
Oxygen-Vacancy-Rich Fe@Fe3O4 Boosting Fenton Chemistry

 and
and 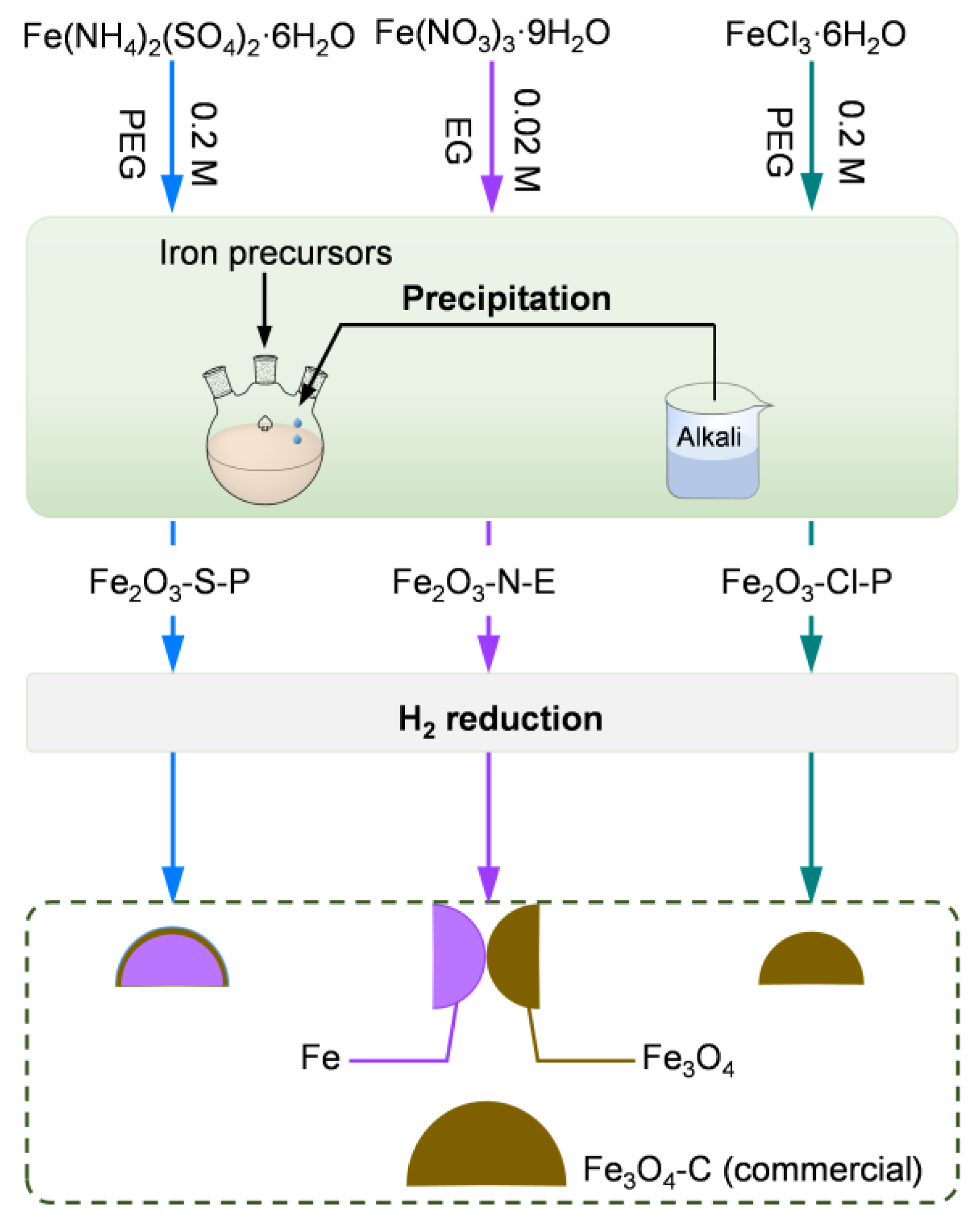{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of the Iron Oxides
2.1.1. Synthesis of the Iron Oxides
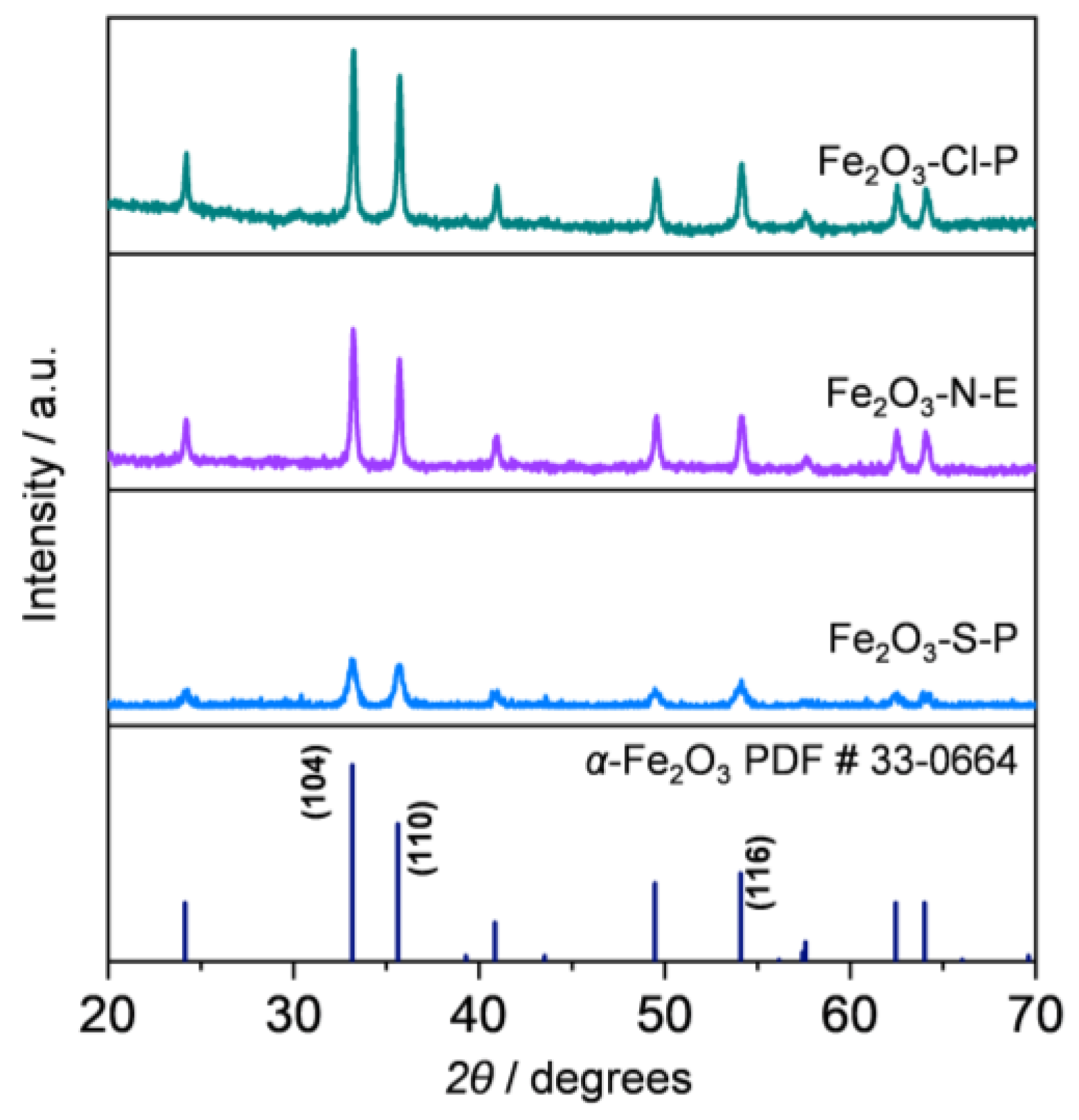
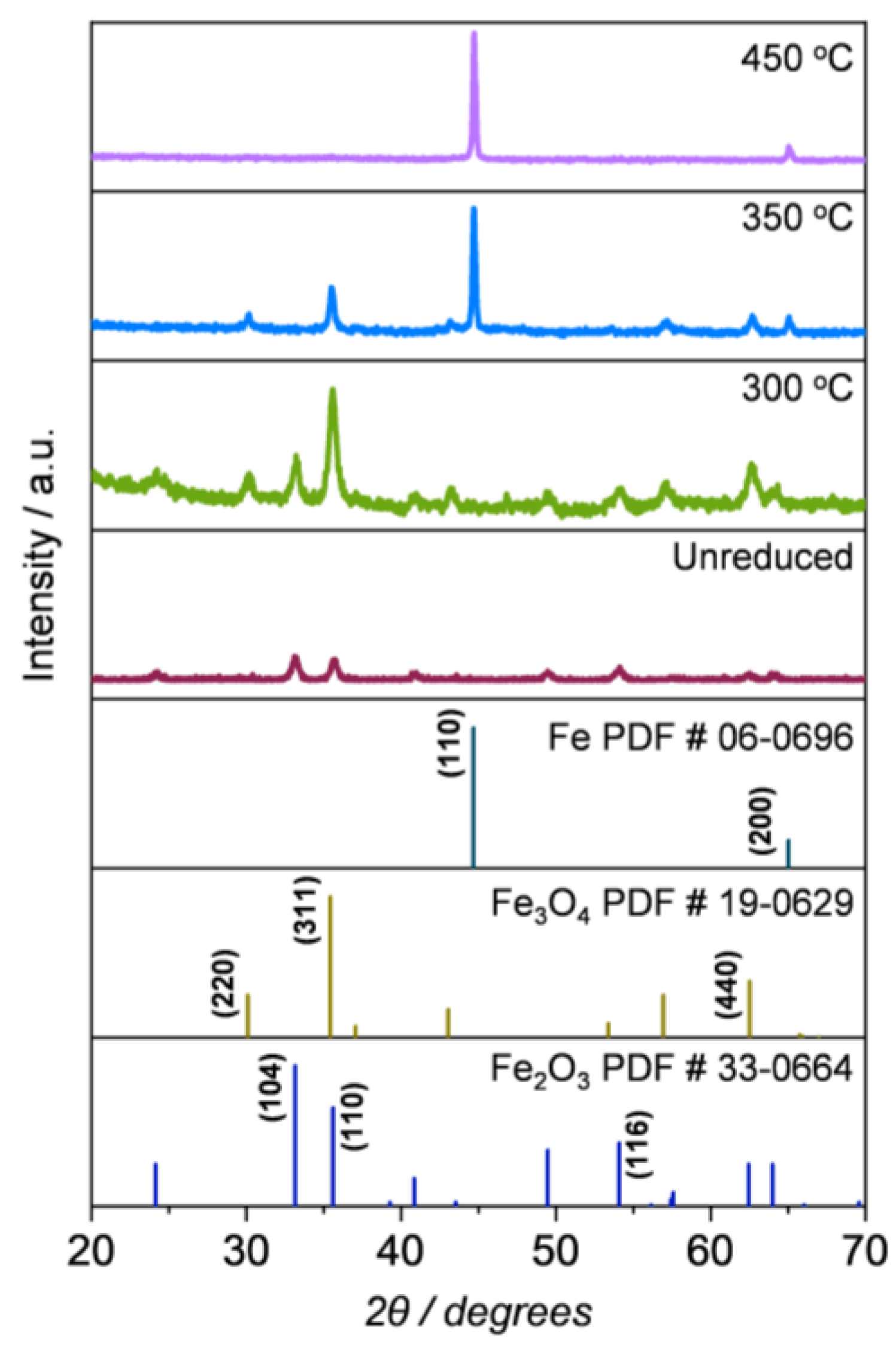
2.1.2. Compositional and Structural Analyses of the Iron Oxides
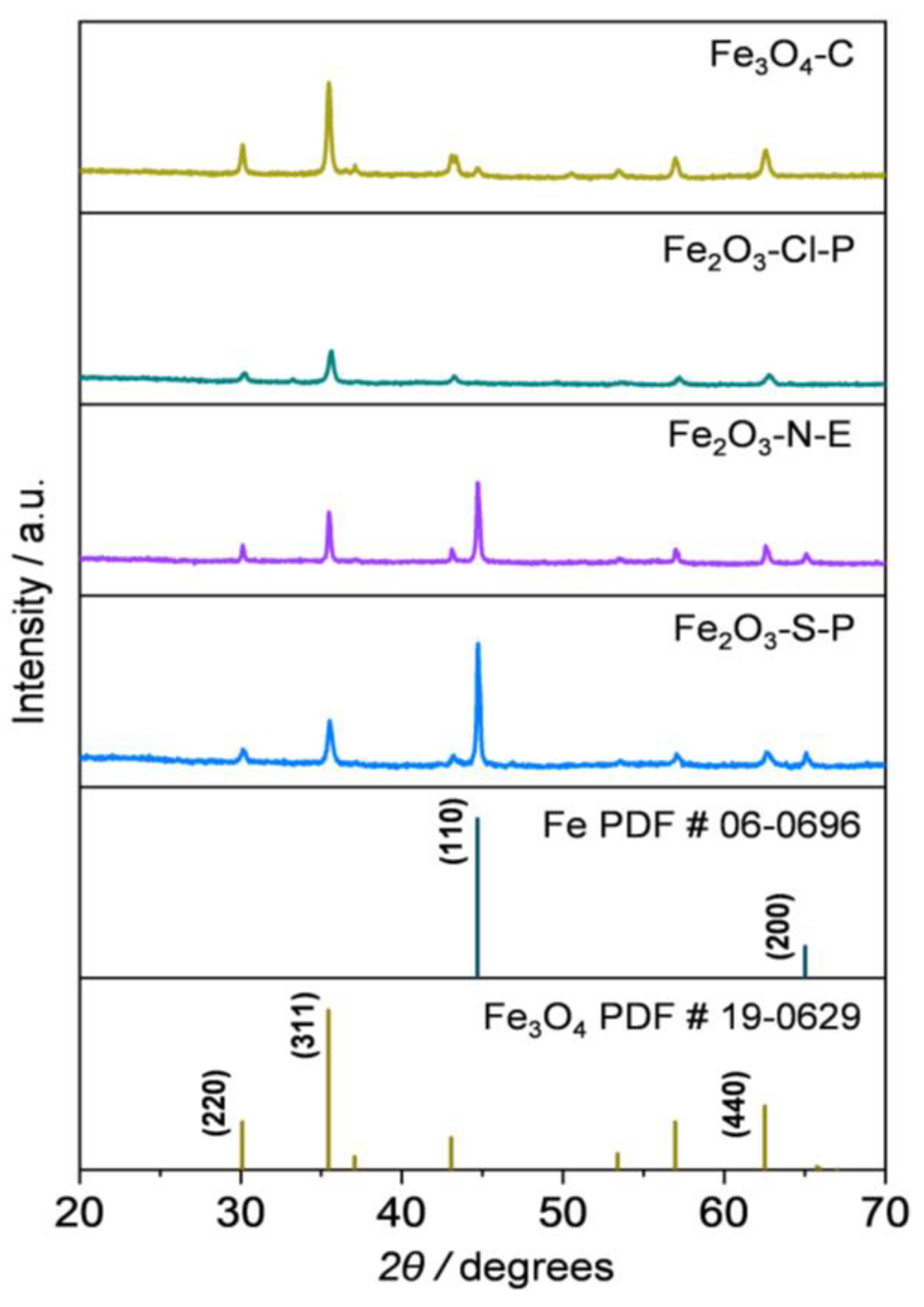
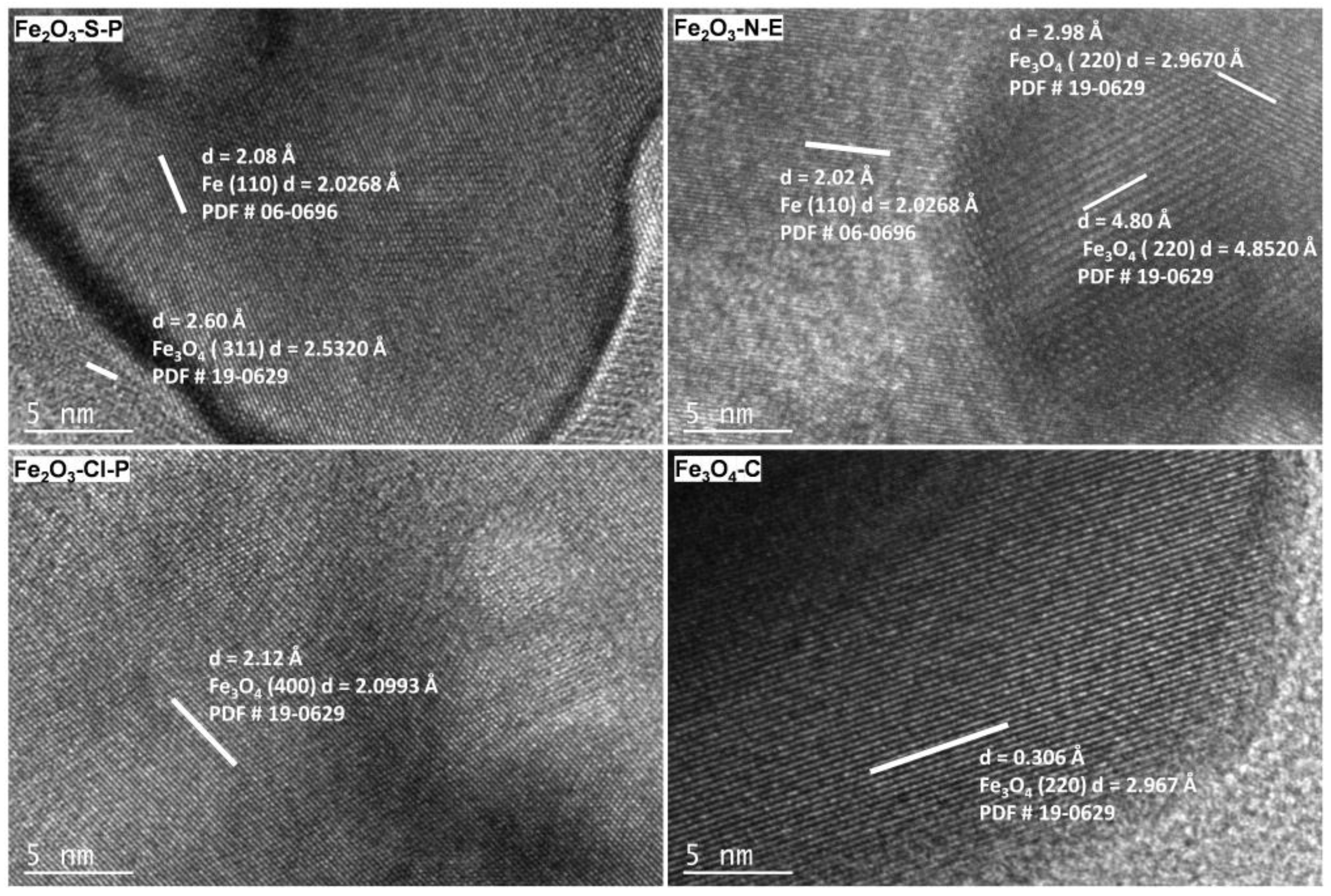
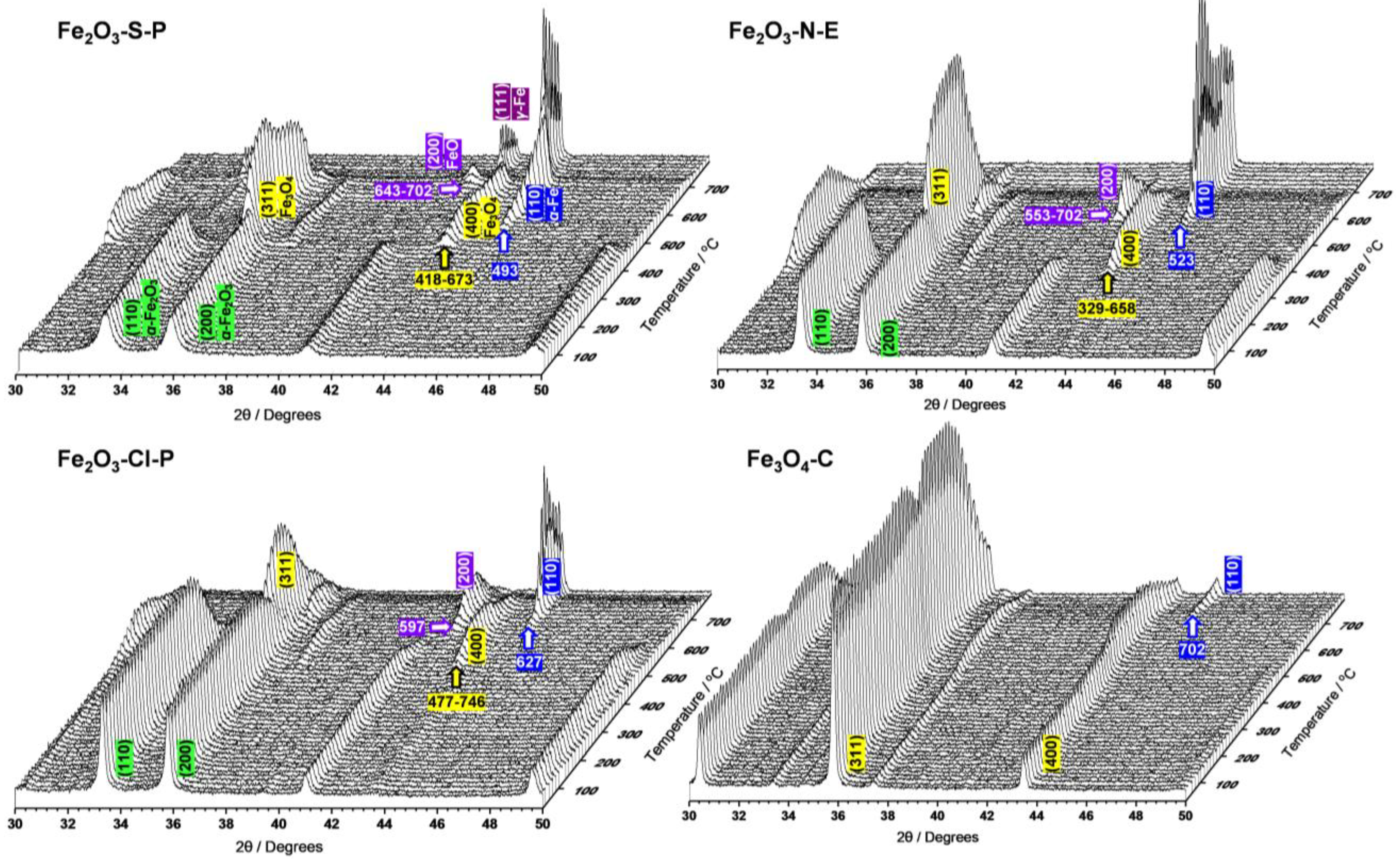
2.1.3. Compositional and Structural Analyses of the Reduced Iron Oxides
2.1.4. Redox Properties of the Iron Oxides
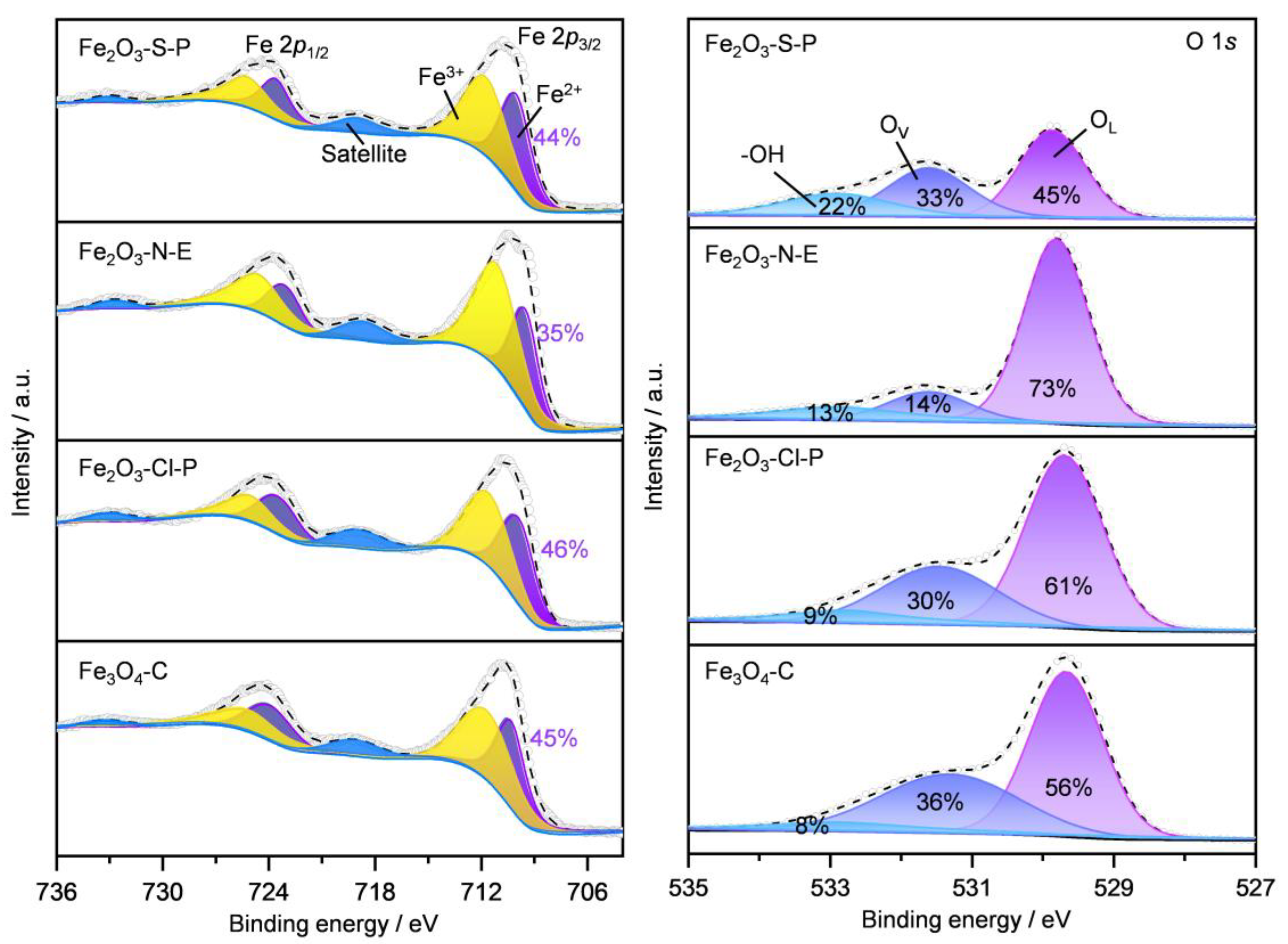
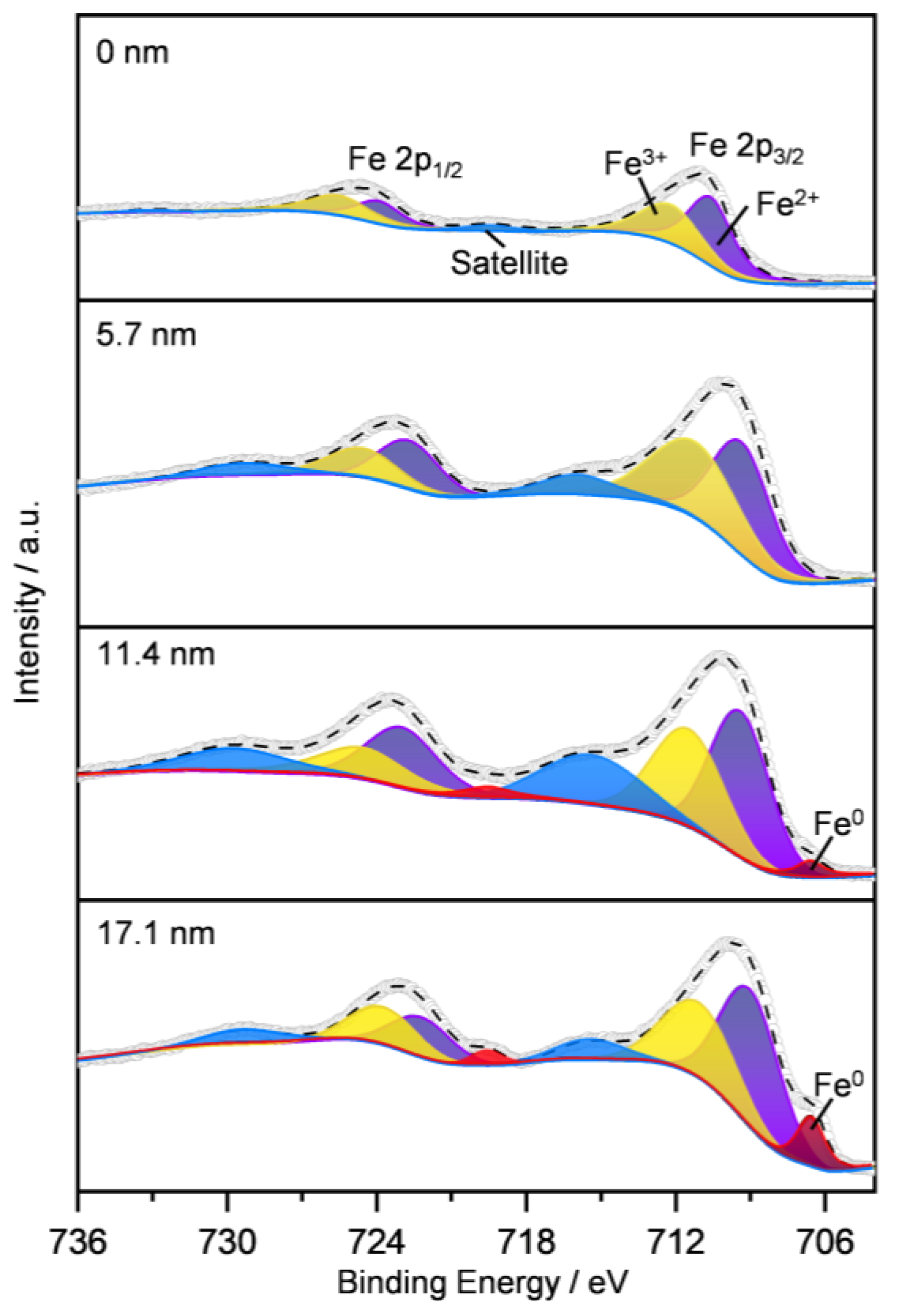
2.1.5. Surface Iron Species and Oxygen Vacancy
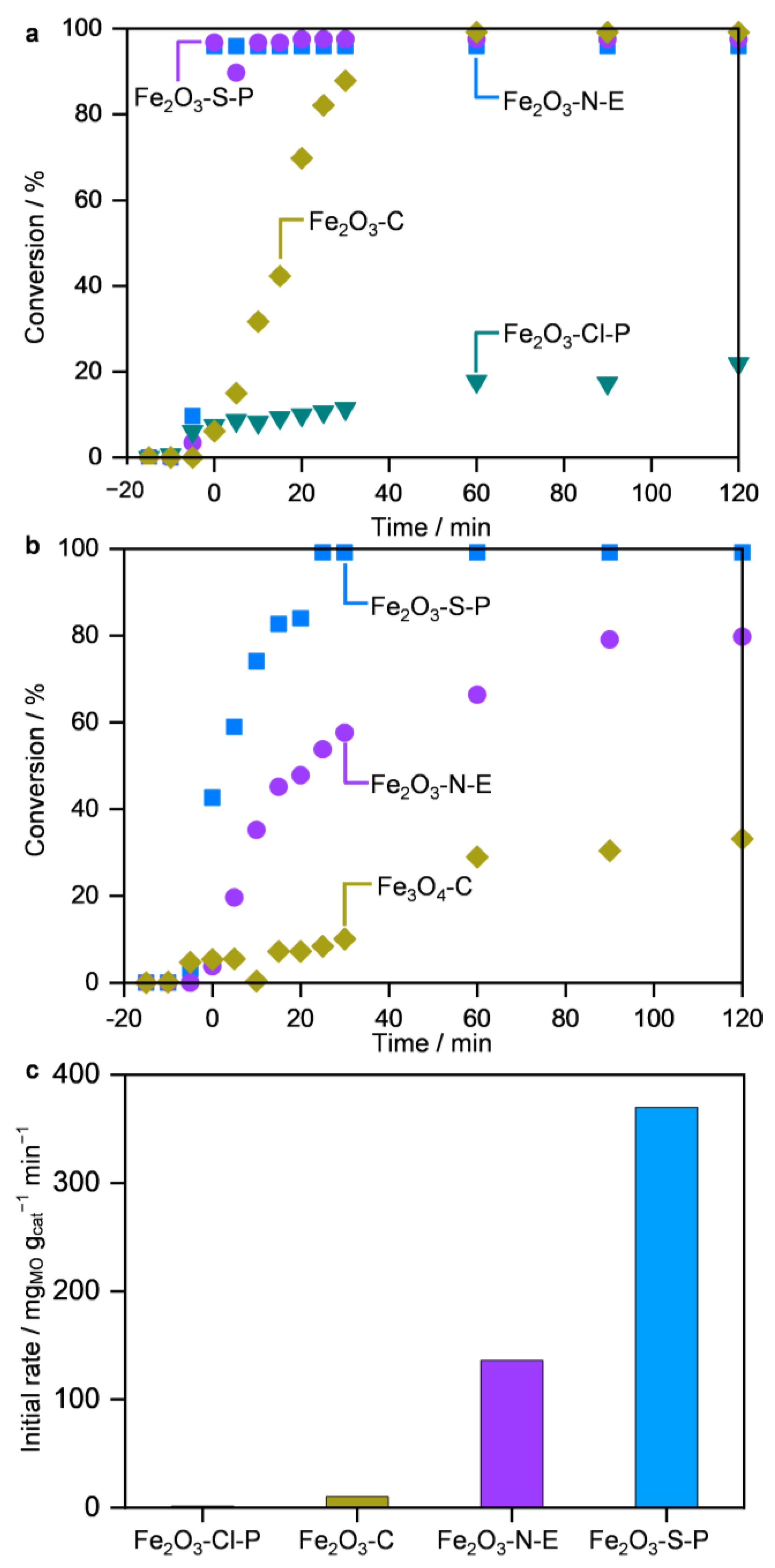
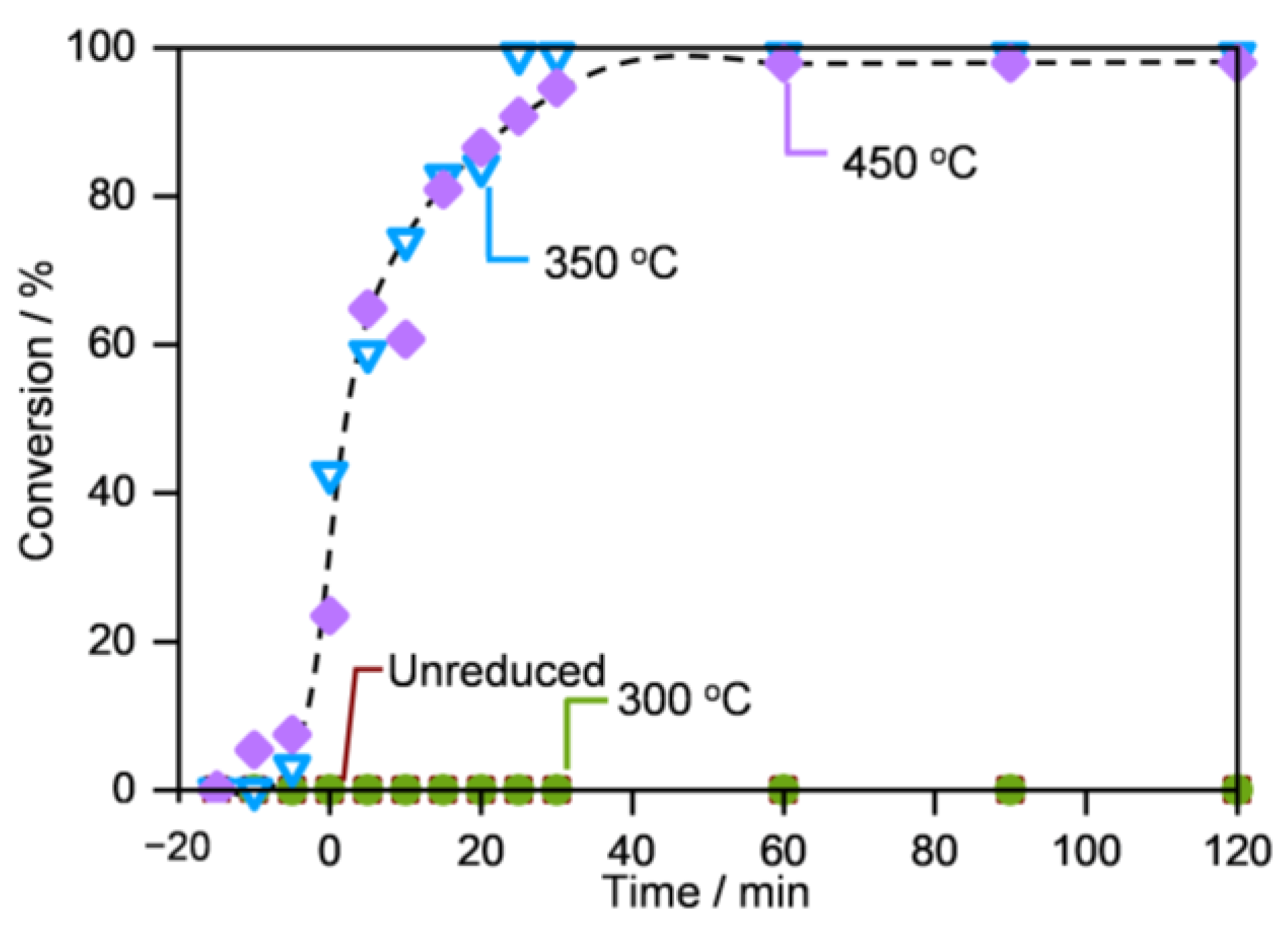
2.2. Performance in MO Decomposition
2.3. Origins of the Divergent Catalytic Performance
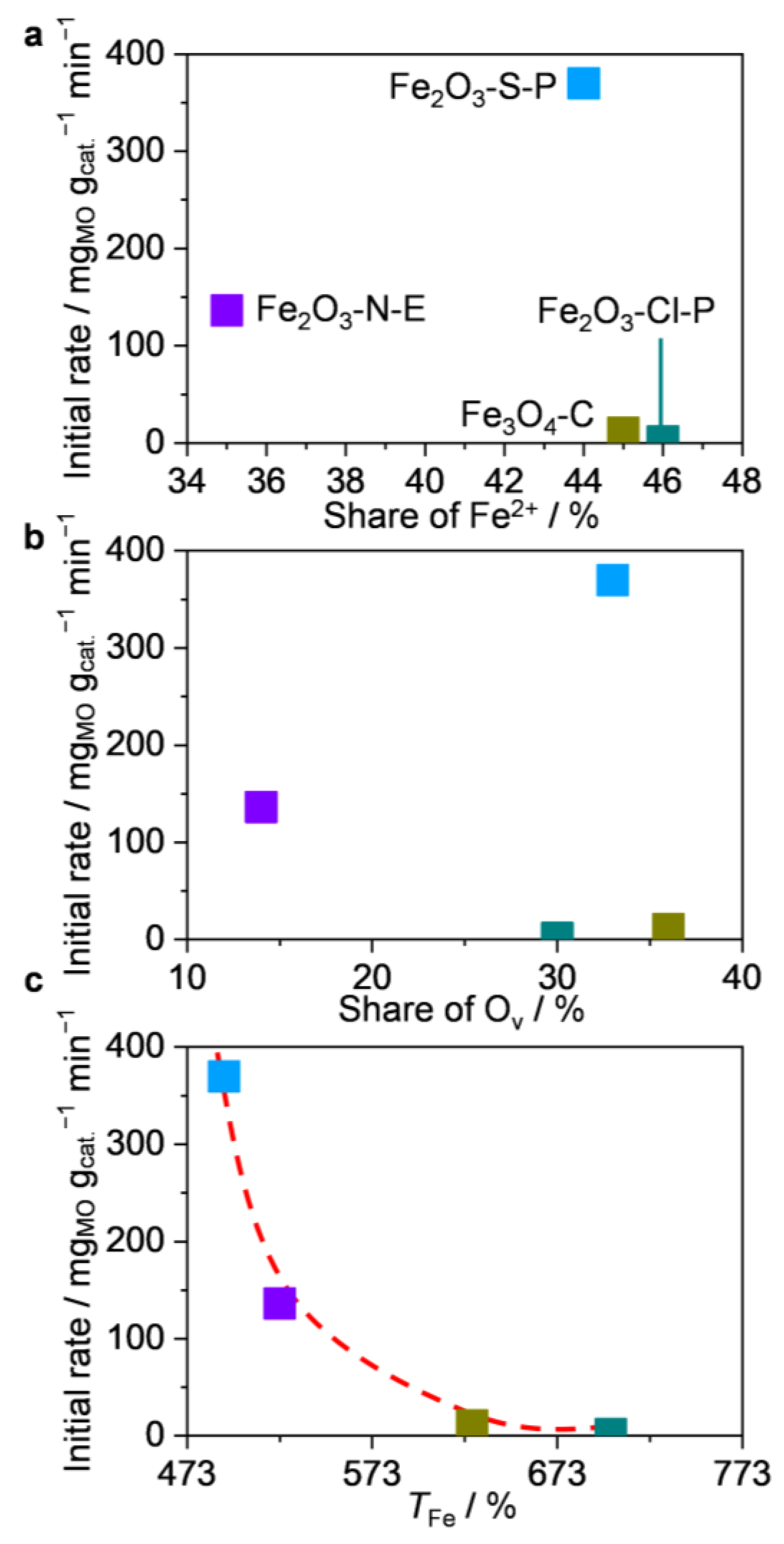
2.3.1. Structure–Performance Relationships
2.3.2. Importance of the Fe@Fe3O4 Interfaces
2.3.3. Discussions on the Origin of Superior Activity
3. Materials and Methods
3.1. Synthesis of the Iron Oxides
3.2. Material Characterizations
3.3. Activity Evaluation of the Iron-Based Materials in Decomposition of Methyl Orange
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, N.H.; Reinhard, M.; Gin, K.Y.-H. Occurrence and fate of emerging contaminants in municipal wastewater treatment plants from different geographical regions-a review. Water Res. 2018, 133, 182–207. [Google Scholar] [CrossRef]
