
Synthesis, Crystal Structure, Hirshfeld Surface Analysis, Energy Framework Calculations, and Halogen Bonding Investigation of Benzene-1,3,5-triyltris((4-chlorophenyl)methanone)
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
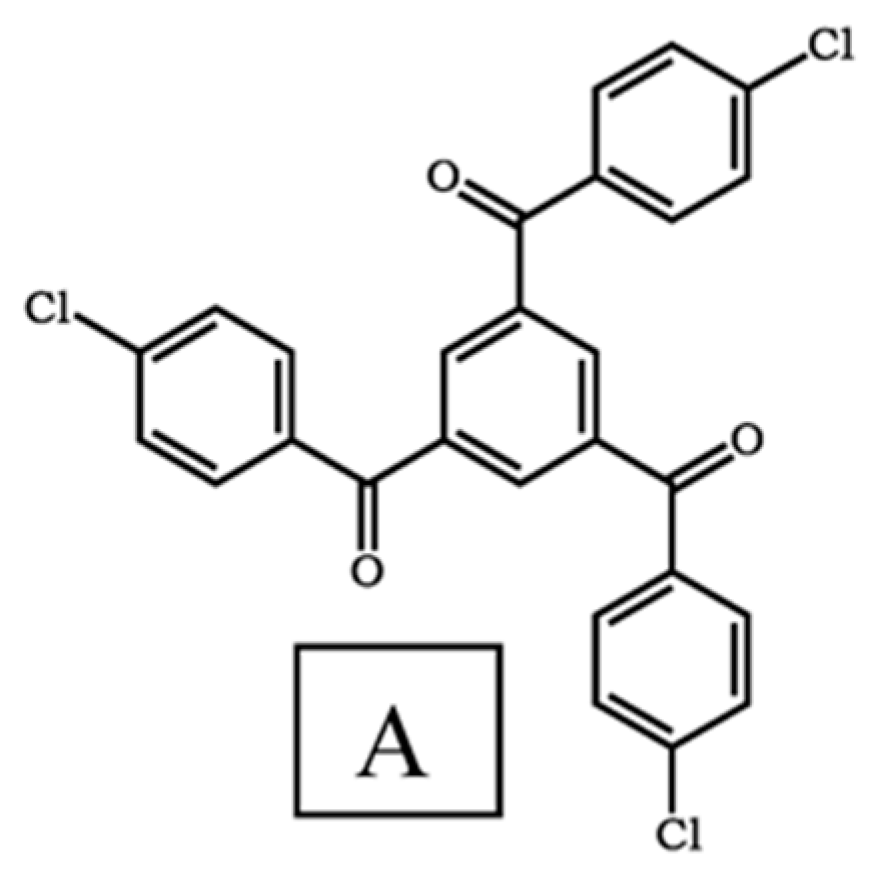
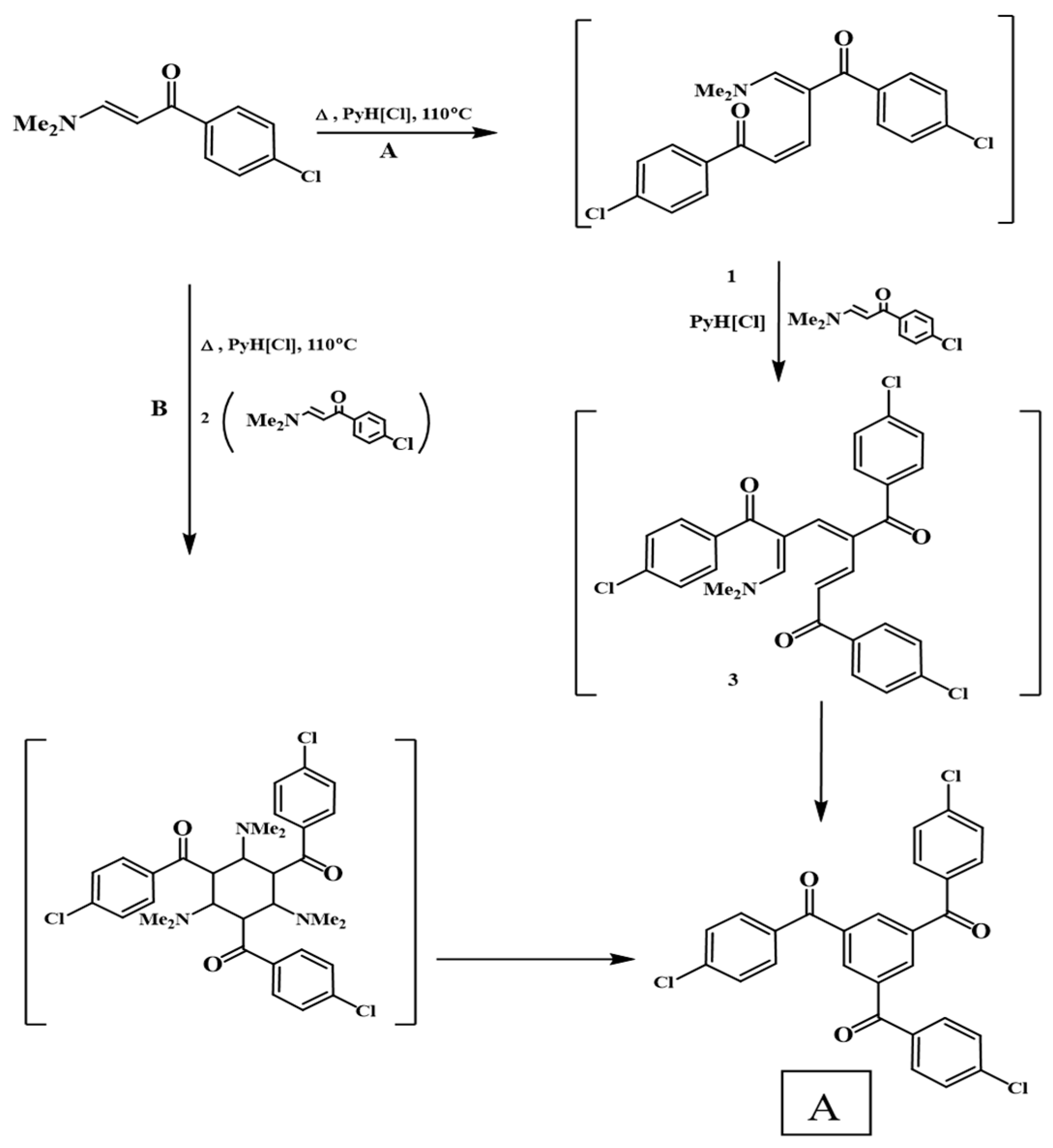
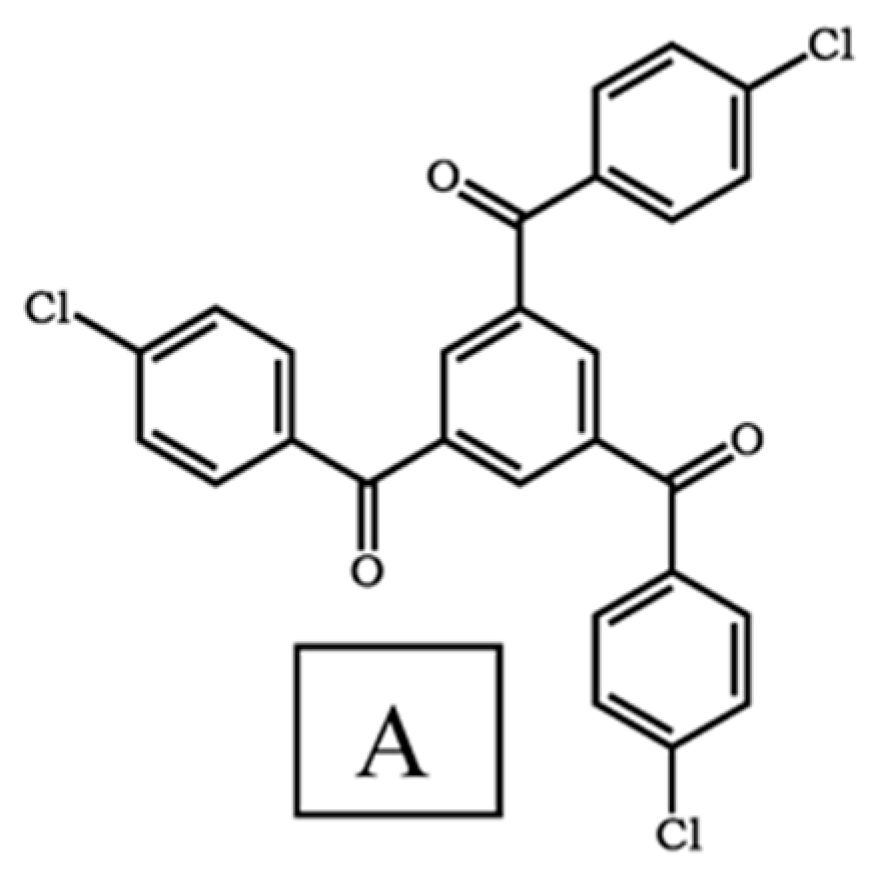
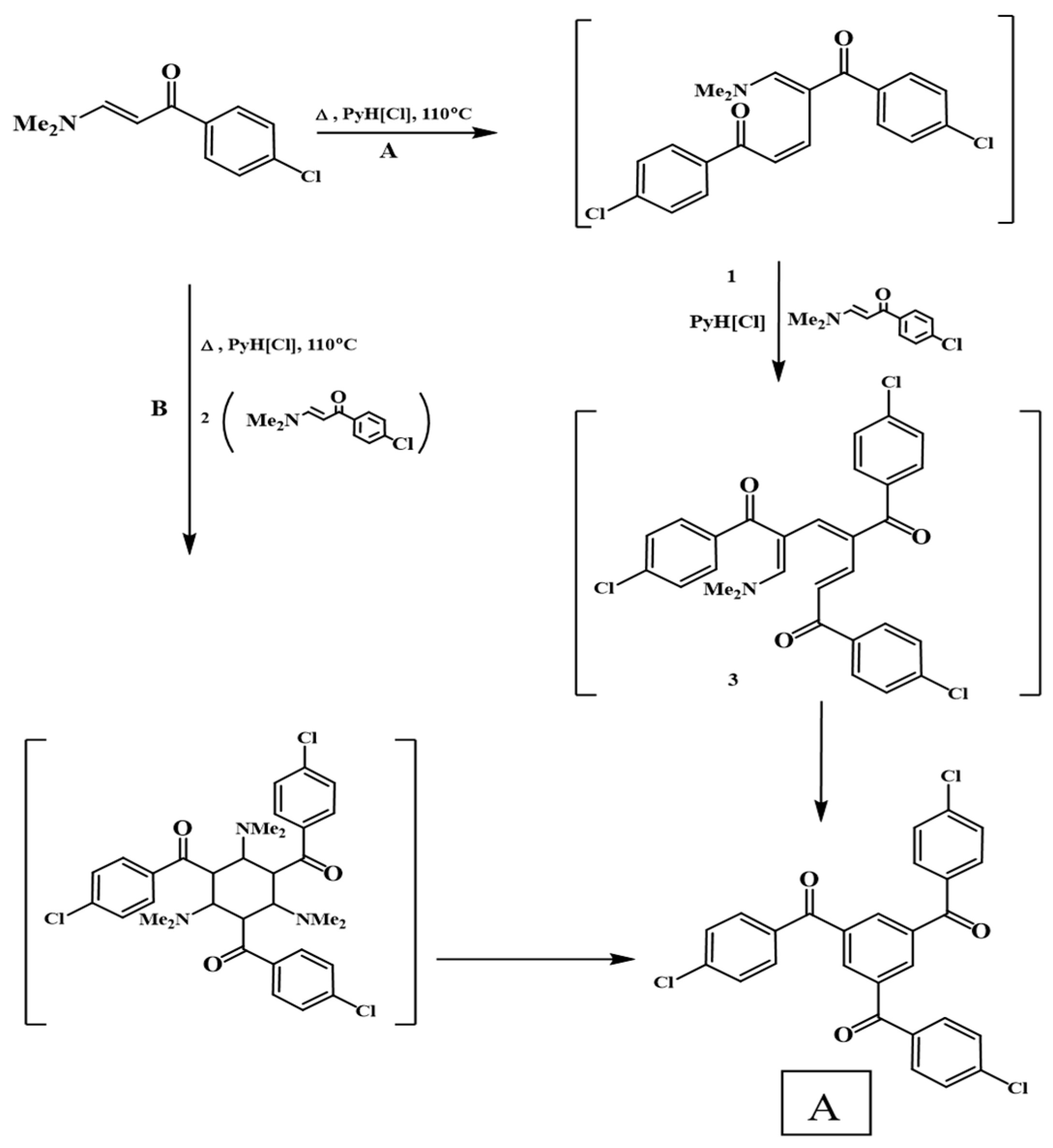
3.1. Synthesis and Characterization
3.2. X-ray Structure and Hirshfeld Surface Analysis
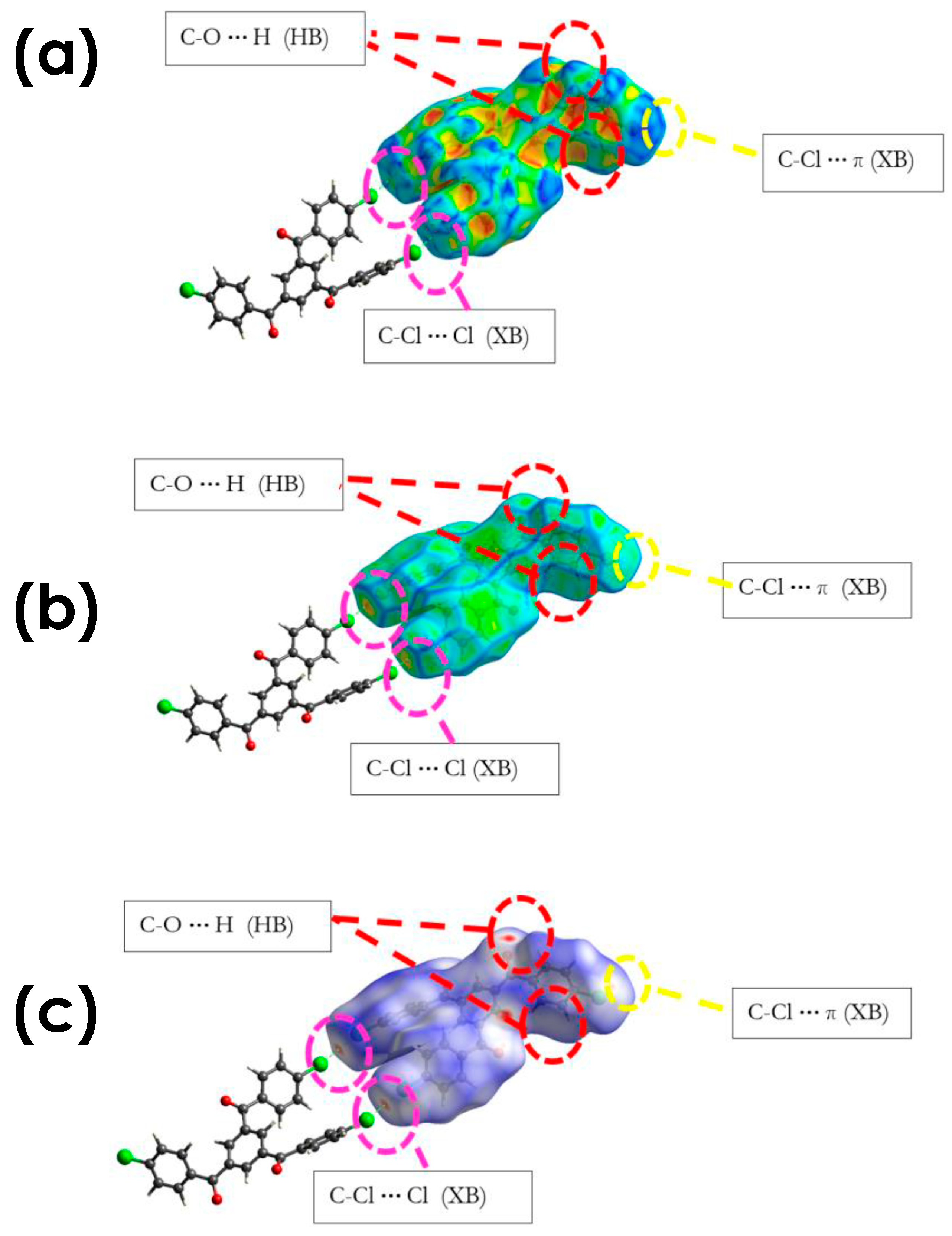
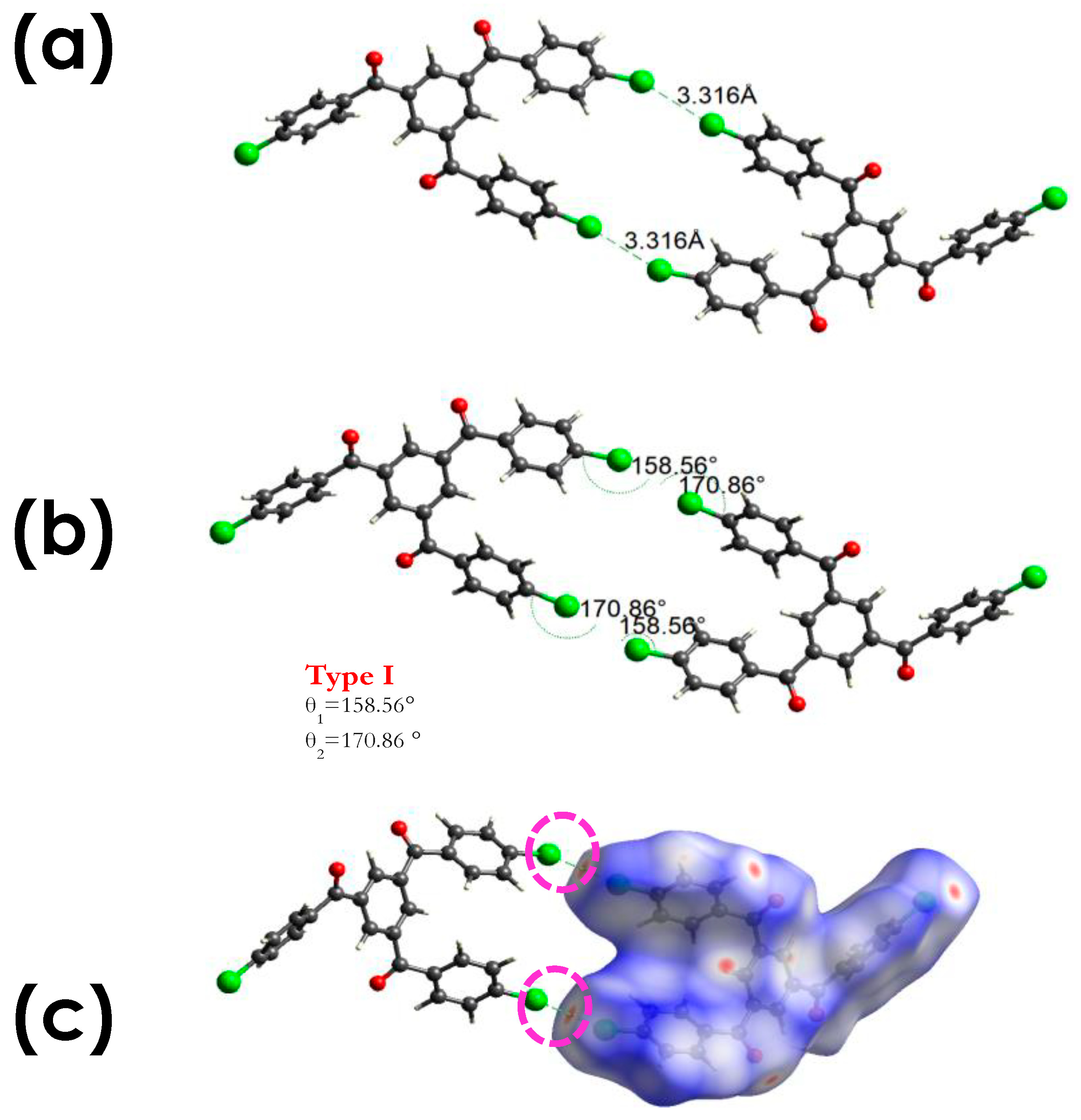
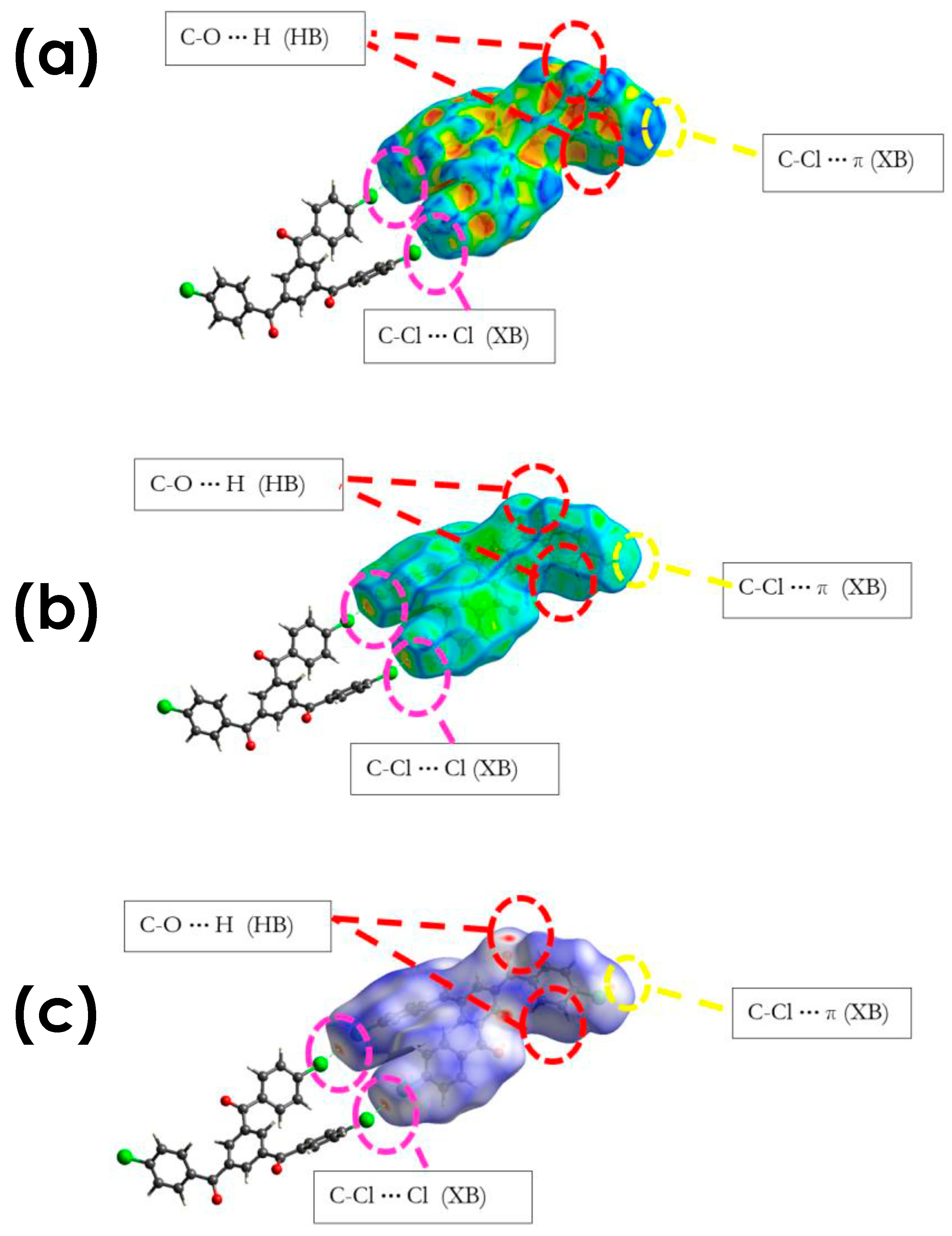
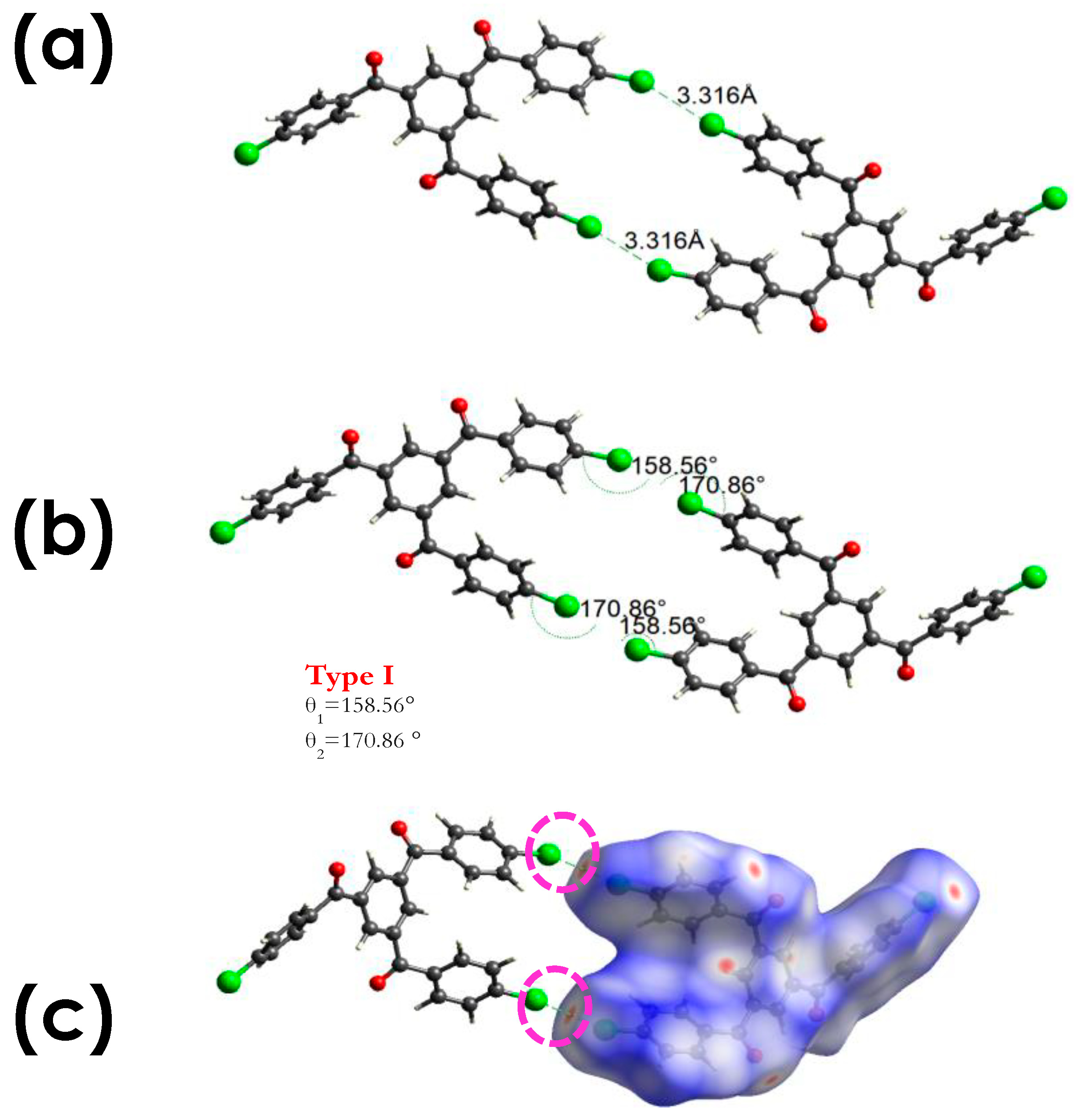
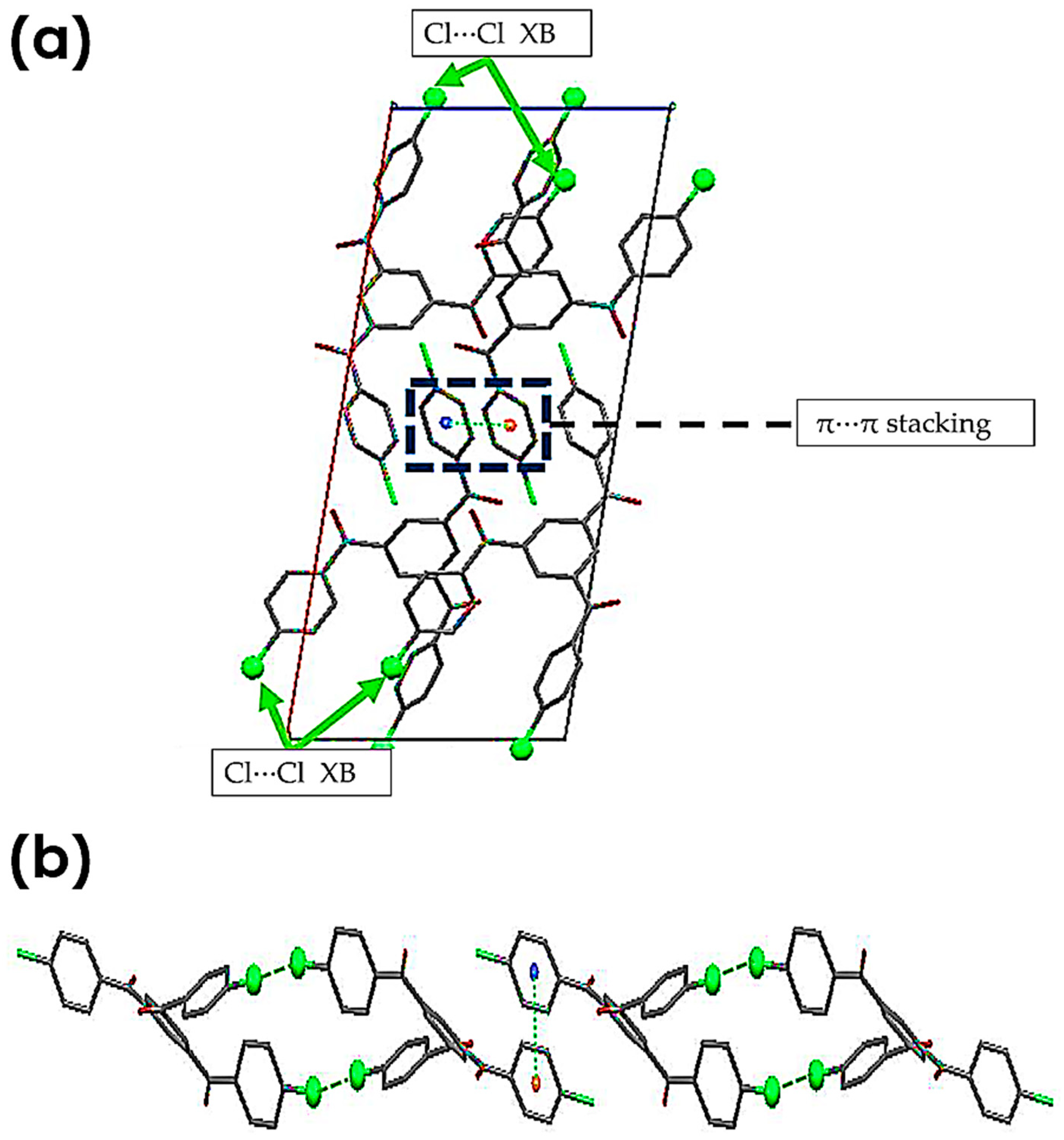
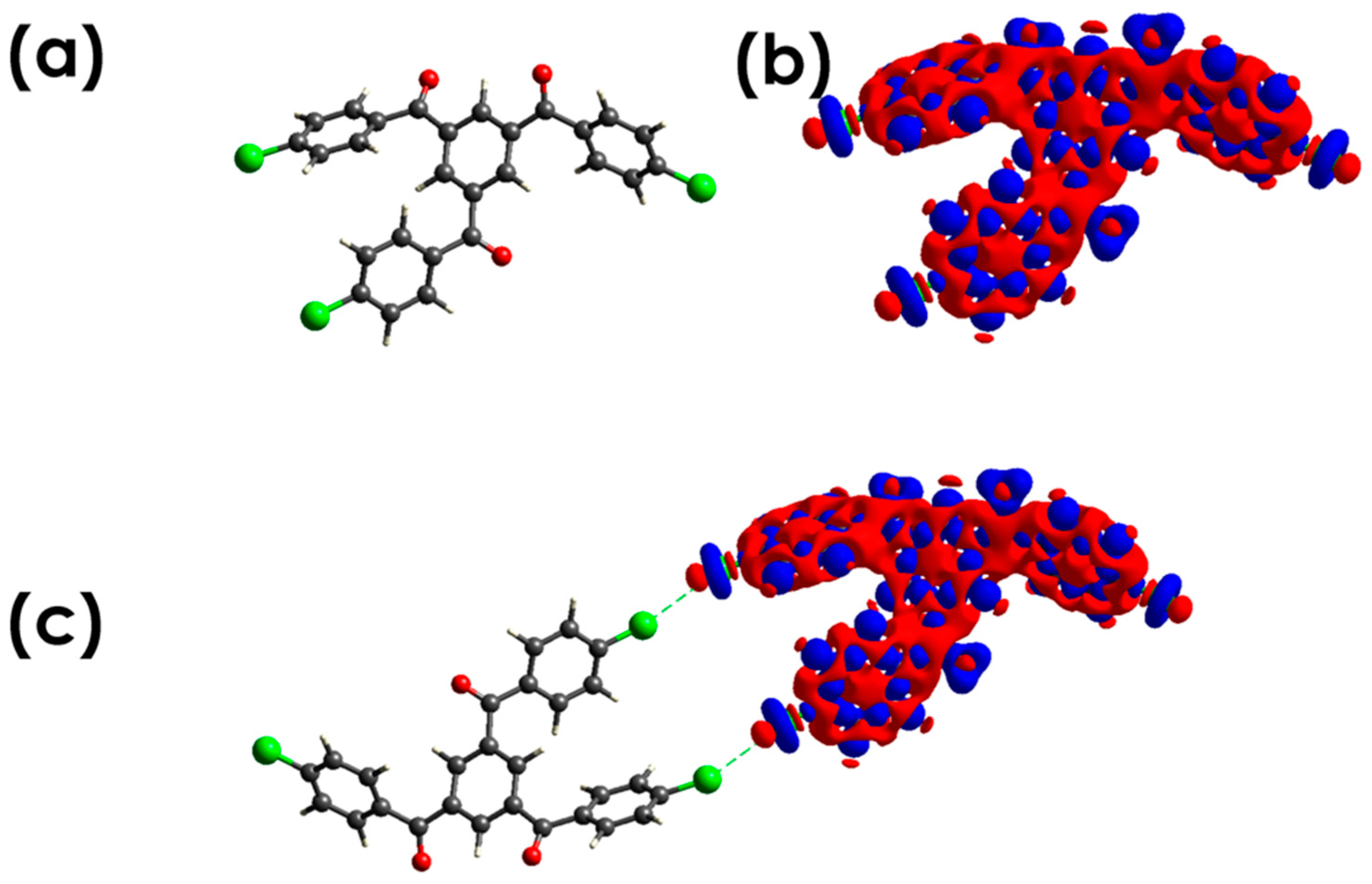
3.2.1. C-Cl···Cl (XB)
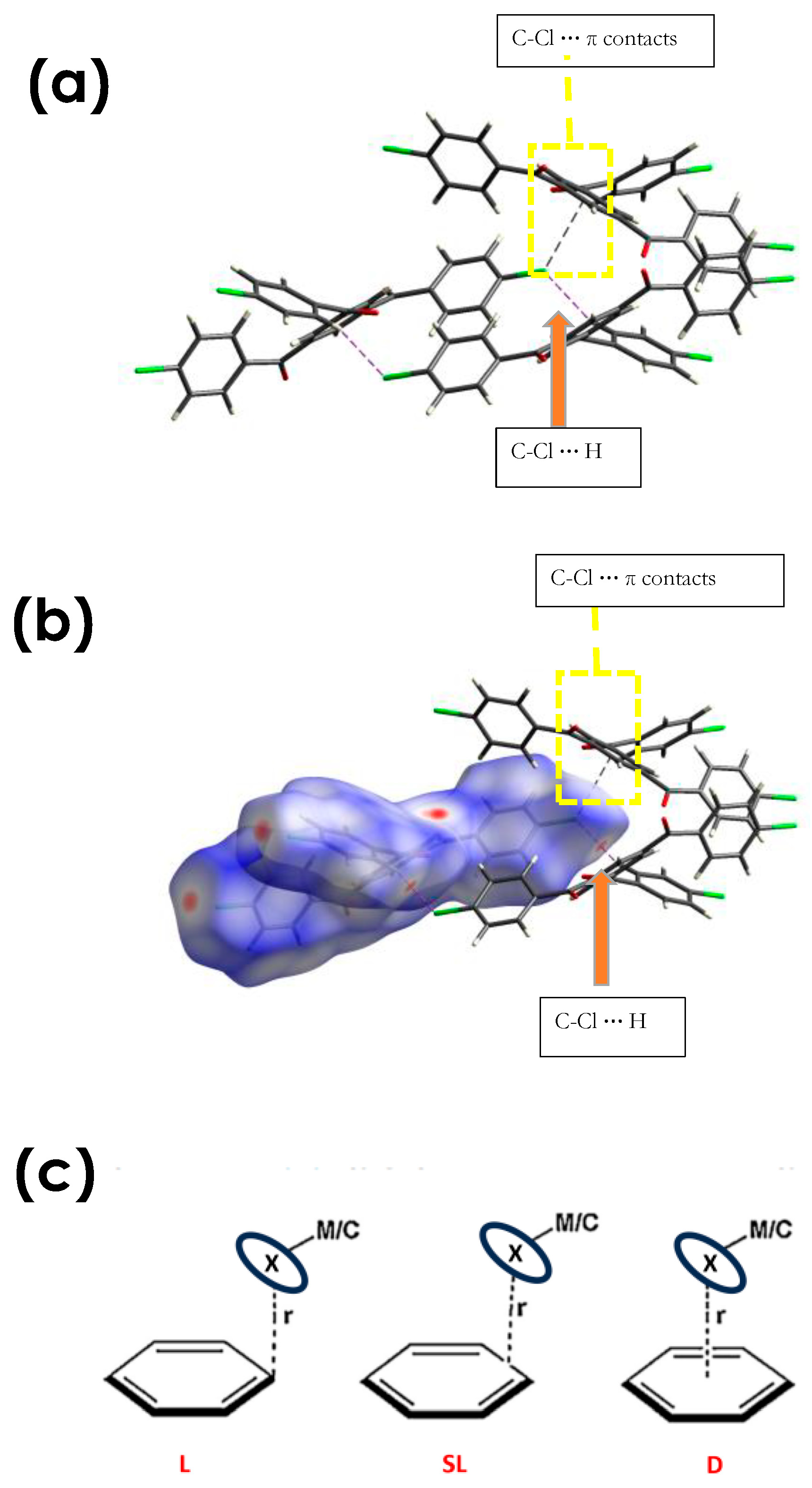
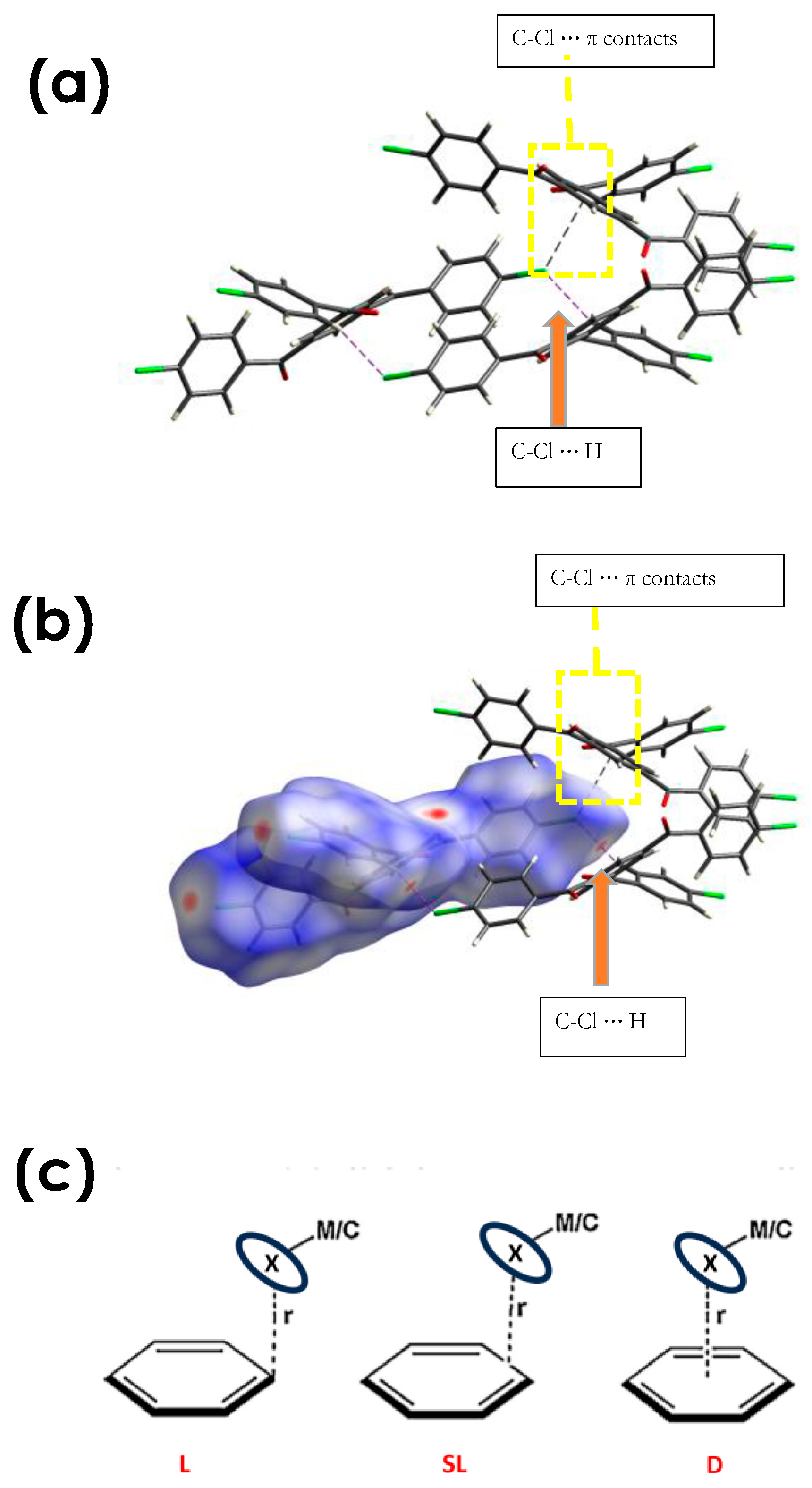
3.2.2. C-Cl···π (XB), and C-Cl···H, and Other Non-Covalent Interactions
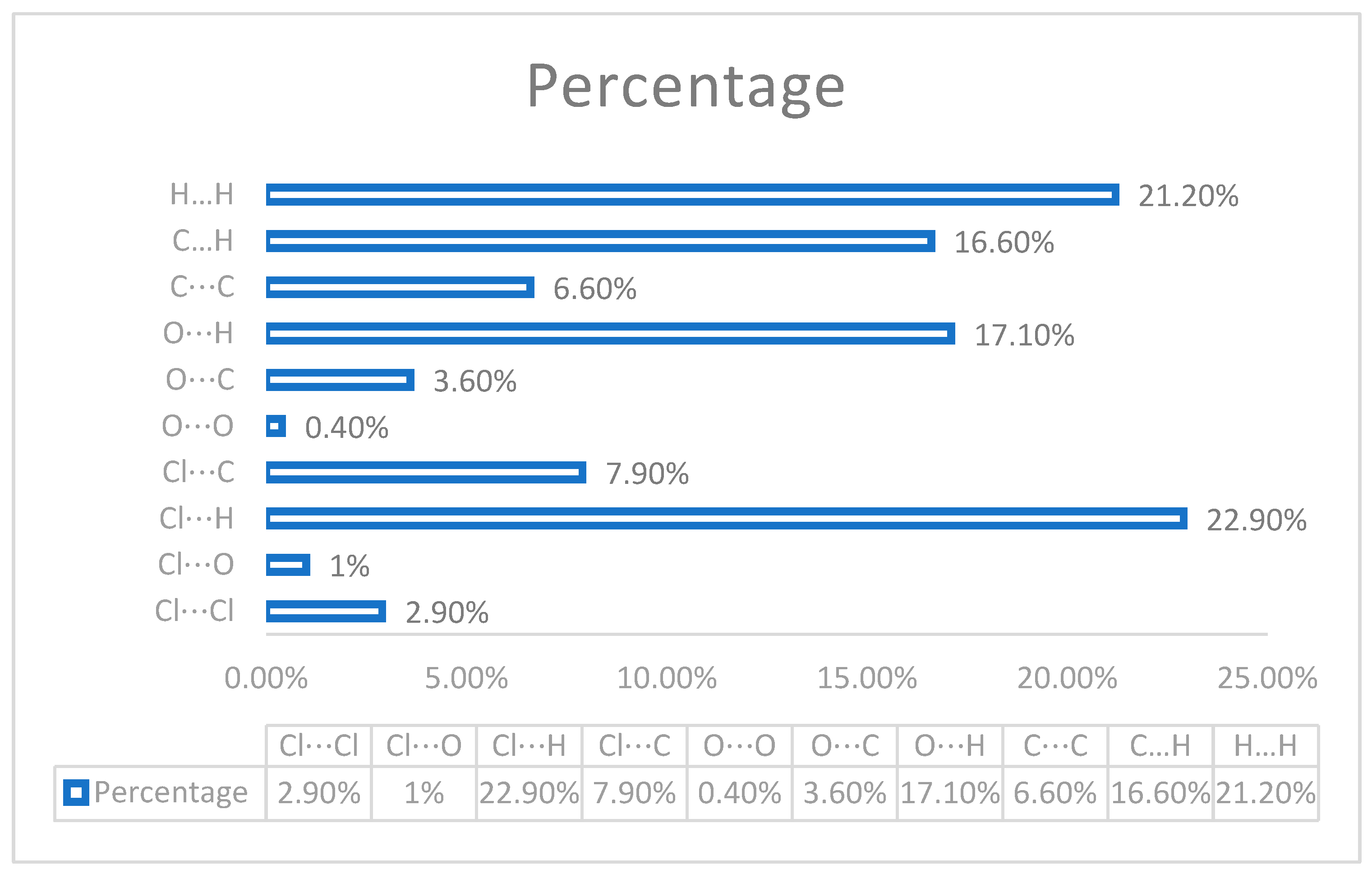
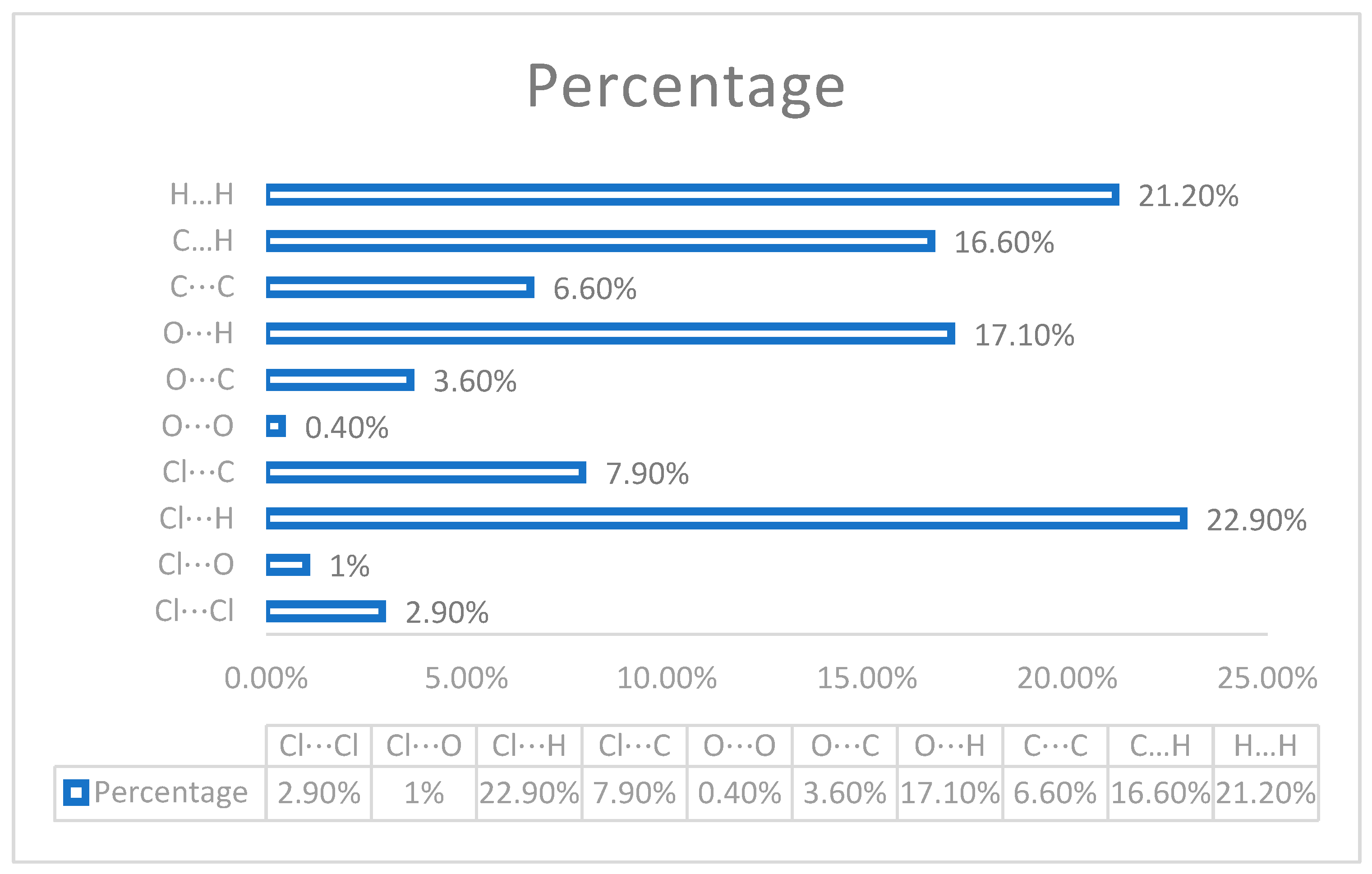
3.3. Fingerprint
3.4. Computational Chemistry
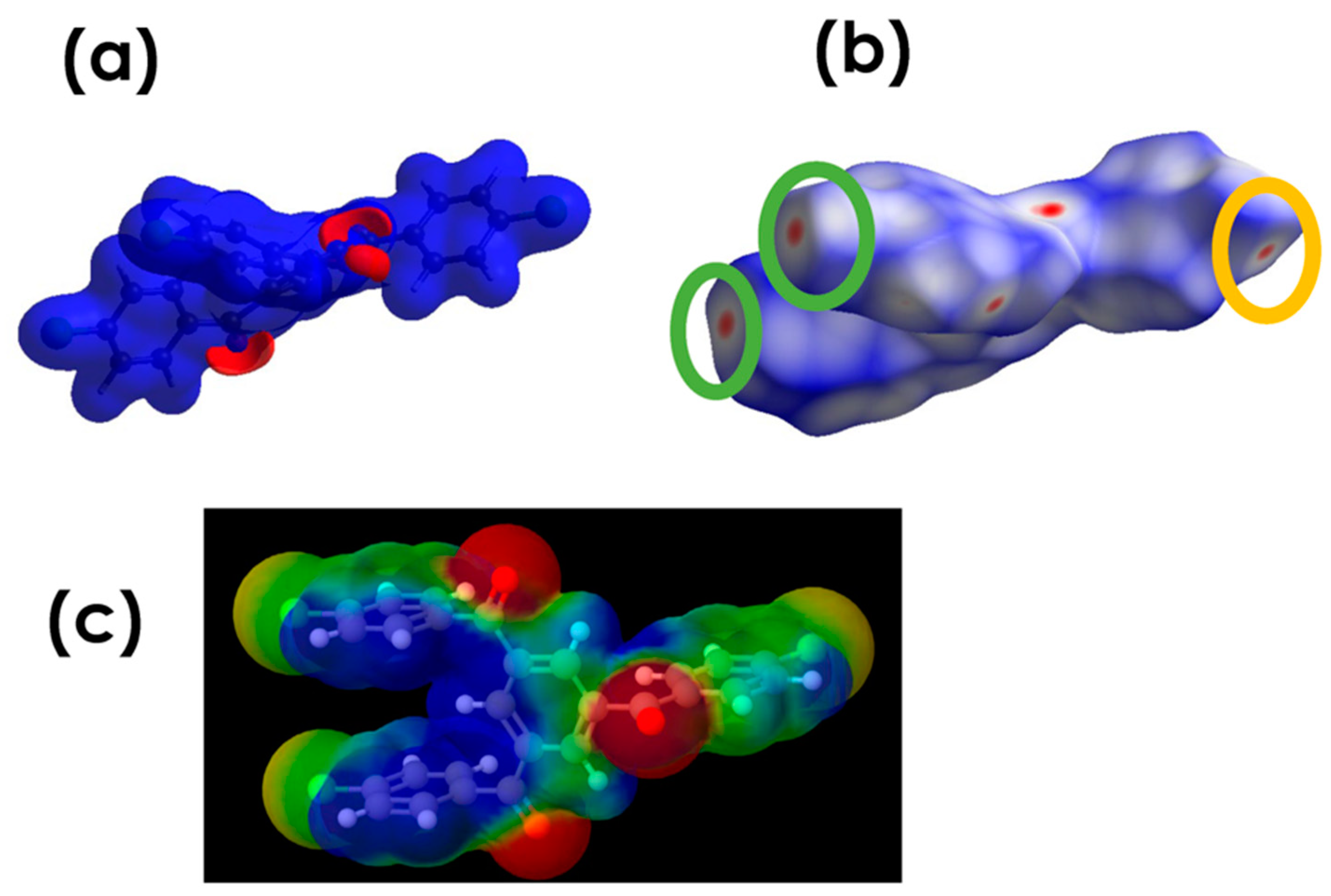
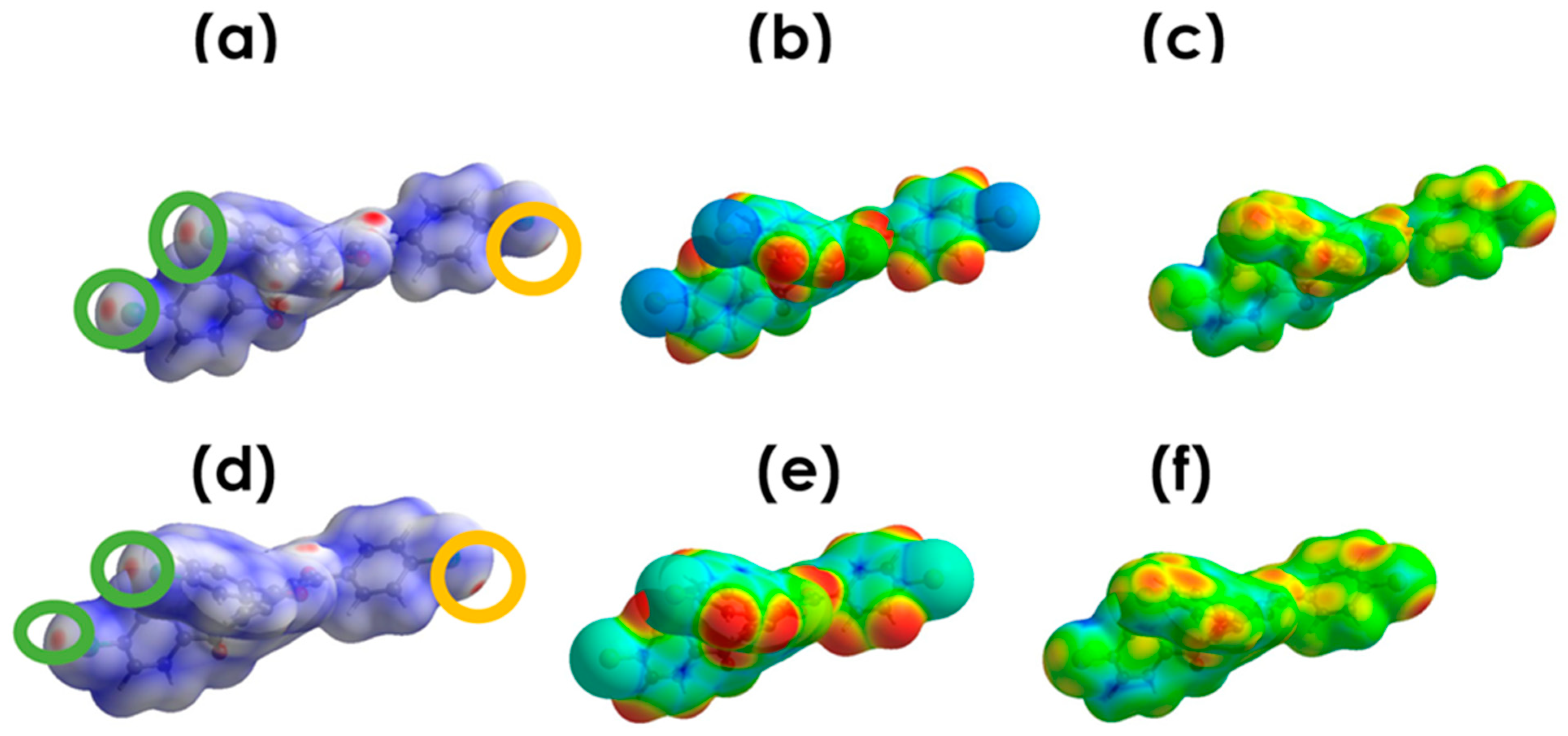
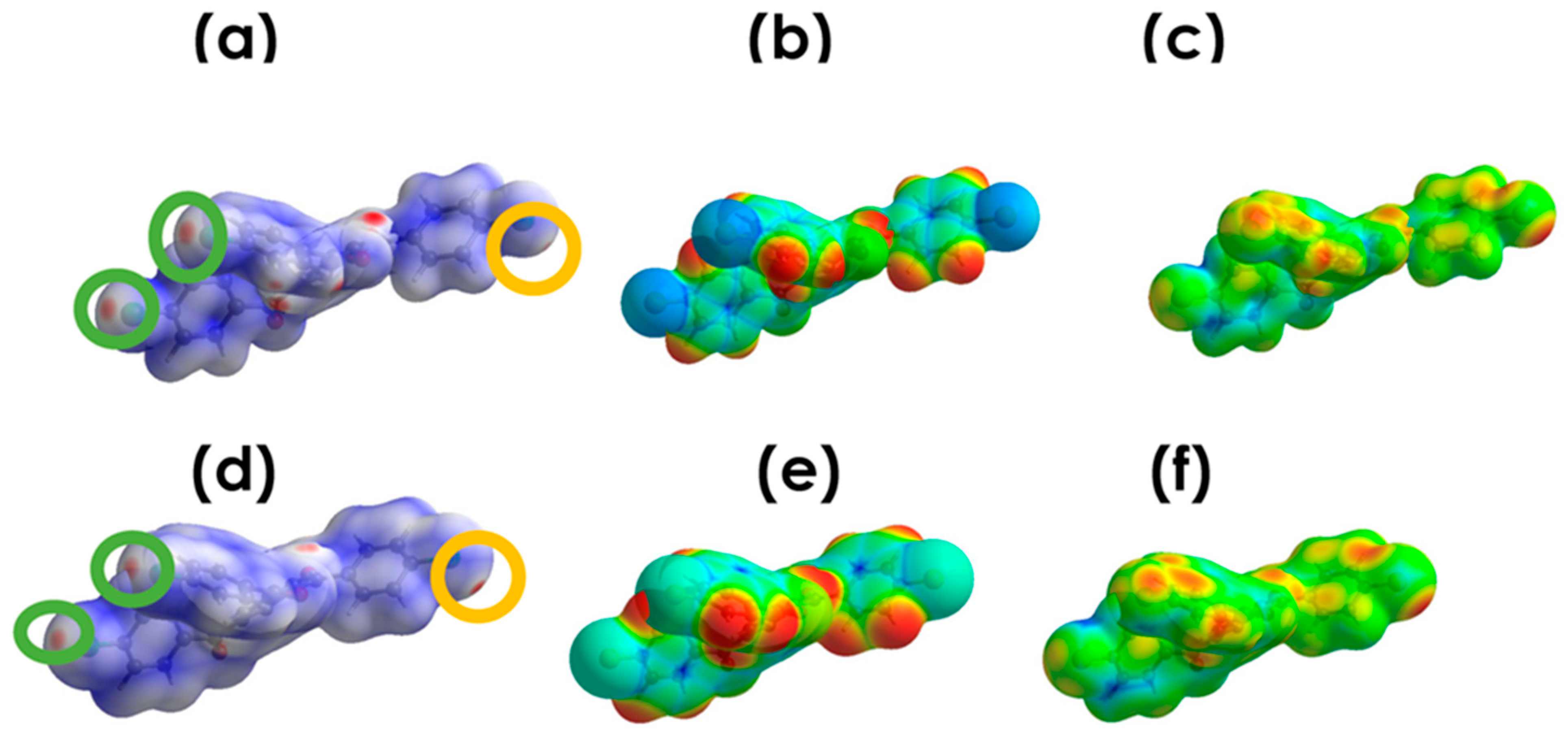
3.4.1. Electrostatic Potential
3.4.2. Deformation Density (DD) Maps
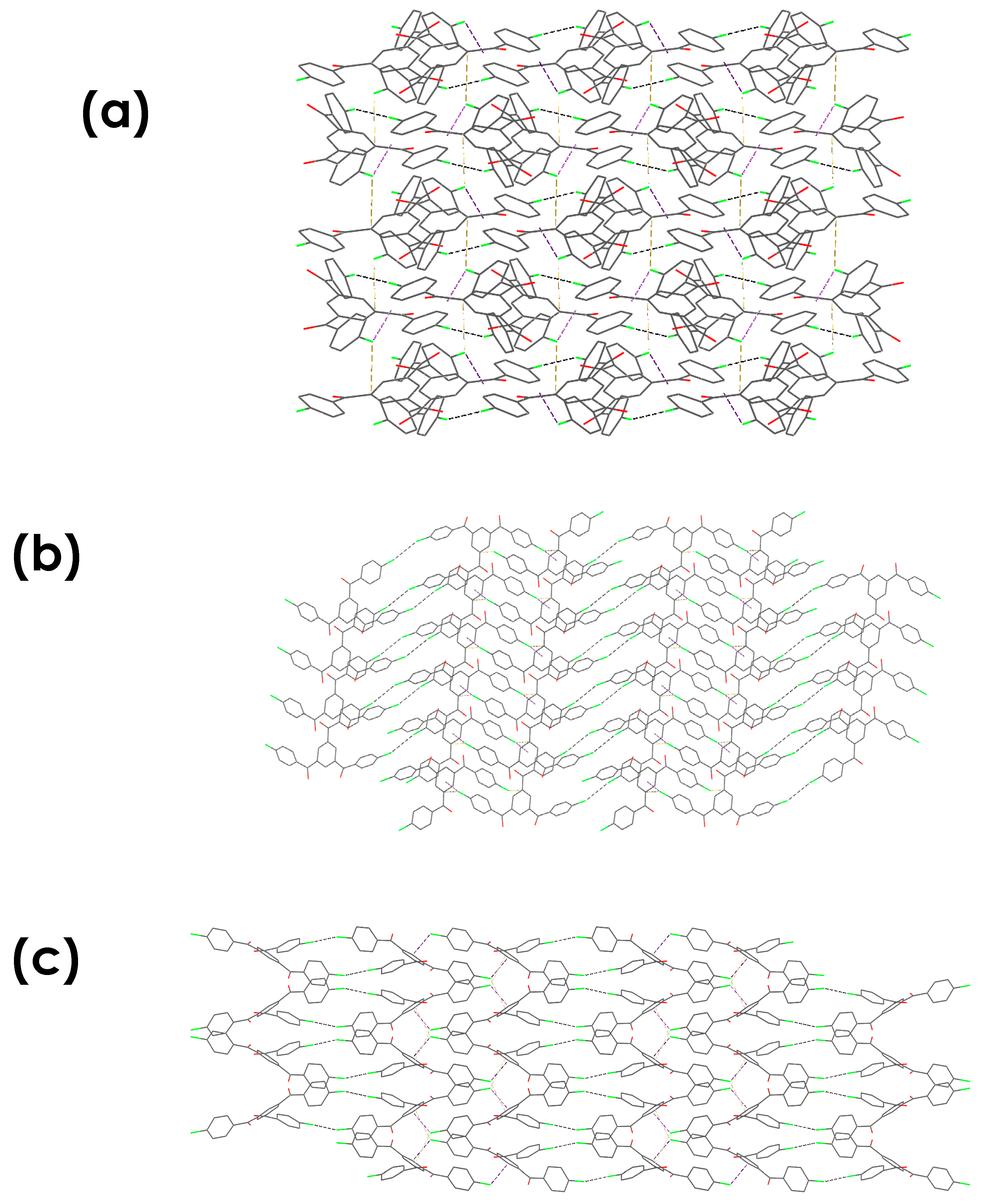
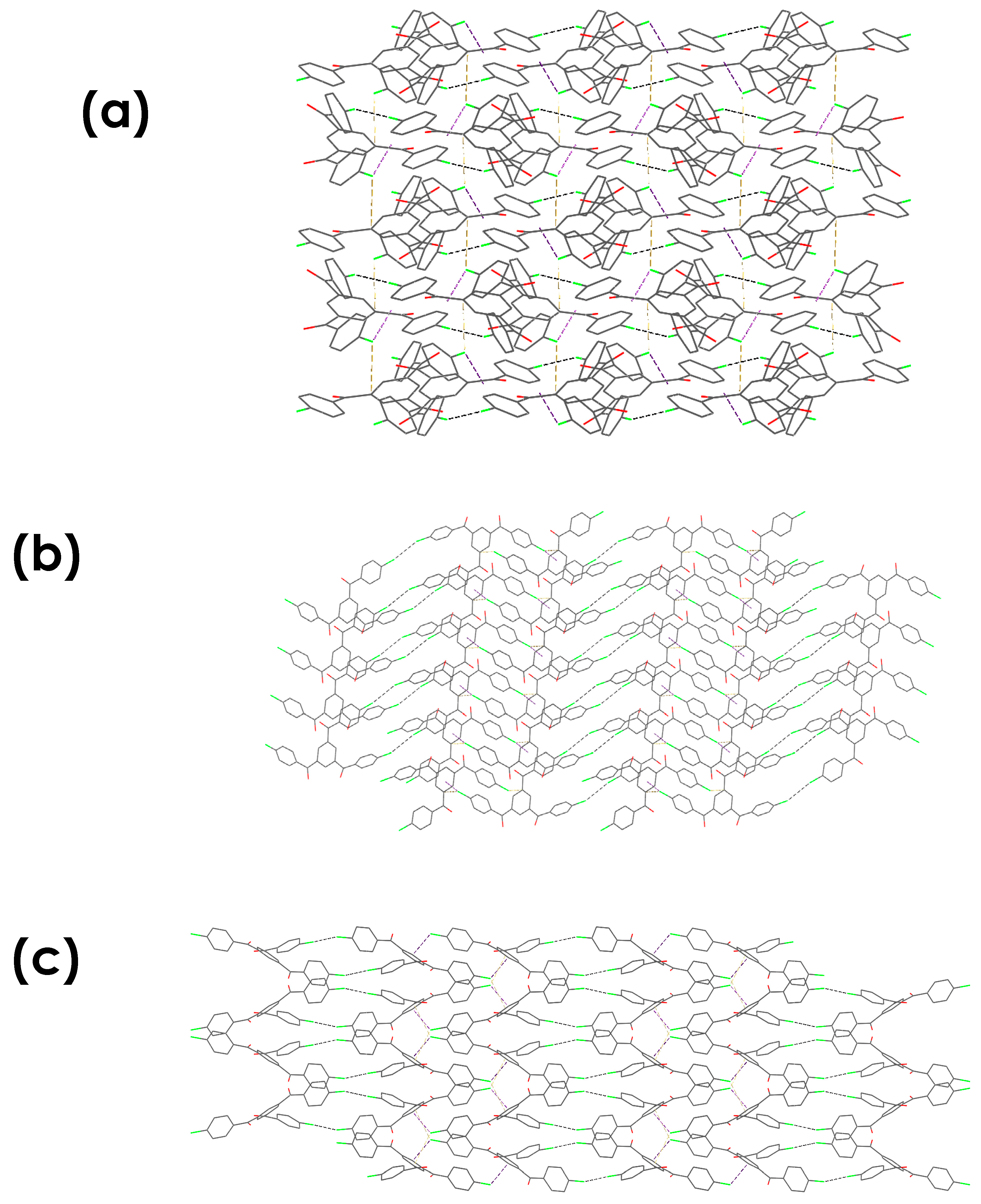
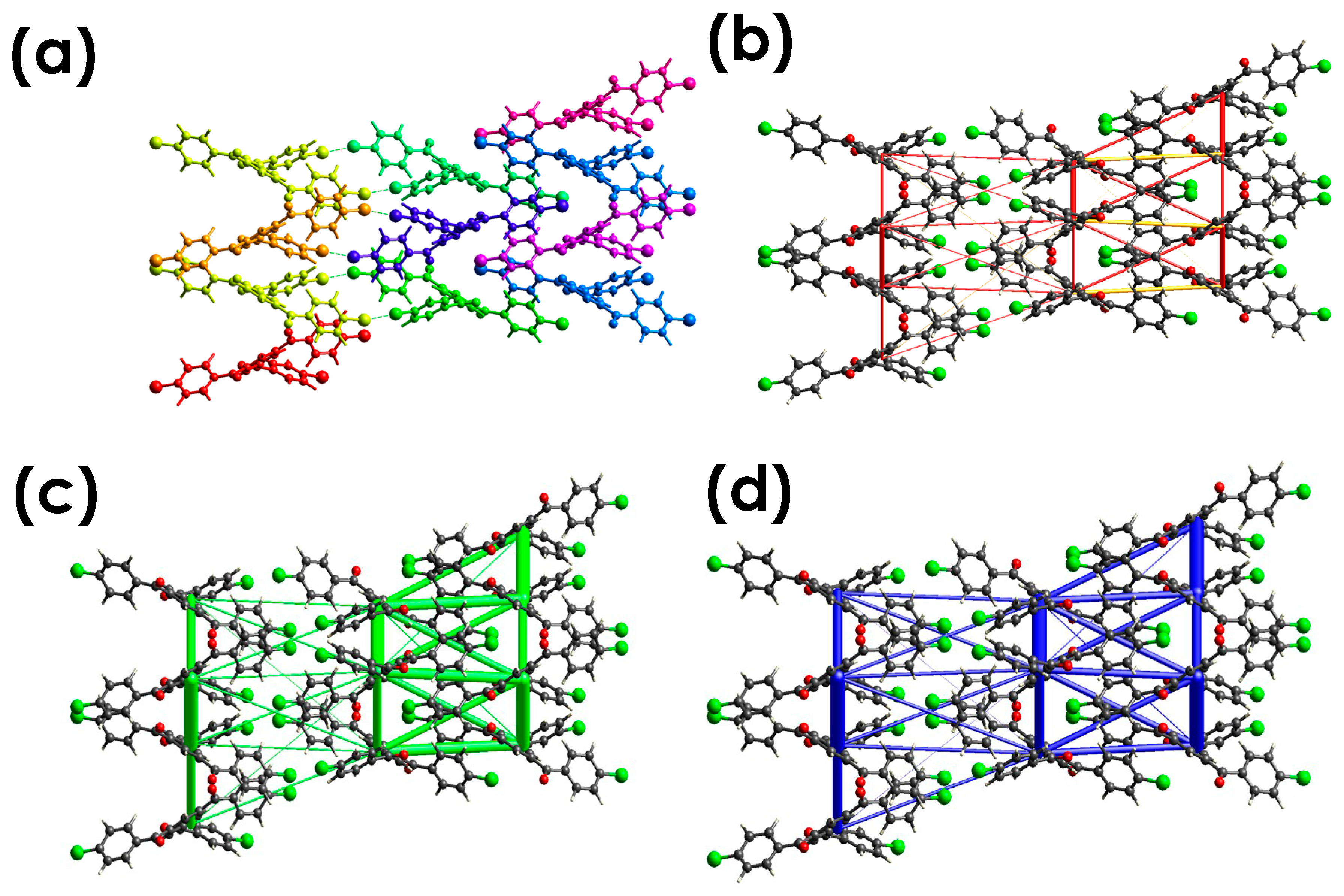
3.4.3. Energy Framework Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R. Crystal engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Champness, N.R.; Janiak, C. Recent advances in crystal engineering. CrystEngComm 2010, 12, 22. [Google Scholar] [CrossRef]
- Blunt, M.O.; Hu, Y.; Toft, C.W.; Slater, A.G.; Lewis, W.; Champness, N.R. Controlling the Two-Dimensional Self-Assembly of Functionalized Porphyrins via Adenine–Thymine Quartet Formation. J. Phys. Chem. C 2018, 122, 26070. [Google Scholar] [CrossRef]
- Räisänen, M.T.; Slater née Phillips, A.G.; Champness, N.R.; Buck, M. Effects of Pore Modification on the Templating of Guest Molecules in a 2D Honeycomb Network. Chem. Sci. 2012, 3, 84–92. [Google Scholar] [CrossRef]
- Theobald, J.A.; Oxtoby, N.S.; Phillips, M.A.; Champness, N.R.; Beton, P.H. Controlling Molecular Deposition and Layer Structure with Supramolecular Surface Assemblies. Nature 2003, 424, 1029. [Google Scholar] [CrossRef]
