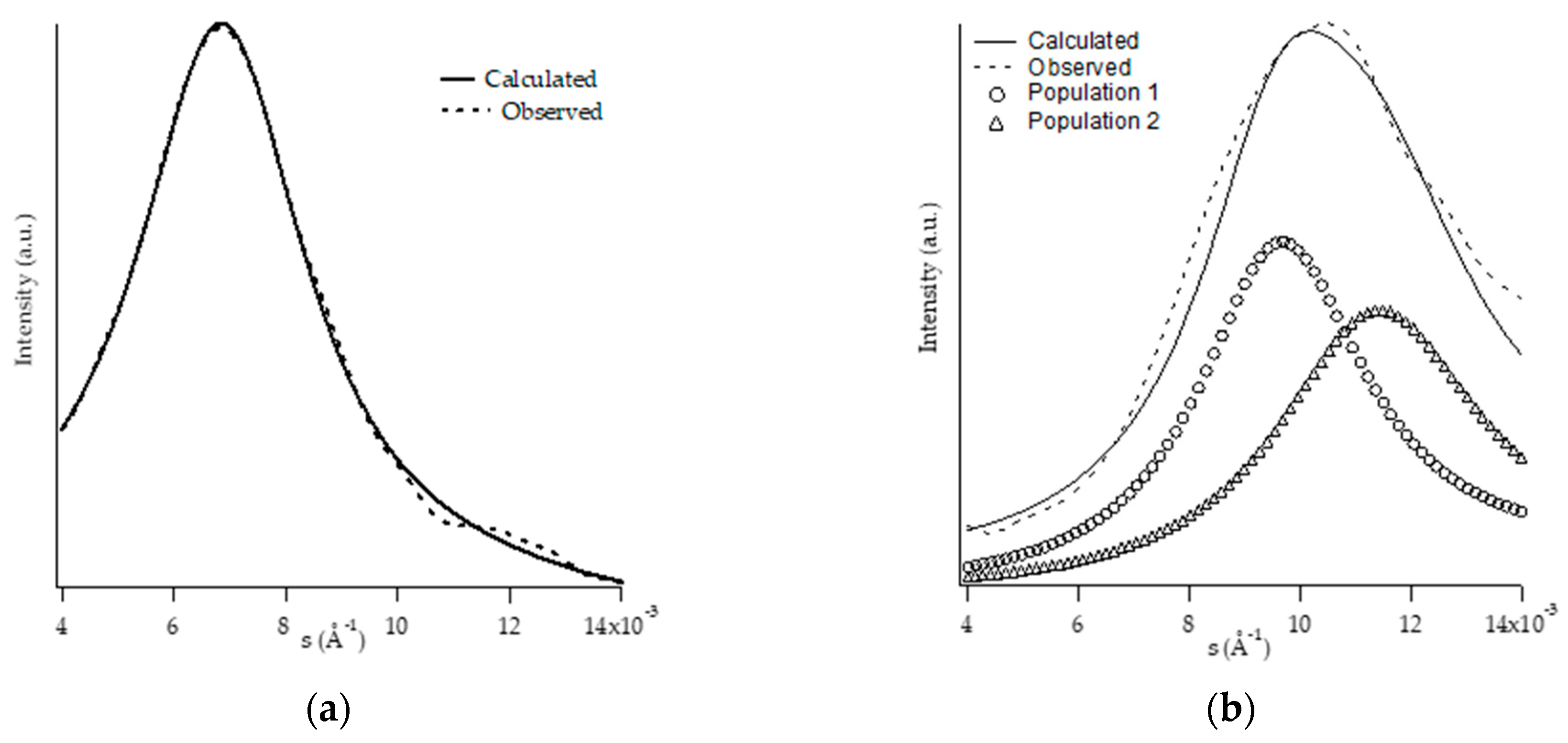
3.2. Morphological Analysis: SAXS and TEM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline High Polymers of alfa-olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Resconi, L. Metalloorganic polymerization catalysis as a tool to probe crystallization properties of polymers: The case of isotactic poly(1-butene). Angew. Chemie Int. Ed. 2009, 48, 9871–9874. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Holland, V. On transformations in isotactic polybutene-1. J. Polym. Sci. Part B Polym. Lett. 1964, 2, 519–521. [Google Scholar] [CrossRef]
- Goldbach, G.; Peitscher, G. Infrared investigations of the polymorphic modifications of polybutene-1. J. Polym. Sci. Part B Polym. Lett. 1968, 6, 783–788. [Google Scholar] [CrossRef]
- Natta, G.; Corradini, P.; Bassi, I.W. Crystal Structure of Isotactic Poly-alfa-butene. Nuovo Cimento Suppl. 1960, 15, 52–67. [Google Scholar] [CrossRef]
- Nakamura, K.; Aoike, T.; Usaka, K.; Kanamoto, T. Phase Transformation in Poly(1-butene) upon Drawing. Macromolecules 1999, 32, 4975–4982. [Google Scholar] [CrossRef]
- Armeniades, C.; Baer, E. Effect of Pressure on the Polymorphism of Melt Crystallized Polybutene-1. J. Macromol. Sci. Part B 1967, B1, 309–334. [Google Scholar] [CrossRef]
- Boor, J.; Youngman, E.A. Polymorphism in poly-1-butene: Apparent direct formation of modification I. Polym. Lett. 1964, 2, 903–907. [Google Scholar] [CrossRef]
- Marega, C.; Causin, V.; Marigo, A.; Saini, R.; Ferrara, G. Crystallization of a (1-butene)-Ethylene Copolymer in Phase I Directly from the Melt in Nanocomposites with Montmorillonite. J. Nanosci. Nanotechnol. 2010, 10, 3078–3084. [Google Scholar] [CrossRef]
- Nakafuku, C.; Miyaki, T. Effect of pressure on the melting and crystallization behaviour of isotactic polybutene-1. Polymer 1983, 24, 141–148. [Google Scholar] [CrossRef]
- Danusso, F.; Gianotti, G. Equilibrium melting temperature: Some experimental data with isotactic polypropylene, polybutene-1, and polypentene-1. Macromol. Chem. Phys. 1964, 80, 1–12. [Google Scholar] [CrossRef]
- Danusso, F.; Gianotti, G.; Polizzotti, G. Isotactic polybutene-1: Modification 3 and its transformations. Macromol. Chem. Phys. 1964, 80, 13–21. [Google Scholar] [CrossRef]
- Geacintov, C.; Schotland, R.S.; Miles, R.B. Phase transition of crystalline polybutene-1 in form III. J. Polym. Sci. Pol. Lett. 1963, 1, 587–591. [Google Scholar] [CrossRef]