- Hodges, B.C.; Cates, E.L.; Kim, J.H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 2018, 13, 642–650. [Google Scholar] [CrossRef]
- Boczkaj, G.; Fernandes, A. Wastewater treatment by means of advanced oxidation processes at basic pH conditions: A review. Chem. Eng. J. 2017, 320, 608–633. [Google Scholar] [CrossRef]
- Altalhi, T.A.; Ibrahim, M.M.; Mersal, G.A.M.; Mahmoud, M.H.H.; Kumeria, T.; El-Desouky, M.G.; El-Bindary, A.A.; El-Bindary, M.A. Adsorption of doxorubicin hydrochloride onto thermally treated green adsorbent: Equilibrium, kinetic and thermodynamic studies. J. Mol. Struct. 2022, 1263, 133160. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes. J. Hazard. Mater. 2014, 275, 121–135. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, R.; Xi, Y.; Zhu, J.; Zhu, G.; He, H. Strategies for enhancing the heterogeneous Fenton catalytic reactivity: A review. Appl. Catal. B 2019, 255, 117739. [Google Scholar] [CrossRef]
- He, J.; Yang, X.; Men, B.; Wang, D. Interfacial mechanisms of heterogeneous Fenton reactions catalyzed by iron-based materials: A review. J. Environ. Sci. 2016, 39, 97–109. [Google Scholar] [CrossRef]
- Rahim Pouran, S.; Abdul Raman, A.A.; Wan Daud, W.M.A. Review on the application of modified iron oxides as heterogeneous catalysts in Fenton reactions. J. Clean. Prod. 2014, 64, 24–35. [Google Scholar] [CrossRef]
