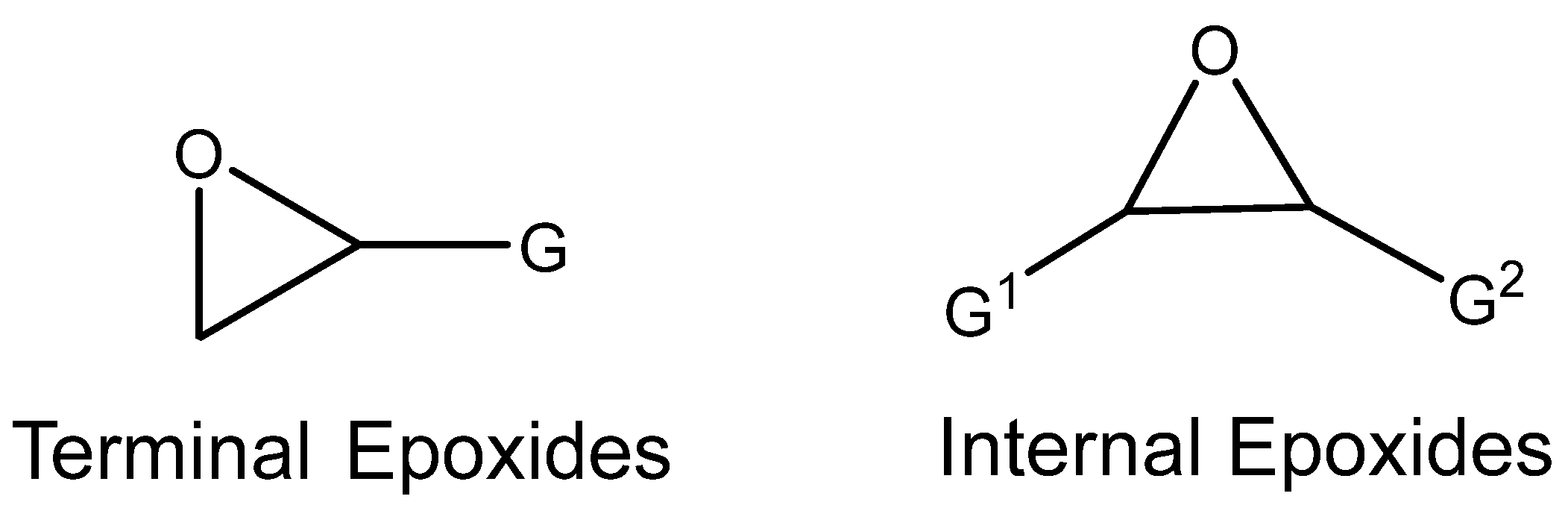
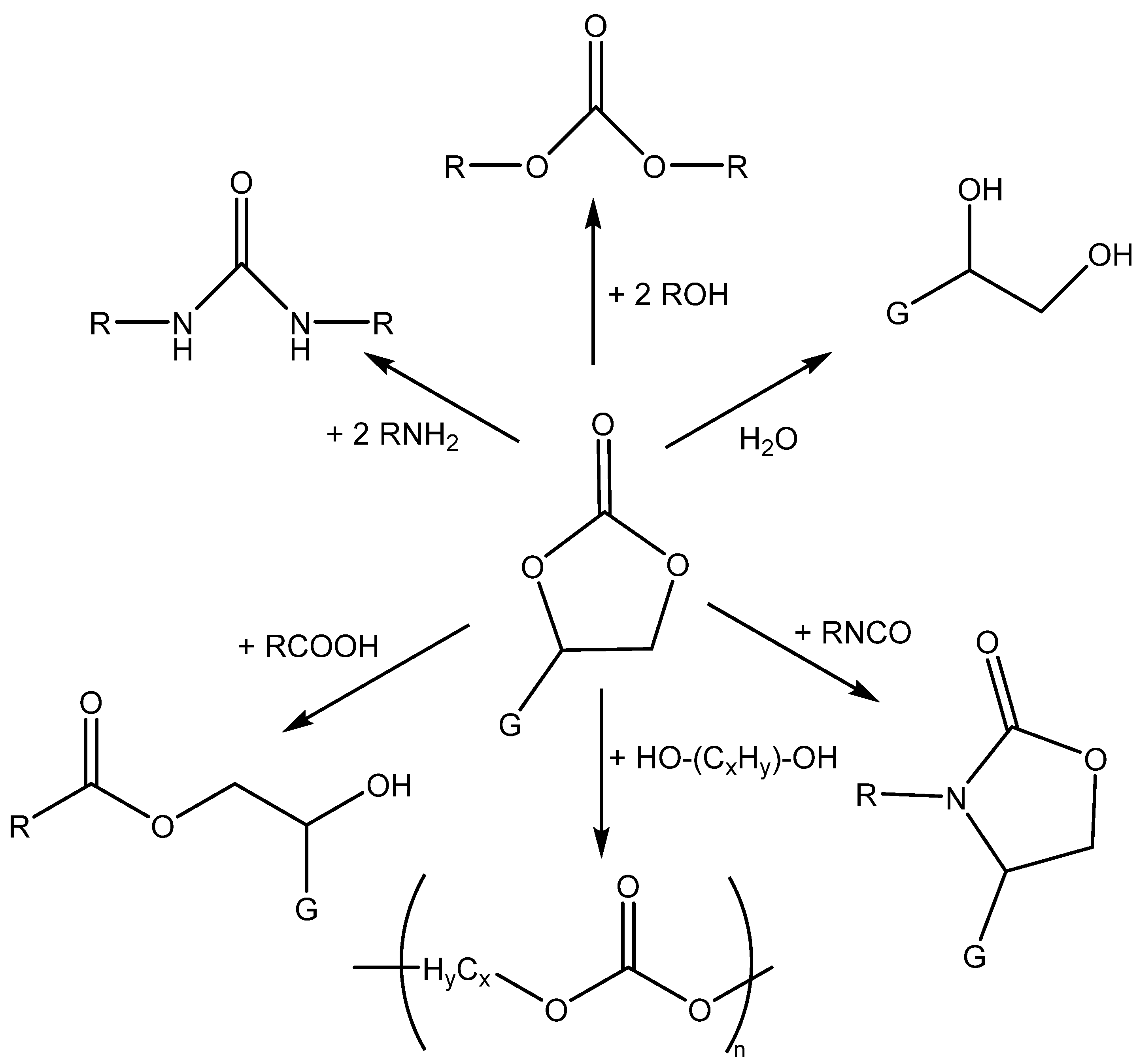
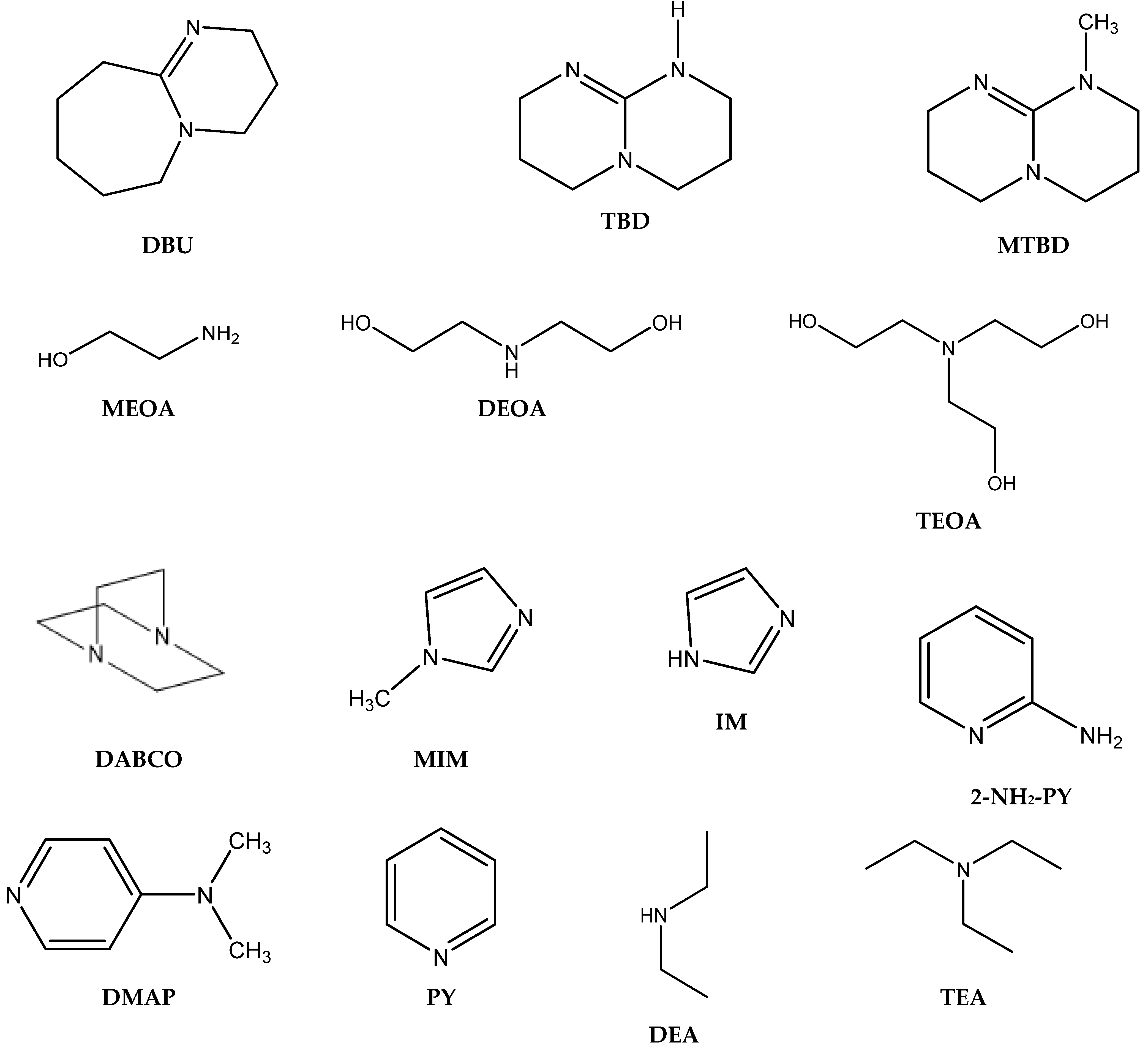
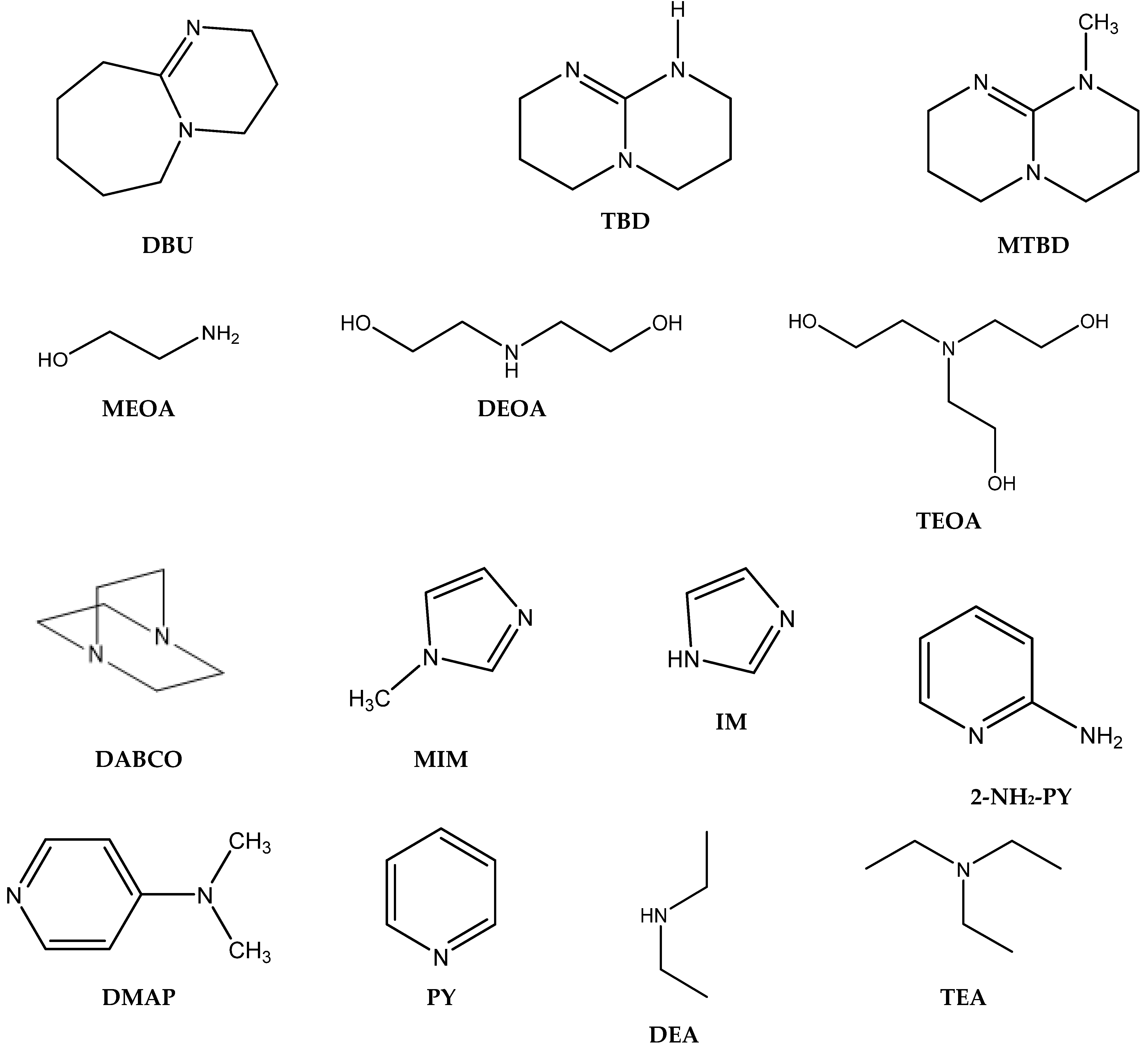
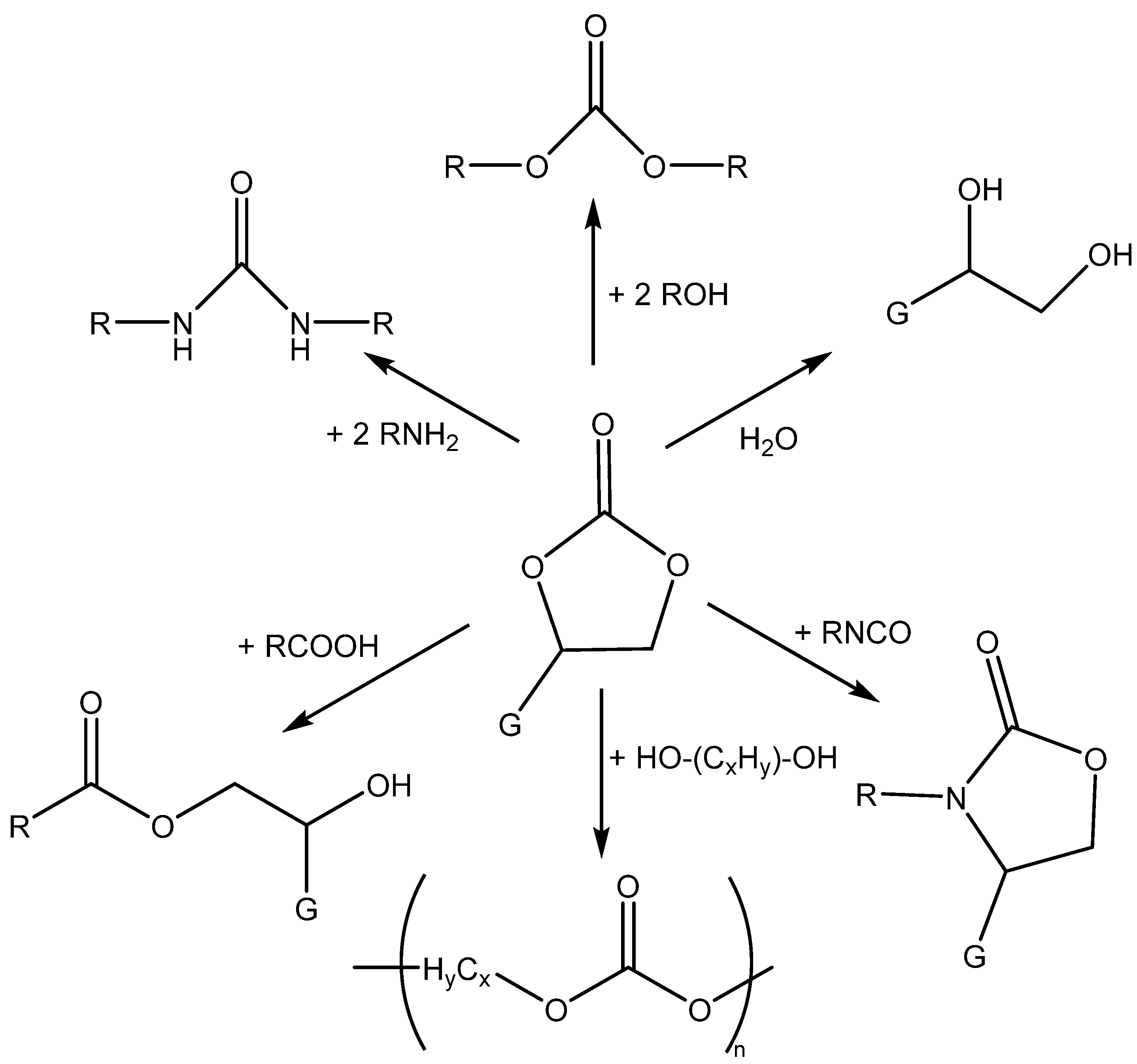
3.1. Catalytically Active Amines and Their Salts
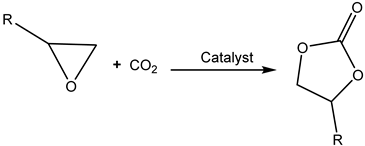
Generally, the effective absorption of CO2 into the liquid phase and its subsequent activation of chemisorbed CO2 are important steps for the subsequent cycloaddition reaction of CO2 with epoxides.
The alkaline absorbents described in the literature as being capable of effectively absorbing CO
2 are different amines [
28], including amidines such as 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) [
28,
29,
30], guanidines such as 1,5,7-triazabicyclo[4.4.0]dec-5-enium (TBD) [
28,
29], and azaheterocycles such as pyridines and imidazoles [
31,
32], which are efficient for the chemisorption and activation of CO
2.