- Geacintov, C.; Miles, R.S.; Schuurmans, H.J.L. On the form III transformation of polybutene-1. J. Polym. Sci. Part A-1 1966, 4, 431–433. [Google Scholar] [CrossRef]
- Luciani, L.; Seppälä, J.; Löfgren, B. Poly-1-butene: Its preparation, properties and challenges. Prog. Polym. Sci. 1988, 37–62. [Google Scholar] [CrossRef]
- Holland, V.F.; Miller, R.L. Isotactic polybutene-1 single crystals: Morphology. J. Appl. Phys. 1964, 35, 3241–3248. [Google Scholar] [CrossRef]
- Chau, K.W.; Geil, P.H.J. Solution history effects in polybutene-1. Macromol. Sci. Phys. 1984, B23, 115–142. [Google Scholar] [CrossRef]
- Turner-Jones, A. Cocrystallization in copolymers of α-olefins II—Butene-1 copolymers and polybutene type II/I crystal phase transition. Polymer 1966, 7, 23–59. [Google Scholar] [CrossRef]
- Gianotti, G.; Capizzi, A. Butene-1/propylene copolymers. Influence of the comonomerie units on polymorphism. Makromol. Chem. 1969, 124, 152–159. [Google Scholar] [CrossRef]
- Azzurri, F.; Alfonso, G.; Gomez, M.; Marti, M.; Ellis, G.; Marco, C. Polymorphic transformation in isotactic1-butene/ethylene copolymers. Macromolecules 2004, 37, 3755–3762. [Google Scholar] [CrossRef]
- Azzurri, F.; Gomez, M.; Alfonso, G.; Ellis, G.; Marco, C. Time-resolved SAXS/WAXS studies of the polymorphic transformation of 1-butene/ethylene copolymers. J. Macromol. Sci. Phys. 2004, B43, 177–189. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; de Ballesteros, O.; Esposito, F.; Laguzza, D.; Di Girolamo, R.; Resconi, L. Crystallization Properties and Polymorphic Behavior of Isotactic Poly(1-Butene) from Metallocene Catalysts: The Crystallization of Form I from the Melt. Macromolecules 2009, 42, 8286–8297. [Google Scholar] [CrossRef]
- De Rosa, C.; de Ballesteros, O.; Auriemma, F.; Di Girolamo, R.; Scarica, C.; Giusto, G.; Esposito, S.; Guidotti, S.; Camurati, I. Polymorphic Behavior and Mechanical Properties of Isotactic 1-Butene-Ethylene Copolymers from Metallocene Catalysts. Macromolecules 2014, 47, 4317–4329. [Google Scholar] [CrossRef]
- Resconi, L.; Camurati, I.; Malizia, F. Metallocene catalysts for 1-butene polymerization. Macromol. Chem. Phys. 2006, 207, 2257–2279. [Google Scholar] [CrossRef]
- Marega, C.; Spataro, S.; Fassone, E.; Camurati, I.; Marigo, A. Self-Welding 1-Butene/Ethylene Copolymers from Metallocene Catalysts: Structure, Morphology, and Mechanical Properties. J. Appl. Polym. Sci. 2014, 131, 40119. [Google Scholar] [CrossRef]
- Davis, K. Hot Melt Adhesive Compositions Including Olefinic Polymer Blends and Articles Including the Same. US Patent 8,623,480 B2m, 7 January 2014. [Google Scholar]