- Zepp, R.G.; Faust, B.C.; Hoigne, J. Hydroxyl radical formation in aqueous reactions (pH 3–8) of iron (II) with hydrogen peroxide: The photo-Fenton reaction. Environ. Sci. Technol. 1992, 26, 313–319. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sánchez-Pérez, J.; Gernjak, W.; Maldonado, M.; Pérez-Estrada, L.; Pulgarín, C. A combined solar photocatalytic-biological field system for the mineralization of an industrial pollutant at pilot scale. Catal. Today 2007, 122, 150–159. [Google Scholar] [CrossRef]
- Malato, S.; Fernández-Ibáñez, P.; Maldonado, M.I.; Blanco, J.; Gernjak, W. Decontamination and disinfection of water by solar photocatalysis: Recent overview and trends. Catal. Today 2009, 147, 1–59. [Google Scholar] [CrossRef]
- Brillas, E.; Sirés, I.; Oturan, M.A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109, 6570–6631. [Google Scholar] [CrossRef] [PubMed]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 1. Combinative and hybrid sonophotochemical oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 3409–3420. [Google Scholar] [CrossRef]
- Cai, W.; Chen, F.; Shen, X.; Chen, L.; Zhang, J. Enhanced catalytic degradation of AO7 in the CeO2–H2O2 system with Fe3+ doping. Appl. Catal. B 2010, 101, 160–168. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Advanced oxidation process based on the Cr (III)/Cr (VI) redox cycle. Environ. Sci. Technol. 2011, 45, 9332–9338. [Google Scholar] [CrossRef]
- Gabriel, J.; Baldrian, P.; Verma, P.; Cajthaml, T.; Merhautová, V.; Eichlerová, I.; Stoytchev, I.; Trnka, T.; Stopka, P.; Nerud, F. Degradation of BTEX and PAHs by Co (II) and Cu (II)-based radical-generating systems. Appl. Catal. B 2004, 51, 159–164. [Google Scholar] [CrossRef]