- Blunt, M.O.; Russell, J.C.; Gimenez-Lopez, M.d.C.; Taleb, N.; Lin, X.; Schröder, M.; Champness, N.R.; Beton, P.H. Guest-Induced Growth of a Surface-Based Supramolecular Bilayer. Nat. Chem. 2011, 3, 74. [Google Scholar] [CrossRef]
- Slater, A.G.; Hu, Y.; Yang, L.; Argent, S.P.; Lewis, W.; Blunt, M.O.; Champness, N.R. Thymine functionalised porphyrins, synthesis and heteromolecular surface-based self-assembly. Chem. Sci. 2015, 6, 1562. [Google Scholar] [CrossRef]
- Gutzler, R.; Fu, C.; Dadvand, A.; Hua, Y.; MacLeod, J.M.; Rosei, F.; Perepichka, D.F. Halogen bonds in 2D supramolecular self-assembly of organic semiconductors. Nanoscale 2012, 4, 5965. [Google Scholar] [CrossRef]
- Yoon, J.K.; Son, W.-J.; Chung, K.-H.; Kim, H.; Han, S.; Kahng, S.-J. Visualizing Halogen Bonds in Planar Supramolecular Systems. J. Phys. Chem. C 2011, 115, 2297. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: A Paradigm in Supramolecular Chemistry. Chem.—Eur. J. 2001, 7, 2511. [Google Scholar] [CrossRef]
- Brammer, L.; Minguez Espallargas, G.; Libri, S. Combining metals with halogen bonds. CrystEngComm 2008, 10, 1712–1727. [Google Scholar] [CrossRef]
- Metrangolo, P.; Pilati, T.; Resnati, G. Halogen bonding and other noncovalent interactions involving halogens: A terminology issue. CrystEngComm 2006, 8, 946. [Google Scholar] [CrossRef]
- Frontera, A.; Quiñonero, D.; Deyà, P.M. Cation–π and anion–π interactions. Wiley Interdisciplinary Reviews: Comput. Mol. Sci. 2011, 1, 440. [Google Scholar] [CrossRef]
- Steed, J.W.; Turner, D.R.; Wallace, K. Core Concepts in Supramolecular Chemistry and Nanochemistry; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Blunt, M.O.; Adisoejoso, J.; Tahara, K.; Katayama, K.; Van der Auweraer, M.; Tobe, Y.; Feyter, S.D. Temperature-Induced Structural Phase Transitions in a Two-Dimensional Self-Assembled Network. J. Am. Chem. Soc. 2013, 135, 12068. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Yokoyama, S.; Kamikado, T.; Okuno, Y.; Mashiko, S. Selective assembly on a surface of supramolecular aggregates with controlled size and shape. Nature 2001, 413, 619. [Google Scholar] [CrossRef]
- Böhringer, M.; Morgenstern, K.; Schneider, W.-D.; Berndt, R. Separation of a Racemic Mixture of Two-Dimensional Molecular Clusters by Scanning Tunneling Microscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 821. [Google Scholar] [CrossRef]