- Hortelano, S.V. Hot Melt Adhesive. EU Patent 2907857 A2, 19 August 2015. [Google Scholar]
- Hindele, A.M.; Johnson, D.J. The resolution of multipeak data in fibre science. J. Phys. D Appl. Phys. 1971, 4, 259–263. [Google Scholar] [CrossRef]
- Vonk, C.G.; Pijpers, A.P. An x-ray diffraction study of nonlinear polyethylene. I. Room-temperature observations. J. Polym. Sci. Part B Polym. Phys. 1985, 23, 2517–2537. [Google Scholar] [CrossRef]
- Vonk, C.G. Investigation of non-ideal two-phase polymer structures by small-angle X-ray scattering. J. Appl. Cryst. 1973, 6, 81–86. [Google Scholar] [CrossRef]
- Vonk, C.G. A procedure for desmearing X-ray small-angle scattering curves. J. Appl. Cryst. 1971, 4, 340–342. [Google Scholar] [CrossRef]
- Marega, C.; Marigo, A.; Cingano, G.; Zannetti, R.; Paganetto, G. Small-angle X-ray scattering from high-density polyethylene: Lamellar thickness distributions. Polymer 1996, 37, 5549–5557. [Google Scholar] [CrossRef]
- Hosemann, R.; Bagchi, S.N. Direct Analysis of Diffraction by Matter; North Holland: Amsterdam, The Netherlands, 1962. [Google Scholar]
- Hatakeyama, T.; Quinn, F.X. Thermal Analysis; John Wiley & Sons: Tokyo, Japan, 1995. [Google Scholar]
- Turner Jones, A. Polybutene-1–type II crystalline form. J. Polym. Sci Part B 1963, 1, 455–456. [Google Scholar] [CrossRef]
- Li, L.; Liu, T.; Zhao, L.; Yuan, W. CO2-Induced Phase Transition of Isotactic Poly-1-butene with Form III upon Heating. Macromolecules 2011, 44, 4836–4844. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Lattice | Helix | a (nm) | b (nm) | c (nm) | |
|---|---|---|---|---|---|
| I/I’ | Hexagonal | 3/1 | 1.77 | 1.77 | 0.65 |
| II/II’ | Tetragonal | 11/3 | 1.46 | 1.46 | 2.12 |
| III | Orthorhombic | 4/1 | 1.25 | 0.89 | 0.76 |
| PB0 | PB1 | PB2.4 | PB2.9 | PB3.8 | PB6.8 | ||
|---|---|---|---|---|---|---|---|
| C2 | %wt | 0 | 0.8 | 2.4 | 2.9 | 3.8 | 6.8 |
| MFI | g/10’ | 40–60 | 40–60 | 40–60 | 40–60 | 40–60 | 40–60 |
| * | 126151 | 126724 | 143923 | 131228 | 135570 | 141250 | |
| * | 61755 | 62100 | 66768 | 61506 | 59634 | 65949 | |
| * | 2.0 | 2.0 | 2.2 | 2.1 | 2.3 | 2.1 | |
| Density | Kg/dm3 | 0.9066 | 0.9028 | 0.8978 | 0.8930 | 0.8903 | 0.8817 |
| Tg ** | °C | −11 | −12 | −21 | −22 | −22 | −27 |
| PB0 | PB1 | PB2.4 | PB2.9 | PB3.8 | PB6.8 | ||
|---|---|---|---|---|---|---|---|
| TmII | °C | 101.3 | 87.3 | 53.5 | - | - | - |
| TmI | °C | 117.3 | 104.6 | 87.1 | 73.9 | 64.1 | 48.9 |
| TmI’ | °C | - | - | 37.2 | 36.1 | 38.8 | 38.3 |