- Gabriel, J.; Shah, V.; Nesměrák, K.; Baldrian, P.; Nerud, F. Degradation of polycyclic aromatic hydrocarbons by the copper (II)-hydrogen peroxide system. Folia Microbiol. 2000, 45, 573–575. [Google Scholar] [CrossRef]
- Han, Y.-F.; Chen, F.; Zhong, Z.; Ramesh, K.; Chen, L.; Jian, D.; Ling, W.W. Complete oxidation of low concentration ethanol in aqueous solution with H2O2 on nanosized Mn3O4/SBA-15 catalyst. Chem. Eng. J. 2007, 134, 276–281. [Google Scholar] [CrossRef]
- Hu, Z.; Leung, C.-F.; Tsang, Y.-K.; Du, H.; Liang, H.; Qiu, Y.; Lau, T.-C. A recyclable polymer-supported ruthenium catalyst for the oxidative degradation of bisphenol A in water using hydrogen peroxide. New J. Chem. 2011, 35, 149–155. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Huang, J.; Jones, A.; Waite, T.D.; Chen, Y.; Huang, X.; Rosso, K.M.; Kappler, A.; Mansor, M.; Tratnyek, P.G.; Zhang, H. Fe(II) Redox chemistry in the environment. Chem. Rev. 2021, 121, 8161–8233. [Google Scholar] [CrossRef]
- Danielsen, K.M.; Hayes, K.F. pH Dependence of carbon tetrachloride reductive dechlorination by magnetite. Environ. Sci. Technol. 2004, 38, 4745–4752. [Google Scholar] [CrossRef]
- Gorski, C.A.; Nurmi, J.T.; Tratnyek, P.G.; Hofstetter, T.B.; Scherer, M.M. Redox behavior of magnetite: Implications for contaminant reduction. Environ. Sci. Technol. 2010, 44, 55–60. [Google Scholar] [CrossRef]
- Zhong, Y.; Yu, L.; Chen, Z.F.; He, H.; Ye, F.; Cheng, G.; Zhang, Q. Microwave-assisted synthesis of Fe3O4 nanocrystals with predominantly exposed facets and their heterogeneous UVA/Fenton catalytic activity. ACS Appl. Mater. Interface. 2017, 9, 29203–29212. [Google Scholar] [CrossRef]