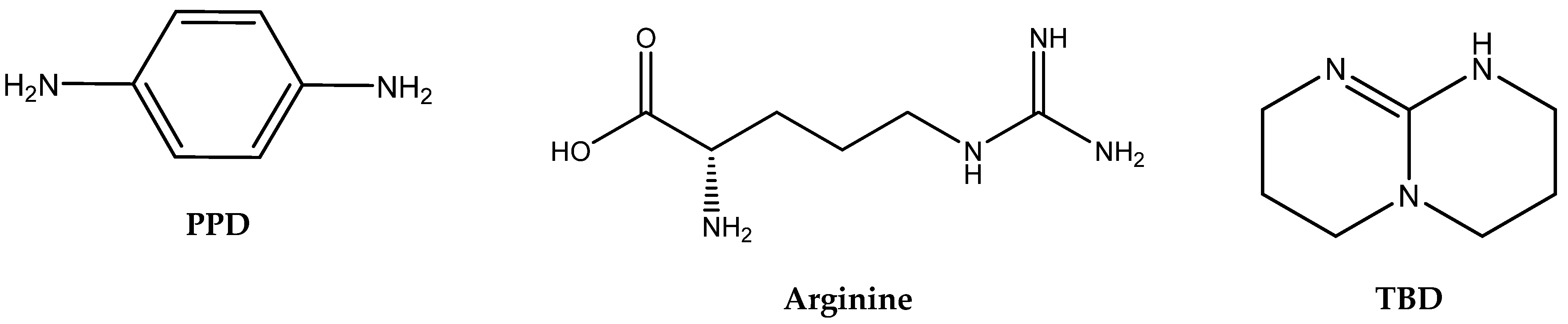
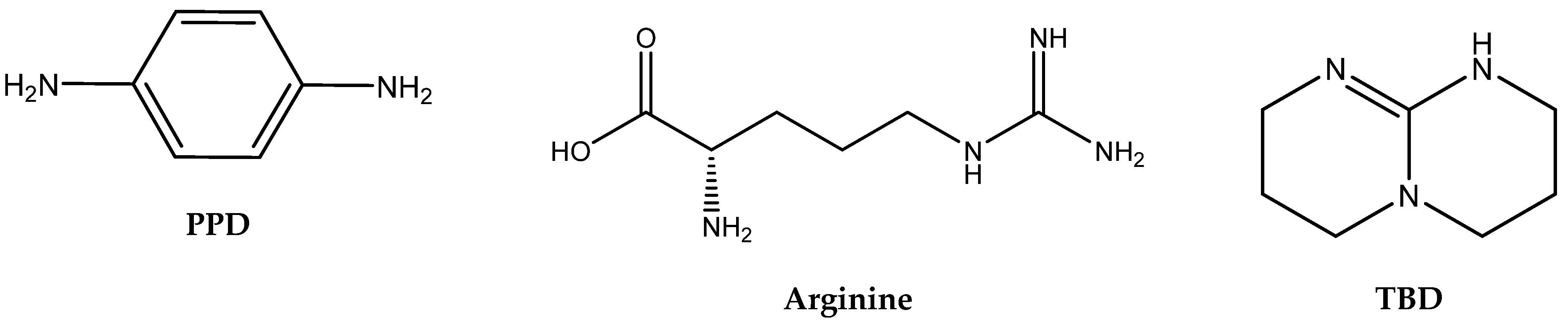
The correlation between the structures of different organic amines and their catalytic activity in the cycloaddition of CO
2 was studied by Yu et al. [
28]. Their article compares the catalytic activity of a variety of nucleophilic aliphatic amines (ethanolamine, bis-(3-aminopropyl)-amine, oleylamine), basic aminoacid arginine, nucleophilic aromatic amines (1,2-phenylenediamine, 2-aminobenzylamine) and non-nucleophilic cyclic amines such as TBD and DBU for the cycloaddition of CO
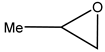
2 to methyloxirane (propylene oxide, PO) [
28].
It is well known that the chemisorption ability of CO
2 (formation of carbamate) increases with the increasing basicity of amine [
33,
34]. Yu et al. observed no apparent relationship of the pK
a value of conjugated acids of the tested amines with respect to their catalytic activity in the case of propylene carbonate (PC) formation (
Figure 2,
Table 1). The authors observed the lowest propylene carbonate conversion when applying aliphatic amines.
Low conversion was even observed in the case of amine forming intramolecular hydrogen bonds (1,2-phenylenediamine and 2-aminobenzylamine).
In contrast, aromatic amines (such as 1,4-phenylenediamine) and amidines or guanidines involving conjugated “N=C–N” structures (arginine, TBD) were found to be the most active ones [
28].
According to the published information [
35,
36], tertiary amines are often higher in activity than primary or secondary ones (
Table 2, Entries 5, 10–12). It has been proven that amidines containing the “N=C–N” bond in their structures are particularly favorable for the cycloaddition of CO
2 with epoxides (
Table 1, Entries 6–8, 17 and
Table 2, Entries 3 and 12) [
2,
3,
4,
6,
32]. The above-mentioned observations are in agreement with the statement of North et al. that for the efficient activation of CO
2, compounds that nucleophilically attack CO
2 but not the epoxide ring are sought out [
12].
Azzouz et al. demonstrated the effective utilization of 2-aminopyridine (2-NH
2-PY) as a catalyst (using 10 molar % of 2-NH
2-PY) for the carbonation of different terminal epoxides at 60–85 °C and 1 MPa of CO
2 [
35]. This carbonation was performed even at pilot scale.
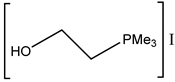
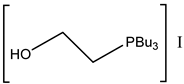
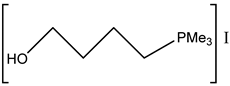
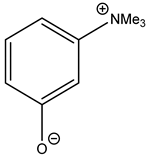
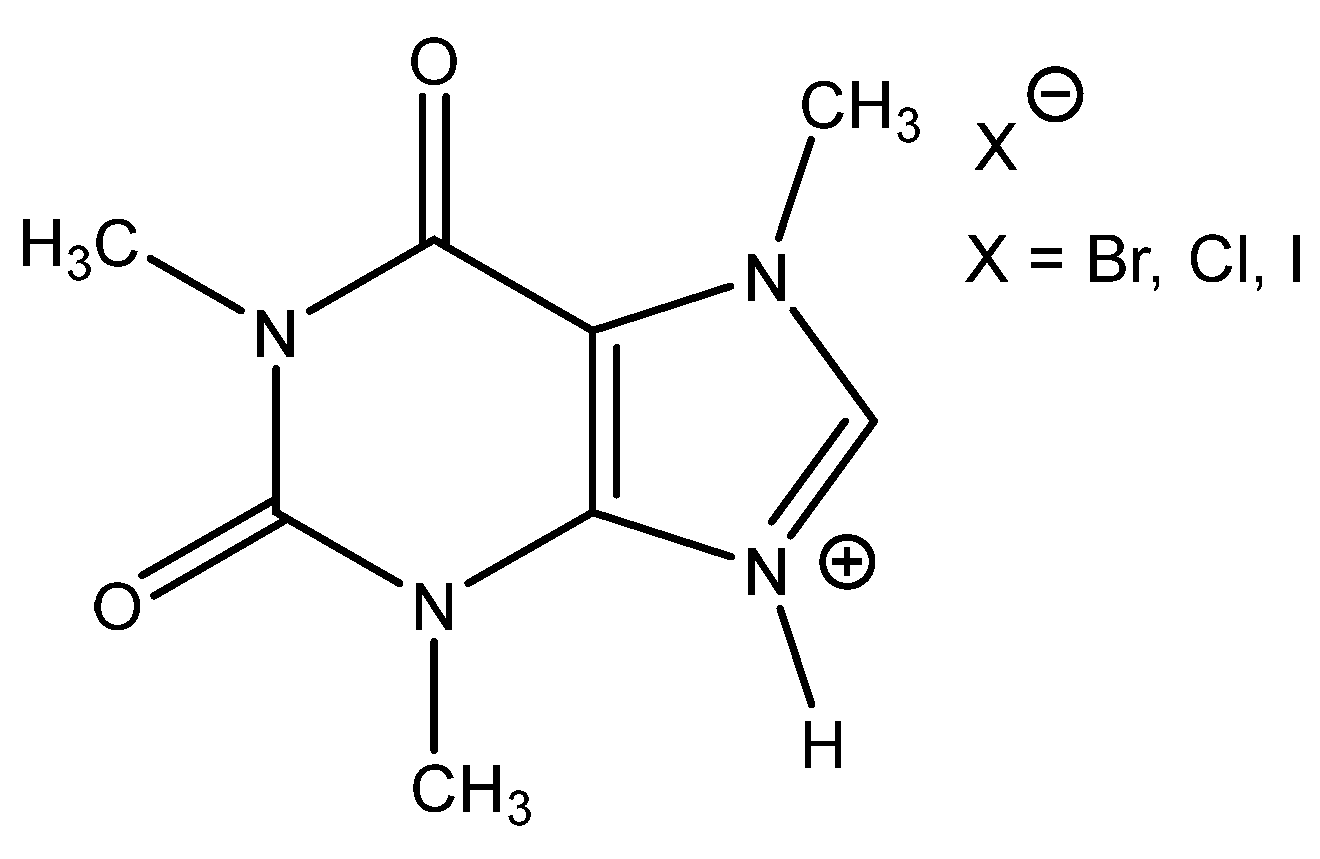
Interestingly, corresponding salts with protic (Bronsted) acids (base.HA) of the above-mentioned non-nucleophilic amines (DBU.HA,
N-methylimidazole (MIM.HA),
N,
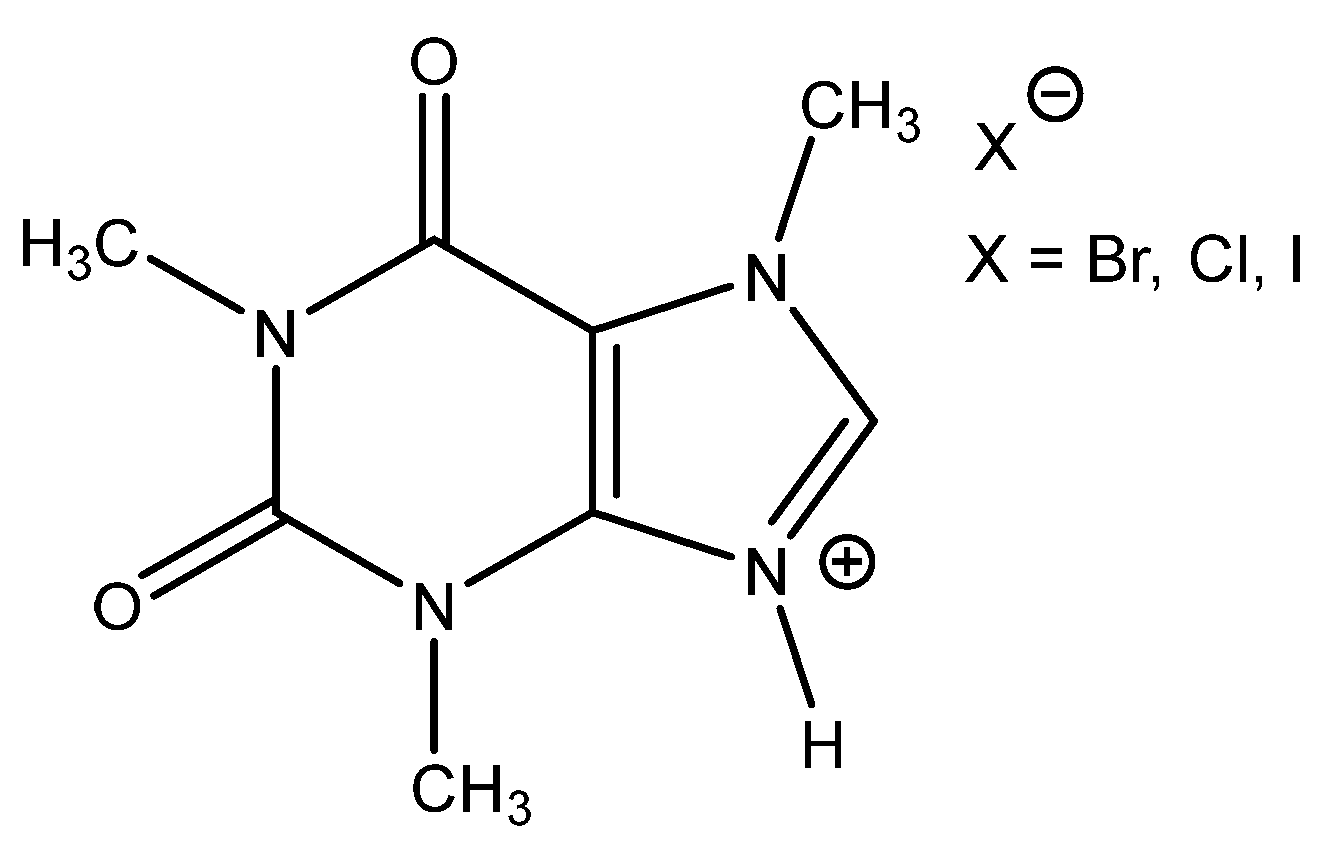
N-dimethylaminopyridine (DMAP.HA)) and, alongside these, even triethanolamine, pyridine and caffeine (
Figure 3), are quite catalytically active in the cycloaddition of CO
2 with epoxides (
Table 3, Entries 3–12, 15–24). Hydrogen halides in particular are the most active in comparison with corresponding free bases [
29,
30,
32,
37,
38,
39] (
Table 3, Entries 3–6, 8–12). The most active seems to be hydroiodides of the corresponding amines (