| ΔHI | J/g | 72.3 | 63.3 | 47.7 | 46.7 | 40.0 | 25.5 |
| CDSC | % | 58 | 51 | 37 | 36 | 31 | 20 |
| CWAXD | % | 52 | 45 | 33 | 31 | 23 | 17 |
| t ½ | h | 27.4 | 11.3 | 8.0 | - | - | - |
| Sample | N | Y1 | Z1 | L1 | Y2 | Z2 | L2 | ΦSAXS |
|---|---|---|---|---|---|---|---|---|
| (nm) | (nm) | (nm) | (nm) | (nm) | (nm) | (%) | ||
| PB0 | ∞ | 7.6 | 6.0 | 13.6 | - | - | - | 56 |
| PB1 | ∞ | 5.3 | 7.3 | 12.6 | - | - | - | 42 |
| PB2.4 | ∞ | 3.2 | 6.5 | 9.7 | 2.6 | 5.5 | 8.1 | 33 |
| PB2.9 | ∞ | 2.8 | 7.1 | 9.9 | 2.2 | 5.5 | 7.7 | 28 |
| PB3.8 | ∞ | 2.9 | 7.6 | 10.5 | 2.5 | 6.4 | 8.9 | 28 |
| PB6.8 | ∞ | 3.3 | 10.7 | 14.0 | 2.7 | 8.7 | 11.4 | 24 |
| σY1/Y1 | σZ1/Z1 | σL1/L1 | σY2/Y2 | σZ2/Z2 | σL1/L2 | |||
| 0.50 | 0.50 | 0.35 | - | - | - | |||
| 0.45 | 0.45 | 0.33 | - | - | - | |||
| 0.35 | 0.35 | 0.26 | 0.36 | 0.36 | 0.27 | |||
| 0.34 | 0.34 | 0.27 | 0.40 | 0.40 | 0.30 | |||
| 0.34 | 0.34 | 0.27 | 0.33 | 0.33 | 0.26 | |||
| 0.40 | 0.40 | 0.32 | 0.41 | 0.41 | 0.41 |
| Sample * | SAXS | TEM | ||||||
|---|---|---|---|---|---|---|---|---|
| Y | L | Ymin | Ymax | |||||
| (nm) | (nm) | (nm) | (nm) | (nm) | (nm) | |||
| PB0 | 7.6 | 13.6 | 9.1 | 7.4 | 12.0 | 13.2 | ||
| PB1 | 5.3 | 12.6 | 5.6 | 4.7 | 6.6 | 12.4 | ||
| PB2.9 | 2.8 | 2.2 | 9.9 | 7.7 | 4.3 | 3.4 | 5.7 | 10.1 |
| PB3.8 | 2.9 | 2.5 | 10.5 | 8.9 | 3.5 | 2.5 | 3.9 | 9.0 |
| PB6.8 | 3.3 | 2.7 | 14.0 | 11.4 | 4.3 | 3.1 | 5.3 | 13.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marega, C.; Malizia, F.; Spataro, S. Poly (1-butene-ran-ethylene) Monomodal Copolymers from Metallocene Catalysts: Structural and Morphological Differences with Increasing Ethylene Content. Polymers 2019, 11, 1133. https://doi.org/10.3390/polym11071133
Marega C, Malizia F, Spataro S. Poly (1-butene-ran-ethylene) Monomodal Copolymers from Metallocene Catalysts: Structural and Morphological Differences with Increasing Ethylene Content. Polymers. 2019; 11(7):1133. https://doi.org/10.3390/polym11071133
Chicago/Turabian StyleMarega, Carla, Federica Malizia, and Stefano Spataro. 2019. "Poly (1-butene-ran-ethylene) Monomodal Copolymers from Metallocene Catalysts: Structural and Morphological Differences with Increasing Ethylene Content" Polymers 11, no. 7: 1133. https://doi.org/10.3390/polym11071133
APA StyleMarega, C., Malizia, F., & Spataro, S. (2019). Poly (1-butene-ran-ethylene) Monomodal Copolymers from Metallocene Catalysts: Structural and Morphological Differences with Increasing Ethylene Content. Polymers, 11(7), 1133. https://doi.org/10.3390/polym11071133