- Jaramillo-Páez, C.; Navío, J.A.; Hidalgo, M.; Bouziani, A.; El Azzouzi, M. Mixed α-Fe2O3/Bi2WO6 oxides for photoassisted hetero-Fenton degradation of Methyl Orange and Phenol. J. Photochem. Photobiol. A 2017, 332, 521–533. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, B.; Wang, Q.; Xing, S. Facile synthesis of α-FeOOH/γ-Fe2O3 by a pH gradient method and the role of γ-Fe2O3 in H2O2 activation under visible light irradiation. Chem. Eng. J. 2018, 354, 75–84. [Google Scholar] [CrossRef]
- Krumina, L.; Lyngsie, G.; Tunlid, A.; Persson, P. Oxidation of a dimethoxyhydroquinone by ferrihydrite and goethite nanoparticles: Iron reduction versus surface catalysis. Environ. Sci. Technol. 2017, 51, 9053–9061. [Google Scholar] [CrossRef]
- Su, S.; Liu, Y.; Liu, X.; Jin, W.; Zhao, Y. Transformation pathway and degradation mechanism of methylene blue through β-FeOOH@ GO catalyzed photo-Fenton-like system. Chemosphere 2019, 218, 83–92. [Google Scholar] [CrossRef]
- He, D.; Chen, Y.; Situ, Y.; Zhong, L.; Huang, H. Synthesis of ternary g-C3N4/Ag/γ-FeOOH photocatalyst: An integrated heterogeneous Fenton-like system for effectively degradation of azo dye methyl orange under visible light. Appl. Surf. Sci. 2017, 425, 862–872. [Google Scholar] [CrossRef]
- Chen, Z.X.; Jin, X.Y.; Chen, Z.; Megharaj, M.; Naidu, R. Removal of methyl orange from aqueous solution using bentonite-supported nanoscale zero-valent iron. J. Colloid Interface Sci. 2011, 363, 601–607. [Google Scholar] [CrossRef]