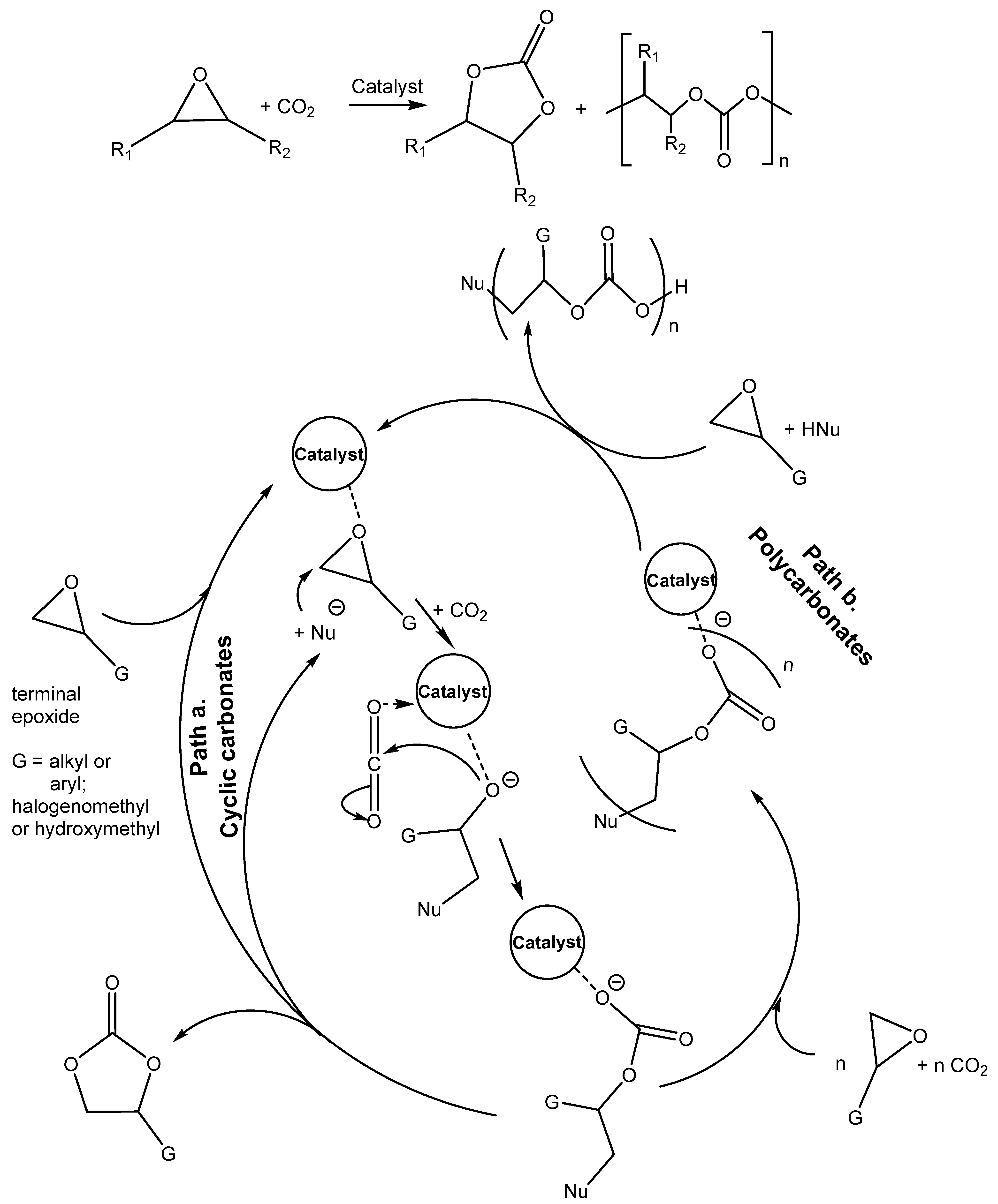
Table 3, Entries 10, 18–24, 26–27). For the above-mentioned catalytic action of amine salts, the reaction mechanism based on the activation of epoxide via the protonation with amine salt in the role of Bronsted acid (HA) is proposed with subsequent anion-based epoxide ring opening.
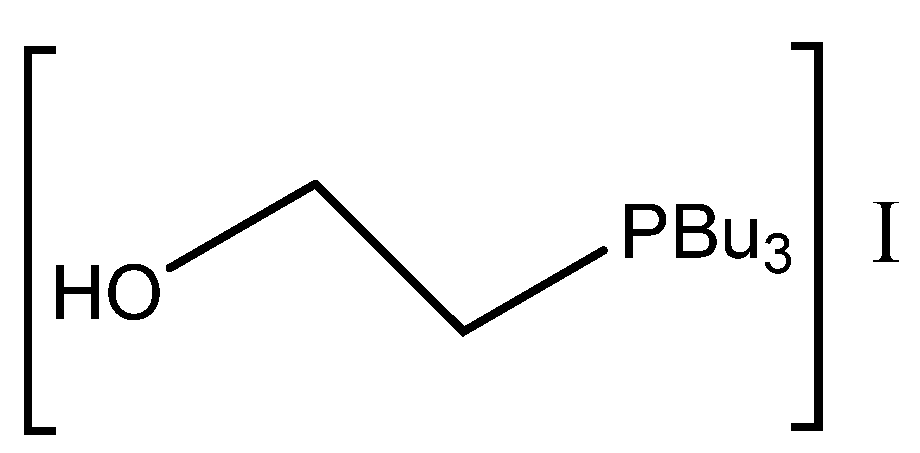
Sun et al. reported very effective carbonation using triethanolamine hydroiodide, including the simple recyclability of this catalyst without loss of activity even after four recycling steps [
38].
The published results indicate that the synergetic effect of hydroxyl groups from protonated aminoalcohol in the role of HBD, together with naked bromide or iodide, significantly influences the cycloaddition of CO
2 to the studied epoxides and makes possible the application of this reaction even at ambient conditions (
Table 3, Entry 17–18).
Catalytically active ammonium halide activates not only the epoxide ring for opening through the H-bond with hydroxyl groups of triethanolammonium cation and the subsequent addition of intermediate 2-halogenoalkoxide to CO
2, but even the next ring closure caused by the facile withdrawal of halide from the produced 2-halogenocarbonate [
38] (
Scheme 3 and
Scheme 4).
Apart from the above-mentioned, in the case of caffeine hydrobromide, potassium halides added to the reaction mixture as an additional source of nucleophiles were successfully tested. With the same reaction conditions, the enhanced efficiency of cyclic carbonate formation was observed utilizing equimolar quantities of KX and caffeine.HBr or even 2: 1 KX: caffeine.HBr using DMSO as the reaction solvent at 70 °C and 0.7 MPa CO
2 pressure [
29,
37]. The yield of cyclic carbonate increased following the trend KF < KCl < KBr < KI, which was in good agreement with nucleophilicity and nucleofugacity of the corresponding halide anions (
Table 4, Entries 17–25).
In
Table 5, the increase in carbonate yields using CAFH.Br/KI in the case of carbonation of terminal epoxides is documented. In the case of internal epoxide (limonene oxide), however, no carbonation was observed using caffeine hydrobromide or even its mixture with KI (
Table 5, Entry 5).
Roshan et al. came to the conclusion that even the addition of a low quantity (a catalytic amount) of H
2O significantly enhances CO
2-based formation of PC over tertiary heterocyclic amines such as IM, PY and DMAP, giving over 98% selectivity of PC formation (at 120 °C, 1.2 MPa, 3 h). The observed results were evaluated by a DFT study comparing energy profiles for free amine in comparison with corresponding amine hydrogencarbonate-mediated cycloadditions of CO
2 to propylene oxide. Ammonium hydrogencarbonates produced in situ from heterocyclic amine, H
2O and CO
2 works similarly to the above-mentioned amine salts as activators of the epoxide ring. As was argued, the HCO
3− anion generated in the water-CO
2-base reaction was the key active species that gave the higher activity of the base-water systems rather than the carbamate salt (produced by a reaction of R
3N with CO
2) [
32].