- Sarkar, N.; Sinha, A.S.; Aakeröy, C.B. Systematic investigation of hydrogen-bond propensities for informing co-crystal design and assembly. CrystEngComm 2019, 21, 6048–6055. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Nanda, A. A review about regulatory status and recent patents of pharmaceutical co-crystals. Adv. Pharm. Bull. 2018, 8, 355. [Google Scholar] [CrossRef]
- Sandhu, B.; Sinha, A.S.; Desper, J.; Aakeröy, C.B. Modulating the physical properties of solid forms of urea using co-crystallization technology. Chem. Comm. 2018, 54, 4657. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: Origins, current status and discussion. Faraday Discuss. 2017, 203, 485. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Tuikka, M.; Haukka, M.; Benedict, J.B. Recent Advances in Crystallography; IntechOpen: London, UK, 2012; p. 143. [Google Scholar]
- Rosokha, S.V.; Neretin, I.S.; Rosokha, T.Y.; Hecht, J.; Kochi, J.K. Charge-Transfer Character of Halogen Bonding: Molecular Structures and Electronic Spectroscopy of Carbon Tetrabromide and Bromoform Complexes with Organic σ- and π-Donors. Heteroat. Chem. 2006, 17, 449. [Google Scholar] [CrossRef]
- Laurence, C.; Graton, J.; Berthelot, M.; El Ghomari, M.J. The Diiodine Basicity Scale: Toward a General Halogen-Bond Basicity Scale. Chem. Eur. J. 2011, 17, 10431. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Karan, N.K.; Arunan, E. Chlorine Bond Distances in ClF and Cl2 Complexes. J. Mol. Struct. 2004, 688, 203–205. [Google Scholar] [CrossRef]
- Raghavendra, B.; Arunan, E. Unpaired and Sigma Bond Electrons as H, Cl, and Li Bond Acceptors: An Anomalous One-Electron Blue-Shifting Chlorine Bond. J. Phys. Chem. A 2007, 111, 9699. [Google Scholar] [CrossRef] [PubMed]
- Chopra, D.; Row, T.N.G. Role of Organic Fluorine in Crystal Engineering. CrystEngComm 2011, 13, 2175. [Google Scholar] [CrossRef]
- Laurence, C.; Graton, J.; Gal, J.F. An Overview of Lewis Basicity and Affinity Scales. J. Chem. Educ. 2011, 88, 1651. [Google Scholar] [CrossRef]
- Lu, Y.X.; Zou, J.W.; Yu, Q.S.; Jiang, Y.J.; Zhao, W.N. Ab Initio Investigation of Halogen Bonding Interactions Involving Fluorine as an Electron Acceptor. Chem. Phys. Lett. 2007, 449, 6–10. [Google Scholar] [CrossRef]