- Xie, S.; Huang, P.; Kruzic, J.J.; Zeng, X.; Qian, H. A highly efficient degradation mechanism of methyl orange using Fe-based metallic glass powders. Sci. Rep. 2016, 6, 21947. [Google Scholar] [CrossRef]
- Yuan, N.; Zhang, G.; Guo, S.; Wan, Z. Enhanced ultrasound-assisted degradation of methyl orange and metronidazole by rectorite-supported nanoscale zero-valent iron. Ultrason. Sonochem. 2016, 28, 62–68. [Google Scholar] [CrossRef]
- Zhu, L.; Ai, Z.; Ho, W.; Zhang, L. Core–shell Fe–Fe2O3 nanostructures as effective persulfate activator for degradation of methyl orange. Sep. Purif. Technol. 2013, 108, 159–165. [Google Scholar] [CrossRef]
- Huang, Q.; Cao, M.; Ai, Z.; Zhang, L. Reactive oxygen species dependent degradation pathway of 4-chlorophenol with Fe@Fe2O3 core–shell nanowires. Appl. Catal. B 2015, 162, 319–326. [Google Scholar] [CrossRef]
- Shen, W.; Lin, F.; Jiang, X.; Li, H.; Ai, Z.; Zhang, L. Efficient removal of bromate with core-shell Fe@Fe2O3 nanowires. Chem. Eng. J. 2017, 308, 880–888. [Google Scholar] [CrossRef]
- Shi, J.; Ai, Z.; Zhang, L. Fe@Fe2O3 core-shell nanowires enhanced Fenton oxidation by accelerating the Fe (III)/Fe(II) cycles. Wat. Res. 2014, 59, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ai, Z.; Cao, M.; Zhang, L. Ferrous ions promoted aerobic simazine degradation with Fe@Fe2O3 core–shell nanowires. Appl. Catal. B 2014, 150, 1–11. [Google Scholar] [CrossRef]