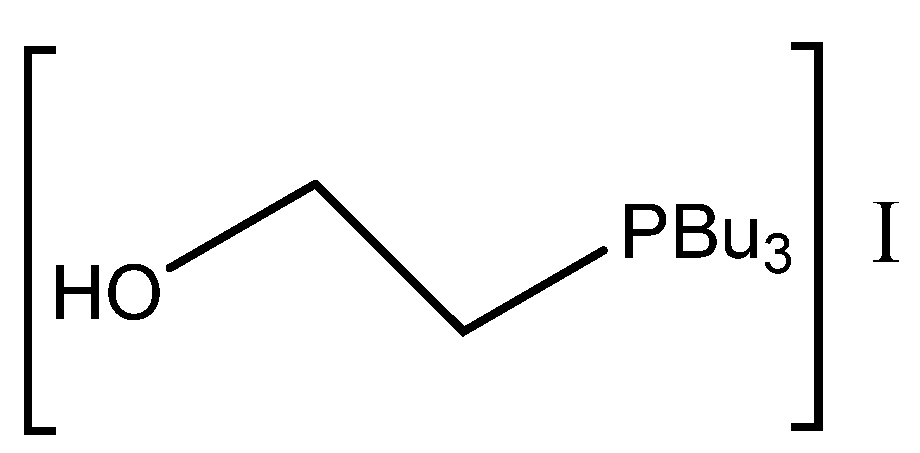
It should be mentioned that low-melting salts (melting point below 100 °C) obtained by the neutralization of organic bases with organic or inorganic acids embodies are called protic ionic liquids (PILs). The melting of PILs could enhance the miscibility of catalytically active PILs with reacting epoxide and CO
2 compared with solid catalysts, as was published by Zhang et al. in the case of DMAP hydroiodide [
42] or by Kumatabara et al. using triethylamine hydroiodide [
41] at ambient pressure (
Table 3, Entry 10 and
Table 4 Entry 26).
Zhang et al. published results obtained even by means of the capture and utilization of CO
2 for the cycloaddition into SO using PIL (DMAP hydrobromide) at ambient pressure and 120 °C [
42]. This PIL has superior catalytic effect compared with other hydrogenhalides of tertiary bases such as DBU, MIM, DABCO or tetramethylguanidine (
Table 6). DMAP.HBr is well reusable with no drop in activity after five recycling steps. DMAP.HBr is able to carbonate even internal epoxides such as ChO, although this cycloaddition is quite sluggish.
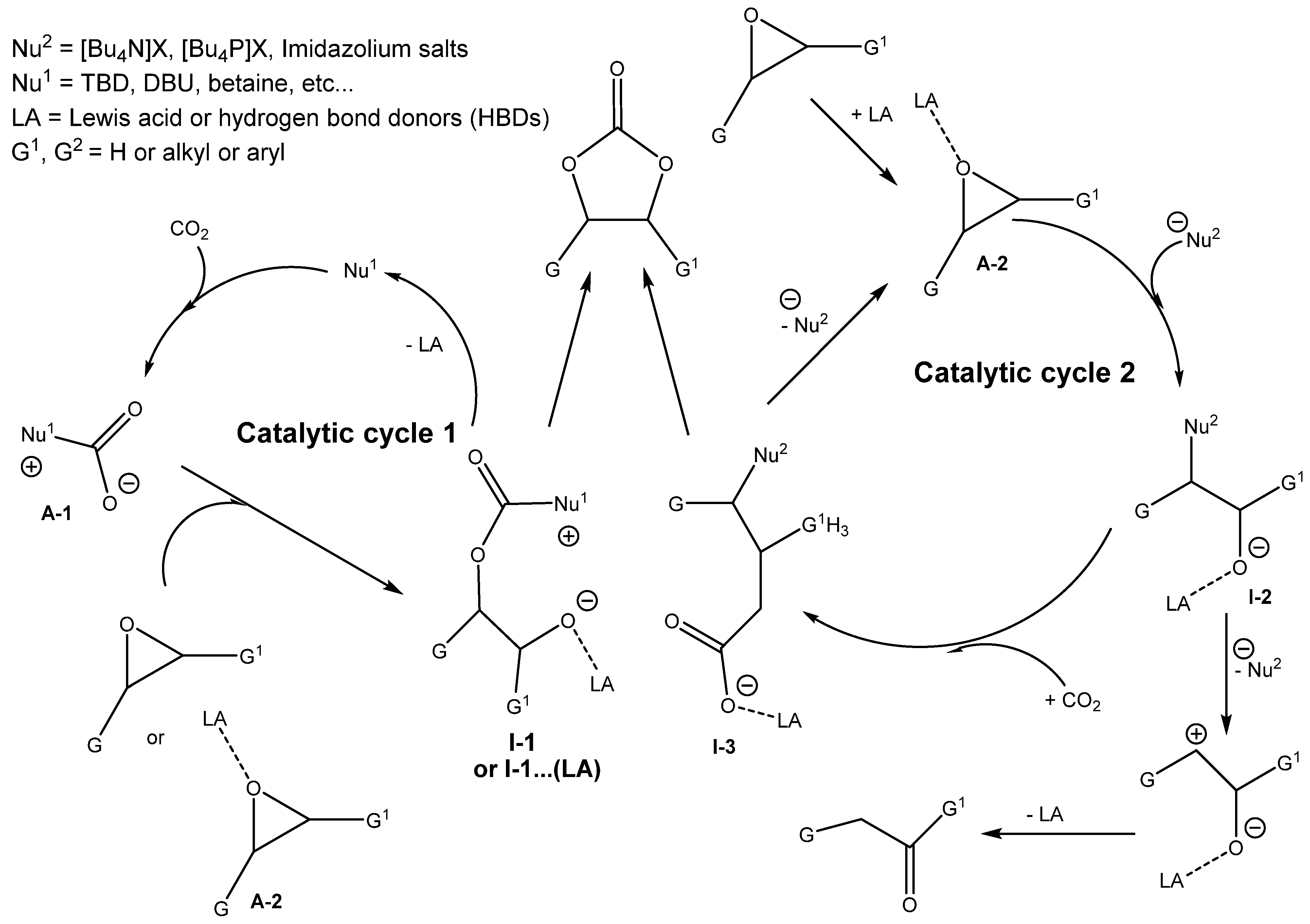
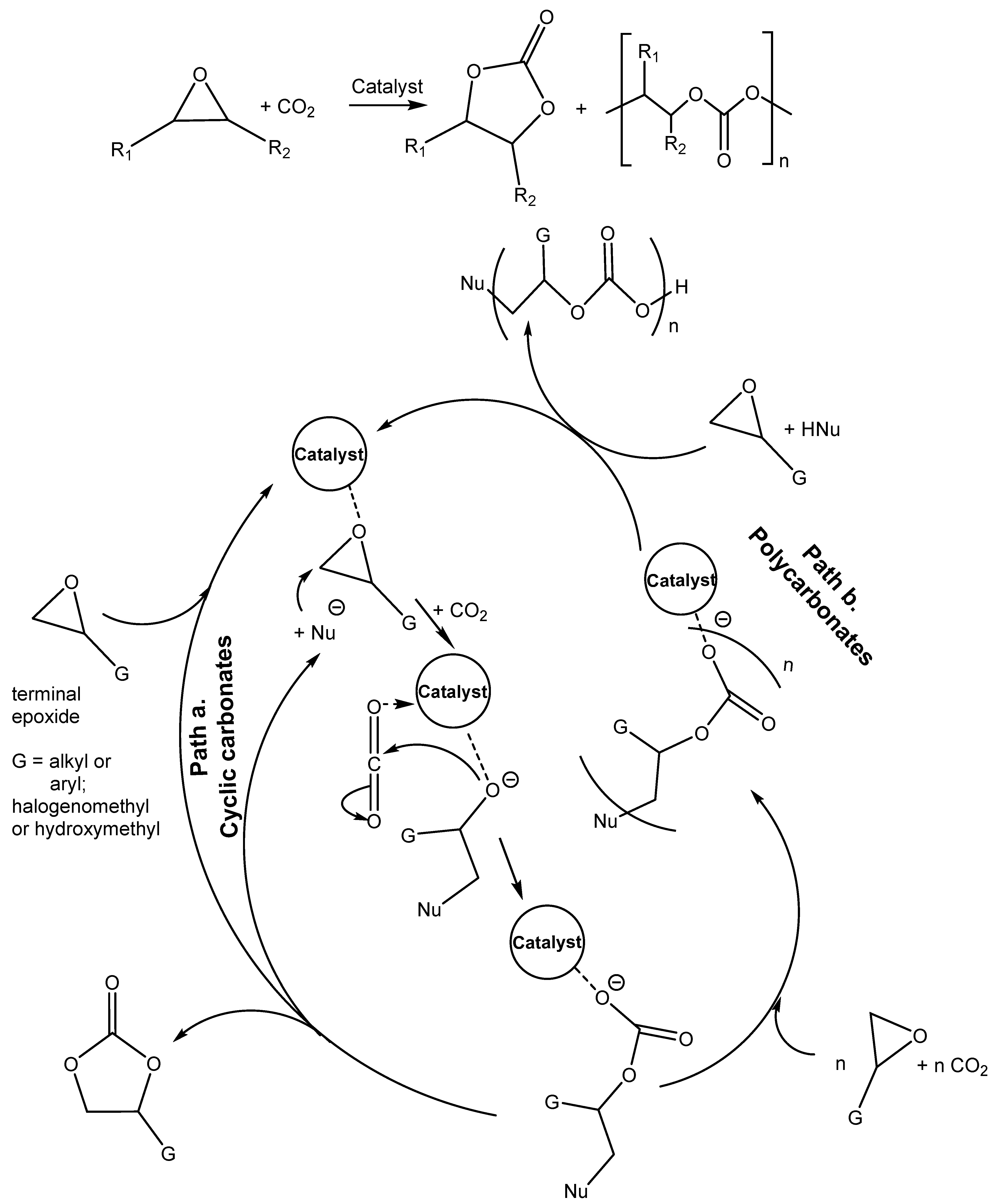
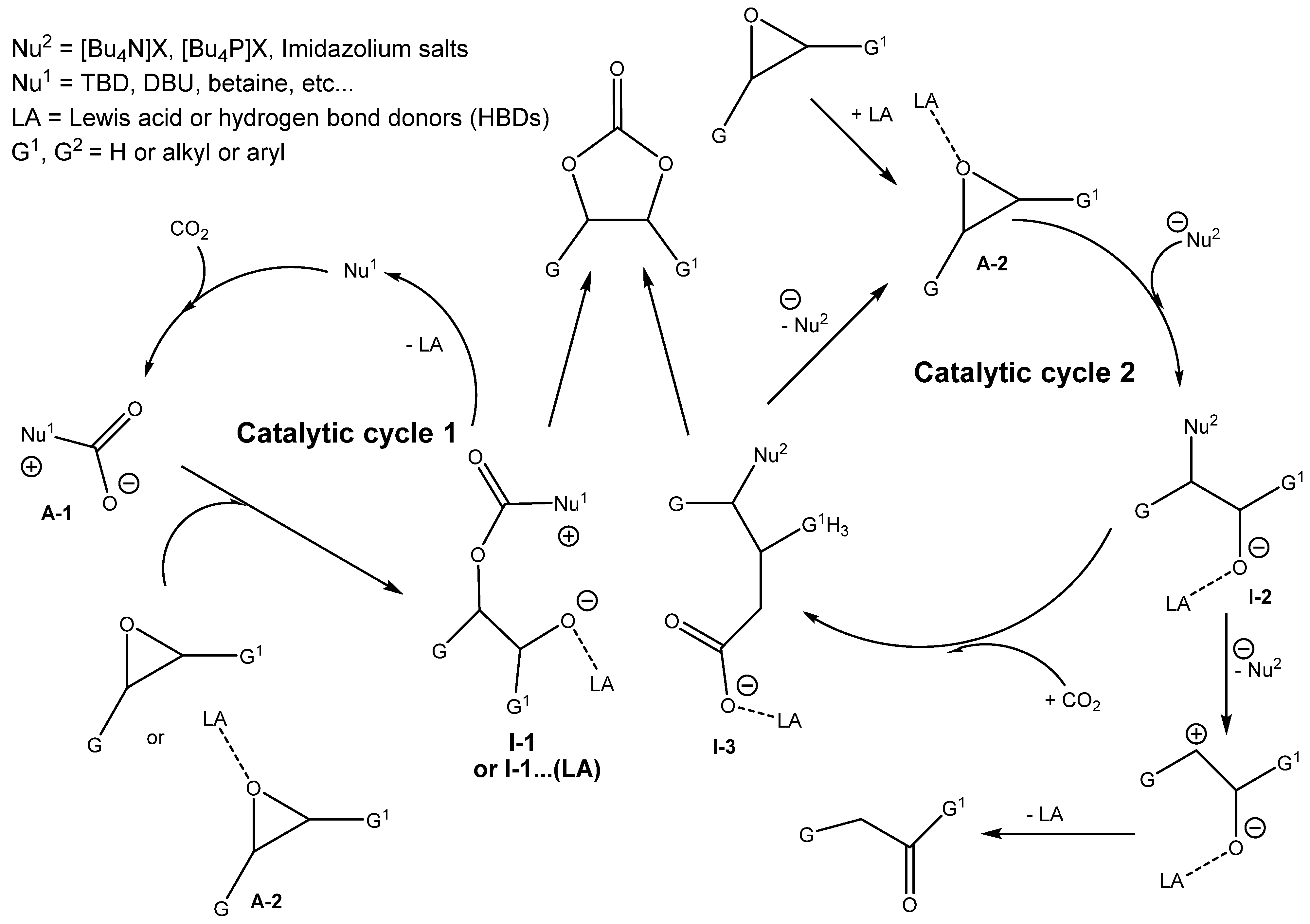
3.2. Two Components Catalysts Based on a Combination of Organic Base and Hydrogen Bond Donor
As was mentioned in
Section 3.1, the combined action of protonated amine with nucleophilic anion positively influences the efficiency of cycloaddition. This synergic action between the Lewis base, such as the amine, and hydrogen bond donors (HBDs) was reported in the literature [
2,
4,
6,
32,
36,
43].
The possible synergic effects of alcohols (glycerol, glycidol, 1,2-propylene glycol (PG), poly(ethylene glycol)-600 (PEG600), poly(ethylene glycol)-400 (PEG400), cellulose, chitosan and β-cyclodextrin (β-CD)) known as HBDs was explored in CO
2-based cyclic carbonates synthesis catalyzed by amines, as mentioned in