- Metrangolo, P.; Pilati, T.; Terraneo, G.; Biella, S.; Resnati, G. Anion coordination and anion-templated assembly under halogen bonding control. CrystEngComm 2009, 11, 1187. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Disney, M.D. Covalent Small-Molecule–RNA Complex Formation Enables Cellular Profiling of Small-Molecule–RNA Interactions. Angew. Chem. Int. Ed. 2013, 52, 10010. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Lang, T.; Caballero, A.; Costa, P.J.; Félix, V.; Beer, P.D. A Catenane Assembled through a Single Charge-Assisted Halogen Bond. Angew. Chem. Int. Ed. 2013, 52, 4356. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Cai, Z.; Zou, H.; Li, J.; Zhang, S.; Ying, L.; Deng, W. Achieving halogen bonding enhanced ultra-highly efficient AIE and reversible mechanochromism properties of TPE-based luminogens: Position of bromine substituents. J. Mater. Chem. C 2022, 10, 8390. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363. [Google Scholar] [CrossRef]
- D’aléo, A.; Saul, A.; Attaccalite, C.; Fages, F. Influence of halogen substitution on aggregation-induced near infrared emission of borondifluoride complexes of 2′-hydroxychalcones. Mater. Chem. Front. 2019, 3, 86. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding. Acc. Chem. Res. 2005, 38, 386. [Google Scholar] [CrossRef]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen bonding vs. halogen bonding: The solvent decides. Chem. Sci. 2017, 8, 5392. [Google Scholar] [PubMed]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-centered halogen bonding: A factor in recognition phenomena and reactivity. Cryst. Growth Des. 2011, 11, 4238. [Google Scholar] [CrossRef]
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2, 20170136. [Google Scholar] [CrossRef]
- Atwood, J.L.; Steed, J.W. Encyclopedia of Supramolecular Chemistry; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Cametti, M.; Crousse, B.; Metrangolo, P.; Milani, R.; Resnati, G. The Fluorous Effect in Biomolecular Applications. Chem. Soc. Rev. 2012, 41, 31. [Google Scholar] [CrossRef] [PubMed]
- Pavan, M.S.; Durga Prasad, K.; Guru Row, T.N. Halogen bonding in fluorine: Experimental charge density study on intermolecular F···F and F···S donor–acceptor contacts. Chem. Comm. 2013, 49, 7558. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, M.; Lucassen, A.C.B.; Shimon, L.J.W.; Van Der Boom, M.E. Cocrystallization of a tripyridyl donor with perfluorinated iodobenzene derivatives: Formation of different n··· I halogen bonds determining network vs plain packing crystals. Cryst. Growth Des. 2008, 8, 786. [Google Scholar] [CrossRef]