- Ai, Z.; Gao, Z.; Zhang, L.; He, W.; Yin, J.J. Core–shell structure dependent reactivity of Fe@Fe2O3 nanowires on aerobic degradation of 4-chlorophenol. Environ. Sci. Technol. 2013, 47, 5344–5352. [Google Scholar] [CrossRef]
- Daliran, S.; Khajeh, M.; Oveisi, A.R. A porous Fe-based porphyrinic metal–organic framework for highly effective removal of organic azo-dye. Appl. Organomet. Chem. 2022, 36, e6830. [Google Scholar] [CrossRef]
- Costa, R.C.; Moura, F.C.; Ardisson, J.; Fabris, J.; Lago, R. Highly active heterogeneous Fenton-like systems based on Fe0/Fe3O4 composites prepared by controlled reduction of iron oxides. Appl. Catal. B 2008, 83, 131–139. [Google Scholar] [CrossRef]
- Leupin, O.X.; Hug, S.J. Oxidation and removal of arsenic (III) from aerated groundwater by filtration through sand and zero-valent iron. Water Res. 2005, 39, 1729–1740. [Google Scholar] [CrossRef]
- Wu, H.; Ai, Z.; Zhang, L. Anoxic and oxic removal of humic acids with Fe@Fe2O3 core–shell nanowires: A comparative study. Water Res. 2014, 52, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, M.; Ai, Z.; Zhang, L. Dramatically enhanced aerobic atrazine degradation with Fe@Fe2O3 core–shell nanowires by tetrapolyphosphate. Environ. Sci. Technol. 2014, 48, 3354–3362. [Google Scholar] [CrossRef]
- Chen, X.; Su, J.; Meng, Y.; Yu, M.; Zheng, M.; Sun, Y.; Xi, B. Oxygen vacancy promoted heterogeneous Fenton-like degradation of sulfamethazine by chlorine-incorporated micro zero-valent iron. Chem. Eng. J. 2023, 463, 142360. [Google Scholar] [CrossRef]