Section 3.1 [
36]. For this purpose, the most catalytically active DBU and DMAP were tested in relation to the co-action of the chosen HBDs (
Table 7).
Out of the set of experiments, cellulose was recognized as the most effective HBD in the addition of CO
2 to propylene oxide [
36].
The effective quantity of cellulose used as HBD in the case of the DBU catalysis of the chemical fixation of CO
2 into propylene carbonate is very low with respect to the optimal quantity of DBU (15 mg of cellulose + 300 mg of DBU per mL of PO). The effect of the DBU excess on the yield of PC in the DBU-cellulose reaction system was studied. Generally, the yield rises with the increasing of the DBU: cellulose ratio with the maximum conversion and selectivity reached at a mass ratio of 25–30:1 [
36]. The high catalytical activity of cellulose was, in all probability, explained by Khiari et al. and Gunnarson et al. [
44,
45]. Cellulose reacts in co-action with a significant excess of non-nucleophilic DBU, with CO
2 producing carbonate by means of a reaction similar to that of cellulose xanthate formation during the production of viscose utilizing a sulfur analogue of CO
2, carbon disulfide. The produced carbonate should be a nucleophilic agent that attacks and opens the epoxide ring rather than the known non-nucleophilic DBU.
Aoyragi et al. described the markedly increased formation of cyclic carbonates in isopropylalcohol using triphenylphosphine hydroiodide as a catalyst. 1H NMR spectra documented the formation of H-bonds between the used isopropylalcohol and the starting epoxide [
46]. The high activity of the above-mentioned hydroiodide (compared with other HXs) was explained by both the high nucleophilicity and even the high leaving ability (nucleofugality) of iodide ion.
Section 3.1 mentioned the significant catalytic activity of triethanolamine [
36], which could be explained by the synergy of HBDs (bound alcoholic OH groups) and the Lewis basicity of the tertiary amine.