- Christopherson, J.C.; Topić, F.; Barrett, C.J.; Friščić, T. Halogen-Bonded Cocrystals as Optical Materials: Next-Generation Control over Light–Matter Interactions. Cryst. Growth Des. 2018, 18, 1245. [Google Scholar] [CrossRef]
- Kitagawa, S.; Uemura, K. Dynamic porous properties of coordination polymers inspired by hydrogen bonds. Chem. Soc. Rev. 2005, 34, 109. [Google Scholar] [CrossRef]
- Raatikainen, K.; Rissanen, K. Breathing molecular crystals: Halogen- and hydrogen-bonded porous molecular crystals with solvent induced adaptation of the nanosized channels. Chem. Sci. 2012, 3, 1235. [Google Scholar] [CrossRef]
- Turunen, L.; Beyeh, N.K.; Pan, F.; Valkonen, A.; Rissanen, K. Tetraiodoethynyl resorcinarene cavitands as multivalent halogen bond donors. Chem. Commun. 2014, 50, 15920. [Google Scholar] [CrossRef]
- Shankar, S.; Chovnik, O.; Shimon, L.J.W.; Lahav, M.; van der Boom, M.E. Directed Molecular Structure Variations of Three-Dimensional Halogen-Bonded Organic Frameworks (XBOFs). Cryst. Growth Des. 2018, 18, 1967. [Google Scholar] [CrossRef]
- Meazza, L.; Foster, J.A.; Fucke, K.; Metrangolo, P.; Resnati, G.; Steed, J.W. Halogen-bonding-triggered supramolecular gel formation. Nat. Chem. 2013, 5, 42. [Google Scholar] [CrossRef]
- Lascialfari, L.; Resnati, G.; Metrangolo, P. 704—Halogen-Bonded Cocrystals. In Comprehensive Supramolecular Chemistry, 2nd ed.; Atwood, J.L., Ed.; Elsevier: Oxford, UK, 2017; pp. 49–72. [Google Scholar]
- Sakurai, T.; Sundaralingam, M.; Jeffrey, G.A. A Nuclear Quadrupole Resonance and X-Ray Study of Crystal Structure of 2, 5- Dichloroaniline. Acta Crystallogr. 1963, 16, 354. [Google Scholar] [CrossRef]
- Sarma, J.A.R.P.; Desiraju, G.R. The chloro-substituent as a steering group: A comparative study of non-bonded interactions and hydrogen bonding in crystalline chloro-aromatics. Chem. Phys. Lett. 1985, 117, 160. [Google Scholar] [CrossRef]
- Nangia, A.; Desiraju, G.R. Supramolecular Synthons and Pattern Recognition. In Design of Organic Solids; Weber, E., Aoyama, Y., Caira, M.R., Desiraju, G.R., Glusker, J.P., Hamilton, A.D., Weber, E., Meléndez, R.E., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 57–95. [Google Scholar]