- Deng, Y.; Tian, X.; Shen, G.; Gao, Y.; Lin, C.; Ling, L.; Cheng, F.; Liao, S.; Zhang, S. Coupling hollow Fe3O4 nanoparticles with oxygen vacancy on mesoporous carbon as a high-efficiency ORR electrocatalyst for Zn-air battery. J. Colloid Interface Sci. 2020, 567, 410–418. [Google Scholar] [CrossRef]
- Li, H.; Shang, J.; Yang, Z.; Shen, W.; Ai, Z.; Zhang, L. Oxygen vacancy associated surface Fenton chemistry: Surface structure dependent hydroxyl radicals generation and substrate dependent reactivity. Environ. Sci. Technol. 2017, 51, 5685–5694. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Puigdollers, A.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The role of metal/oxide Interfaces in the formation of oxygen vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, R.; Tan, R.; Lv, Y.; Mou, X.; Qian, J.; Lin, R.; Fang, P.; Kan, W. Oxygen-Vacancy-Rich Fe@Fe3O4 Boosting Fenton Chemistry. Catalysts 2023, 13, 1057. https://doi.org/10.3390/catal13071057
Zheng R, Tan R, Lv Y, Mou X, Qian J, Lin R, Fang P, Kan W. Oxygen-Vacancy-Rich Fe@Fe3O4 Boosting Fenton Chemistry. Catalysts. 2023; 13(7):1057. https://doi.org/10.3390/catal13071057
Chicago/Turabian StyleZheng, Rongwei, Ruifan Tan, Yali Lv, Xiaoling Mou, Junqiao Qian, Ronghe Lin, Ping Fang, and Weidong Kan. 2023. "Oxygen-Vacancy-Rich Fe@Fe3O4 Boosting Fenton Chemistry" Catalysts 13, no. 7: 1057. https://doi.org/10.3390/catal13071057
APA StyleZheng, R., Tan, R., Lv, Y., Mou, X., Qian, J., Lin, R., Fang, P., & Kan, W. (2023). Oxygen-Vacancy-Rich Fe@Fe3O4 Boosting Fenton Chemistry. Catalysts, 13(7), 1057. https://doi.org/10.3390/catal13071057