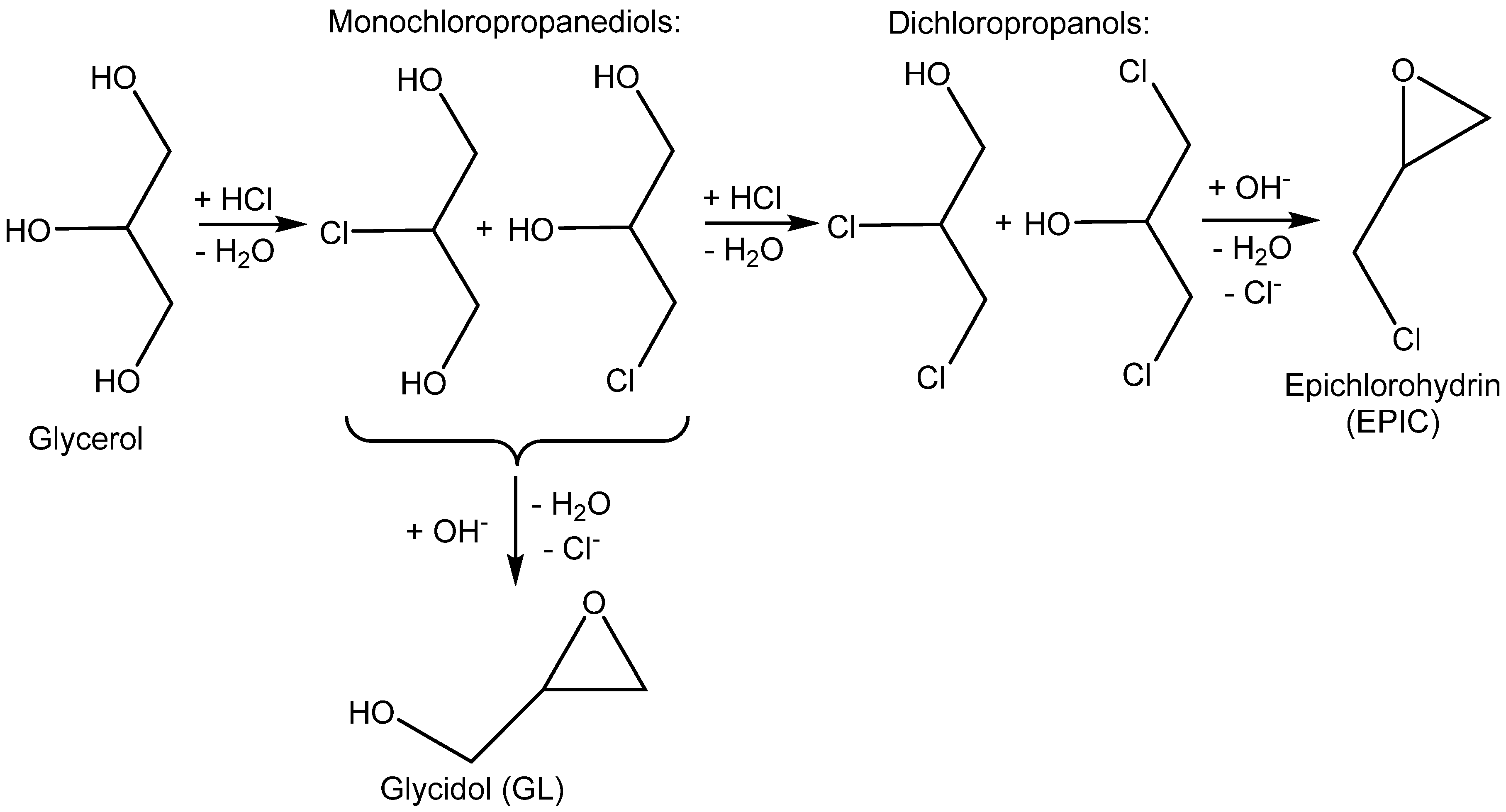
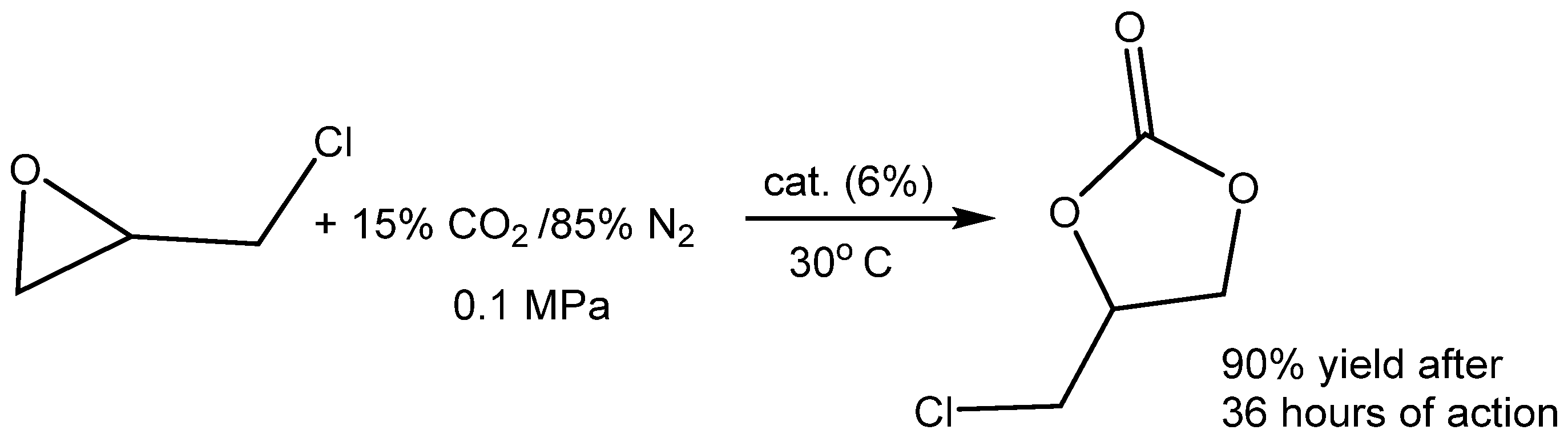
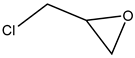
More advanced catalysts such as 2-hydroxymethylpyridine (2-PY-CH
2OH) and 2,6-bis(hydroxymethyl)pyridine (2,6-PY-CH
2OH)
2 were developed for the high-efficiency cycloaddition of CO
2 with epichlorohydrin (EPIC) under a slightly elevated temperature and ambient pressure (T = 25–60 °C, 0.1 MPa of CO
2; see
Table 4, Entry 4) [
40]. The high catalytic effect was demonstrated by
1H NMR spectroscope observing the formation of a stable H-bond between the PY-CH
2OH and oxygen of epichlorohydrin. The authors demonstrated that the tested compounds with either heterocyclic nitrogen (benzylalcohol PhCH
2OH) or hydroxymethyl (CH
2OH) groups (PY) catalyzed EC formation only sparingly (PY) or not at all (PhCH
2OH) (
Table 4, Entries 1–5).
The catalytic activity of nitrogen-doped charcoal for CO
2 cycloaddition reactions could be explained by the co-operation of OH groups working as HBDs and tertiary amines bound in the graphitic structure of specially prepared
N-doped carbons together with the ability of active carbon to adsorb CO
2 [
47].
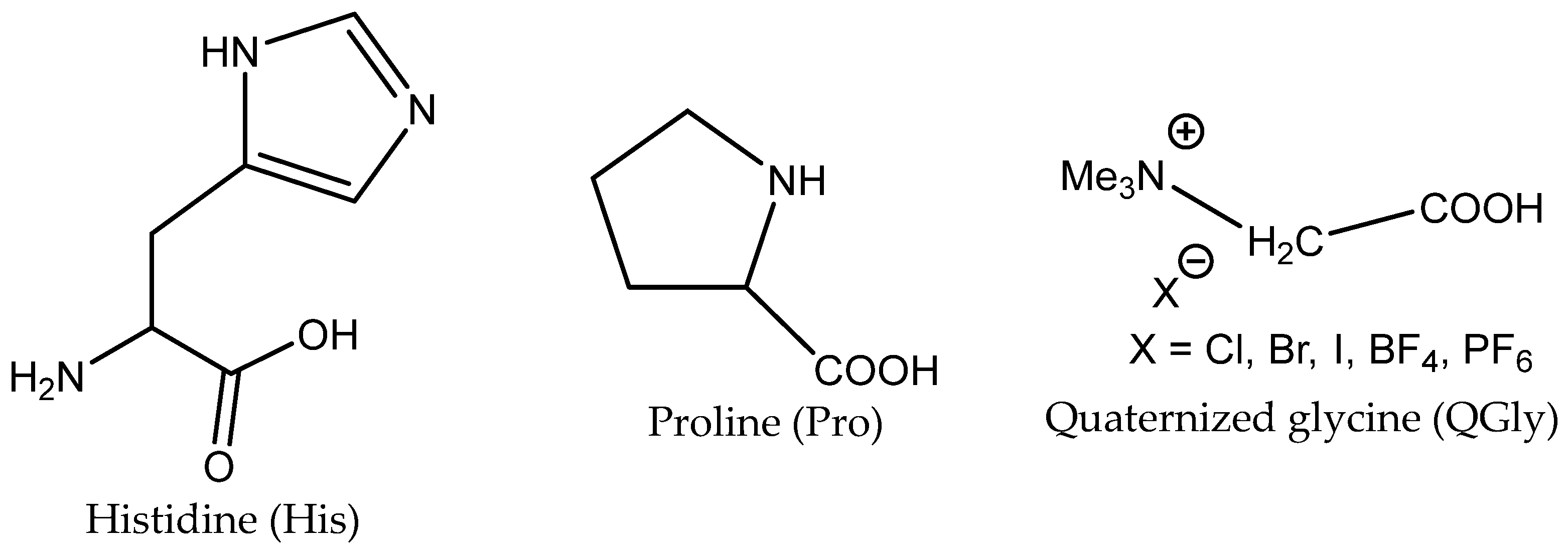
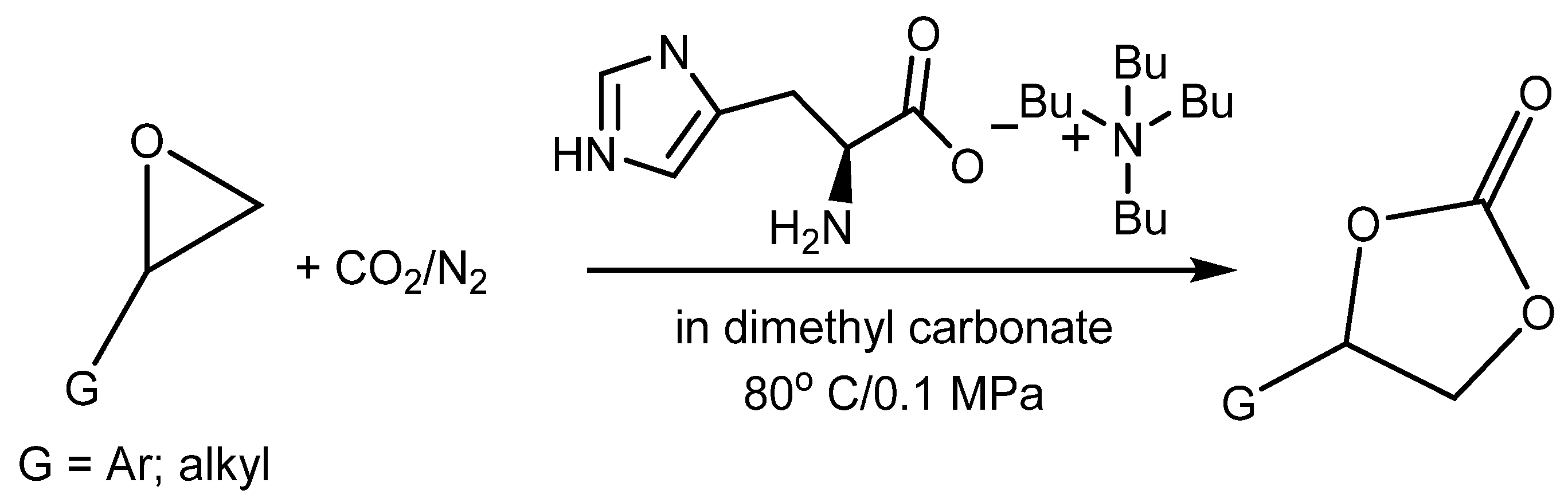
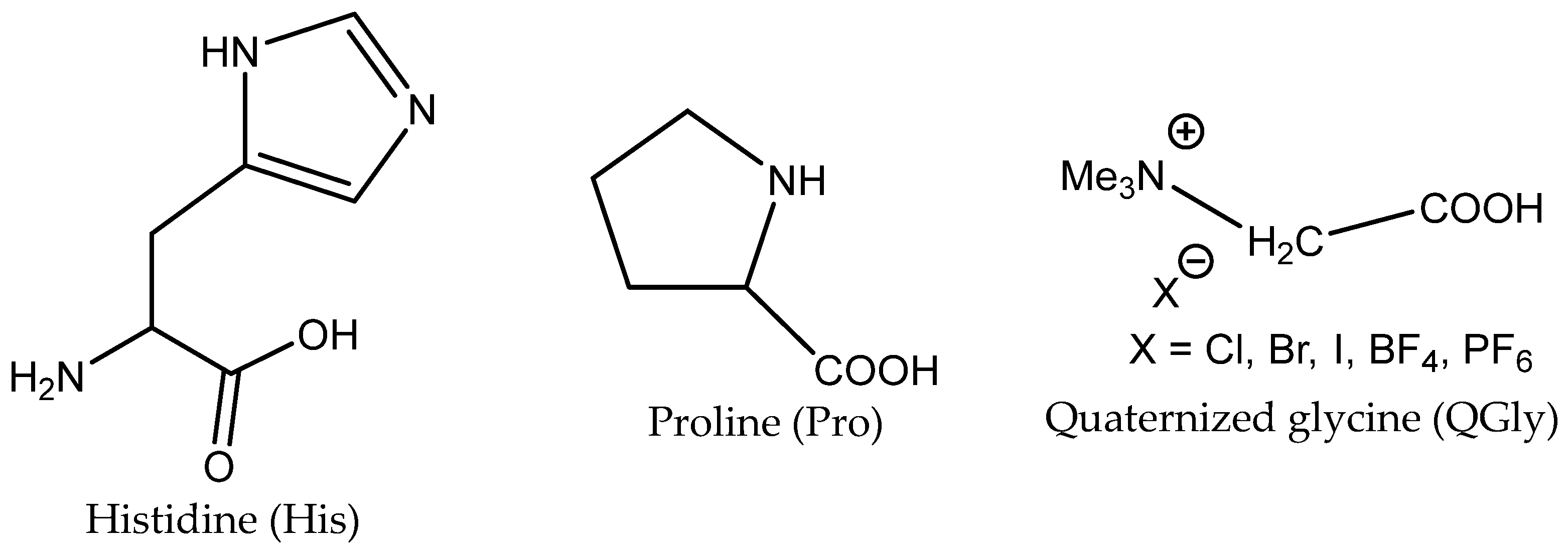
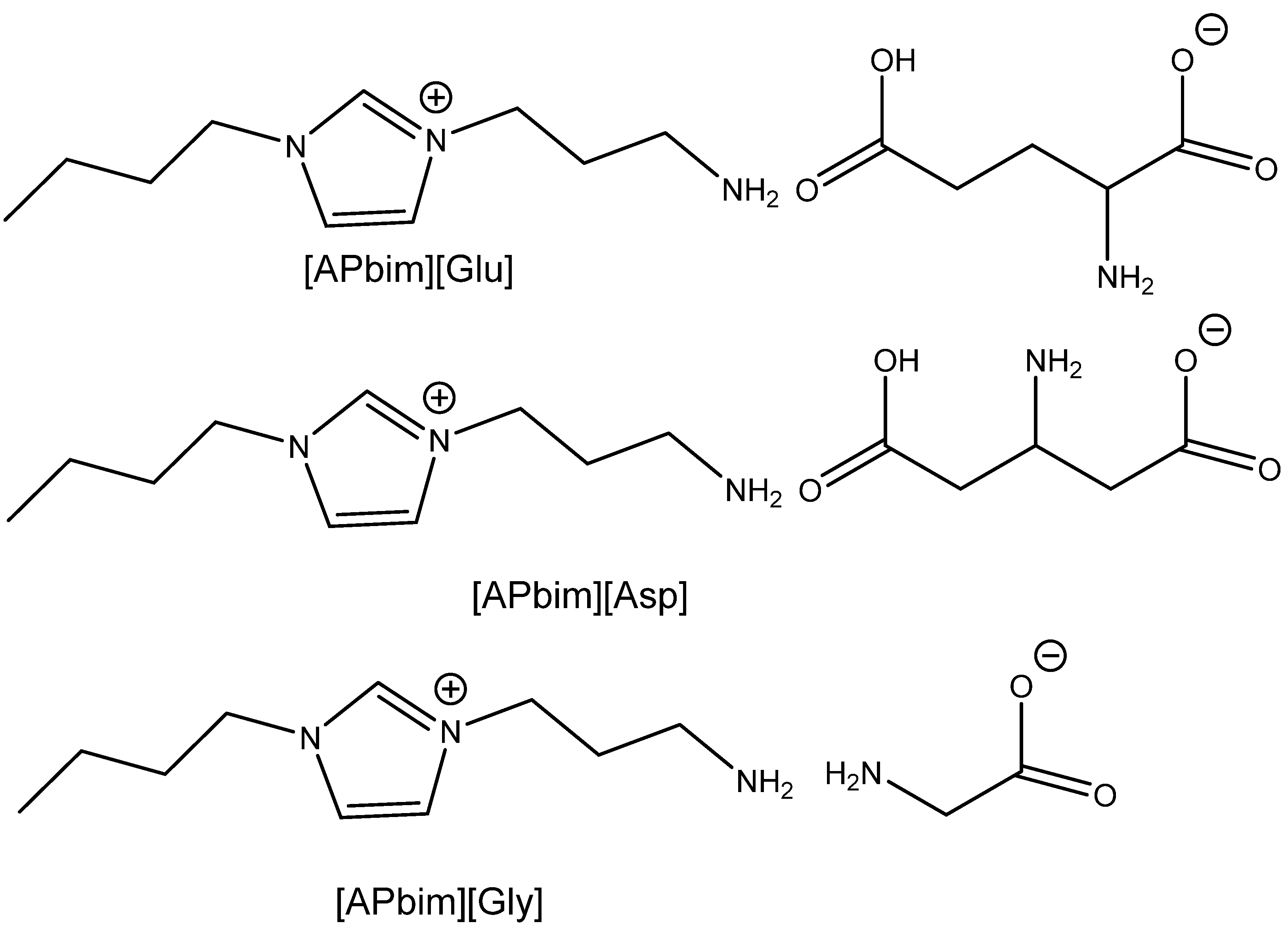
3.3. Aminoacids (AAs) as Catalysts
AAs contain amino and carboxylic groups in their structures. Amino groups can react with CO
2 to form
N-COO
− (carbamate) products with low binding energy, which can catalyze the transfer of CO
2 to the 2-halogenoalcoholate produced by the halide-based opening of the epoxide ring. The carboxylate group (–COOH) can catalyze the oxirane ring opening as effective HBD analogously to the amino group (–NHR) and hydroxyl (–OH). Some AA salts have been successfully tested in the capture of CO