- Li, B.; Zang, S.-Q.; Wang, L.-Y.; Mak, T.C.W. Halogen bonding: A powerful, emerging tool for constructing high-dimensional metal-containing supramolecular networks. Coord. Chem. Rev. 2016, 308, 1–21. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 345, 54. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M. Unconventional type III halogen··· halogen interactions: A quantum mechanical elucidation of σ-hole··· σ-hole and di-σ-hole interactions. ACS Omega 2020, 5, 21824. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, H. Development of Novel Fluoreescent Dye Molecules Based on BODIPY. Ph.D. Thesis, University of Nottingham, Nottingham, UK, March 2019. [Google Scholar]
- Liu, F.-S.; Liu, X.-H.; Ye, K.-Z.; Shen, D.-S. Facile Synthesis of 1,3,5-Triaroylbenzenes by Direct Cyclotrimerization of Ketone Enolates. Synth. Commun. 2013, 43, 1640. [Google Scholar] [CrossRef]
- Pigge, F.C.; Vangala, V.R.; Swenson, D.C.; Rath, N.P. Examination of halogen bonding interactions in electronically distinct but structurally related tris (haloarenes). Cryst. Growth Des. 2010, 10, 224. [Google Scholar] [CrossRef]
- Pigge, F.C.; Dighe, M.K.; Rath, N.P. Self-assembly of sticky TABs: Inclusion complexes and hydrates from 1,3,5-tris (4-hydroxybenzoyl) benzene. Cryst. Growth Des. 2006, 6, 2732. [Google Scholar] [CrossRef]
- Kumar, V.S.S.; Pigge, F.C.; Rath, N.P. Polymorphism in 1, 3, 5-triaroylbenzenes: Structural characterization of concomitant polymorphs obtained from 1, 3, 5-tris (4-chlorobenzoyl) benzene. CrystEngComm 2004, 6, 102. [Google Scholar] [CrossRef]
- Kumar, V.S.S.; Pigge, F.C.; Rath, N.P. Polymorphism and pseudopolymorphism in the triaroylbenzene derivative 1,3,5-tris (4-cyanobenzoyl) benzene. Cryst. Growth Des. 2004, 4, 1217. [Google Scholar] [CrossRef]
- van Terwingen, S.; Wang, R.; Englert, U. Three for the Price of One: Concomitant I···N, I···O, and I···π Halogen Bonds in the Same Crystal Structure. Molecules 2022, 27, 7550. [Google Scholar] [CrossRef]
- Hassanain, H.; Davies, E.S.; Lewis, W.; Kays, D.L.; Champness, N.R. Morpholino-Substituted BODIPY Species: Synthesis, Structure and Electrochemical Studies. Crystals 2020, 10, 36. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Chem.—Eur. J. 1998, 4, 2136. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Comm. 2007, 3814, 2136–2141. [Google Scholar] [CrossRef]
- Smith, T.J. MOLView: A program for analyzing displaying atomic structures on the Macintosh personal computer. J. Mol. Graph. 1995, 13, 122. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E Crystallogr. 2019, 75, 308. [Google Scholar] [CrossRef] [PubMed]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. Hirshfeld Surface Analysis and Density Functional Theory Calculations of 2-Benzyloxy-1,2,4-triazolo [1,5-a] quinazolin-5 (4 H)-one: A Comprehensive Study on Crystal Structure, Intermolecular Interactions, and Electronic Properties. Crystals 2023, 13, 1410. [Google Scholar] [CrossRef]
- Kamat, V.; Kumara, K.; Naik, K.; Kotian, A.; Netalkar, P.; Shivalingegowda, N.; Neratur, K.L.; Revankar, V. [Dichlorido (2-(2-(1H-benzo [d] thiazol-2-yl) hydrazono) propan-1-ol) Cu (II)]: Crystal structure, Hirshfeld surface analysis and correlation of its ESI-MS behavior with [Dichlorido 3-(hydroxyimino)-2-butanone-2-(1H-benzo [d] thiazol-2-yl) hydrazone Cu (II)]. J. Mol. Struct. 2017, 1149, 357. [Google Scholar]
- Kumara, K.; Jyothi, M.; Kouser, S.; Kumar, A.H.U.; Warad, I.; Khanum, S.A.; Lokanath, N.K. Structural investigations and theoretical insights of a polymethoxy chalcone derivative: Synthesis, crystal structure, 3D energy frameworks and SARS CoV-2 docking studies. J. Mol. Struct. 2023, 1272, 134226. [Google Scholar] [CrossRef]
- Abdel-Khalik, M.M.; Elnagdi, M.H. Enaminones in organic synthesis: A novel synthesis of 1,3,5-trisubstituted benzene derivatives and of 2-substituted-5-aroylpyridines. Synth. Commun. 2002, 32, 159. [Google Scholar] [CrossRef]
- Schollmeyer, D.; Shishkin, O.V.; Rühl, T.; Vysotsky, M.O. OH–π and halogen–π interactions as driving forces in the crystal organisations of tri-bromo and tri-iodo trityl alcohols. CrystEngComm 2008, 10, 715. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 3.0; University of Western Australia: Perth, Australia, 2012. [Google Scholar]
- Spackman, M.A.; McKinnon, J.J.; Jayatilaka, D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377. [Google Scholar] [CrossRef]
- Thomas, S.P.; Dikundwar, A.G.; Sarkar, S.; Pavan, M.S.; Pal, R.; Hathwar, V.R.; Guru Row, T.N. The relevance of experimental charge density analysis in unraveling noncovalent interactions in molecular crystals. Molecules 2022, 27, 3690. [Google Scholar] [CrossRef]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals. J. Phys. Chem. Lett. 2014, 5, 4249. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N a | Symop b | R c | Electron Density | E_ele | E_pol | E_dis | E_rep | E_tot | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | −x, −y, −z | 16.62 | B3LYP/6-31G(d,p) | 1.3 | −0.2 | −2.7 | 0.0 | −1.0 | |
| 0 | −x, −y, −z | 15.48 | B3LYP/6-31G(d,p) | −6.5 | −0.0 | −7.2 | 0.0 | −13.2 | |
| 1 | −x, y + 1/2, −z + 1/2 | 14.27 | B3LYP/6-31G(d,p) | −4.4 | −0.4 | −9.1 | 0.0 | −12.9 | |
| 2 | x, −y + 1/2, z + 1/2 | 7.36 | B3LYP/6-31G(d,p) | −9.4 | −2.8 | −39.5 | 18.9 | −34.8 | |
| 0 | x, −y + 1/2, z + 1/2 | 6.67 | B3LYP/6-31G(d,p) | −19.3 | −6.1 | −63.2 | 40.5 | −54.9 | |
| 1 | −x, −y, −z | 12.43 | B3LYP/6-31G(d,p) | 14.9 | −1.7 | −45.9 | 0.0 | −25.5 | |
| 1 | −x, y + 1/2, −z + 1/2 | 11.65 | B3LYP/6-31G(d,p) | −9.9 | 0.0 | −30.6 | 23.2 | −22.7 | |
| 1 | x, y, z | 10.52 | B3LYP/6-31G(d,p) | −2.0 | 0.0 | −17.3 | 9.9 | −11.1 | |
| 0 | −x, −y, −z | 11.39 | B3LYP/6-31G(d,p) | −11.2 | −1.1 | −15.8 | 19.5 | −14.4 | |
| 0 | −x, −y, −z | 15.18 | B3LYP/6-31G(d,p) | 0.5 | −0.2 | −5.0 | 0.0 | −4.0 |
| Energy Model | K_ele | K_pol | K_disp | K_rep |
|---|---|---|---|---|
| CE-HF … HF/3-21G electron densities | 1.019 | 0.651 | 0.901 | 0.811 |
| CE-B3LYP … B3LYP/6-31G(d,p) electron densities | 1.057 | 0.740 | 0.871 | 0.618 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanain, H.M.; Al-Sharif, S.; Al-Ghamdi, H.A.; Nahari, L.M.; Al-Sulami, A.I.; Mousally, S.M.; Al-Zaydi, K.M. Synthesis, Crystal Structure, Hirshfeld Surface Analysis, Energy Framework Calculations, and Halogen Bonding Investigation of Benzene-1,3,5-triyltris((4-chlorophenyl)methanone). Crystals 2024, 14, 17. https://doi.org/10.3390/cryst14010017
Hassanain HM, Al-Sharif S, Al-Ghamdi HA, Nahari LM, Al-Sulami AI, Mousally SM, Al-Zaydi KM. Synthesis, Crystal Structure, Hirshfeld Surface Analysis, Energy Framework Calculations, and Halogen Bonding Investigation of Benzene-1,3,5-triyltris((4-chlorophenyl)methanone). Crystals. 2024; 14(1):17. https://doi.org/10.3390/cryst14010017
Chicago/Turabian StyleHassanain, Hawazen M., Samah Al-Sharif, Huda A. Al-Ghamdi, Layla M. Nahari, Ahlam I. Al-Sulami, Sameera M. Mousally, and Khadijah M. Al-Zaydi. 2024. "Synthesis, Crystal Structure, Hirshfeld Surface Analysis, Energy Framework Calculations, and Halogen Bonding Investigation of Benzene-1,3,5-triyltris((4-chlorophenyl)methanone)" Crystals 14, no. 1: 17. https://doi.org/10.3390/cryst14010017
APA StyleHassanain, H. M., Al-Sharif, S., Al-Ghamdi, H. A., Nahari, L. M., Al-Sulami, A. I., Mousally, S. M., & Al-Zaydi, K. M. (2024). Synthesis, Crystal Structure, Hirshfeld Surface Analysis, Energy Framework Calculations, and Halogen Bonding Investigation of Benzene-1,3,5-triyltris((4-chlorophenyl)methanone). Crystals, 14(1), 17. https://doi.org/10.3390/cryst14010017