2 from flue gas. In addition, the amino groups can also be utilized in quaternization with the aim of introducing halide as a nucleophile into the engineered catalytically active molecules (
Table 8).
The yield of PO is dependent on the AA structure; basic AAs such as L-histidine (L-His) and proline (Pro) containing basic additional groups (imidazolium and amino, respectively) provided higher yields than acidic aspartic or glutamic acids (
Figure 4).
In addition, the combination of an amino acid with an HBD results in a higher catalytic activity under milder reaction conditions than in the absence of an HBD. The binary catalytic systems formed with the amino acid and H
2O produced, for example, more active systems for the synthesis of PO than in the absence of H
2O [
53]. In the case of L-His, the time taken for the nearly total conversion was reduced from 48 h to 3 h by adding H
2O as an HBD (L-His:H
2O ratio = 1:29) under similar conditions (
Table 8, Entries 1 and 2). The low H
2O concentration was used to avoid the hydrolysis of the produced PC [
49,
53].
Roshan et al. showed that the combination of halide ions as nucleophiles (added in the form of KI, for example) with L-histidine produced highly active catalytic systems for the cycloaddition of CO
2 to epoxides [
51] (
Table 8, Entry 4).
The most effective binary catalytic systems contained KI with basic AAs such as His/KI (
Table 8, Entry 4). In a related work, Yang et al. proved the high stability of AA/KI catalytic systems, namely L-Trp/KI, for the cycloaddition of CO
2 to PO to form propylene carbonate. After carbonate separation conducted by means of distillation, the catalytic system was reused five times without loss of activity [
54].
The catalytic activity of the different KX salts followed the order Cl < Br < I corresponding to the increasing nucleophilicity and leaving group ability [
54]. This confirmed the role of these anions in the opening of the epoxide ring [
54].
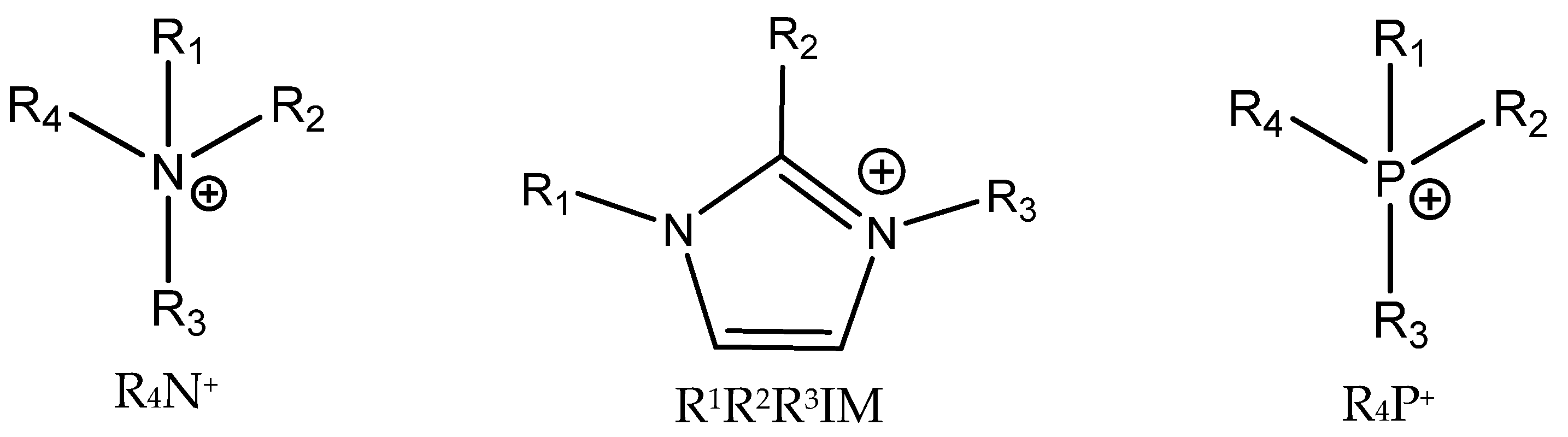
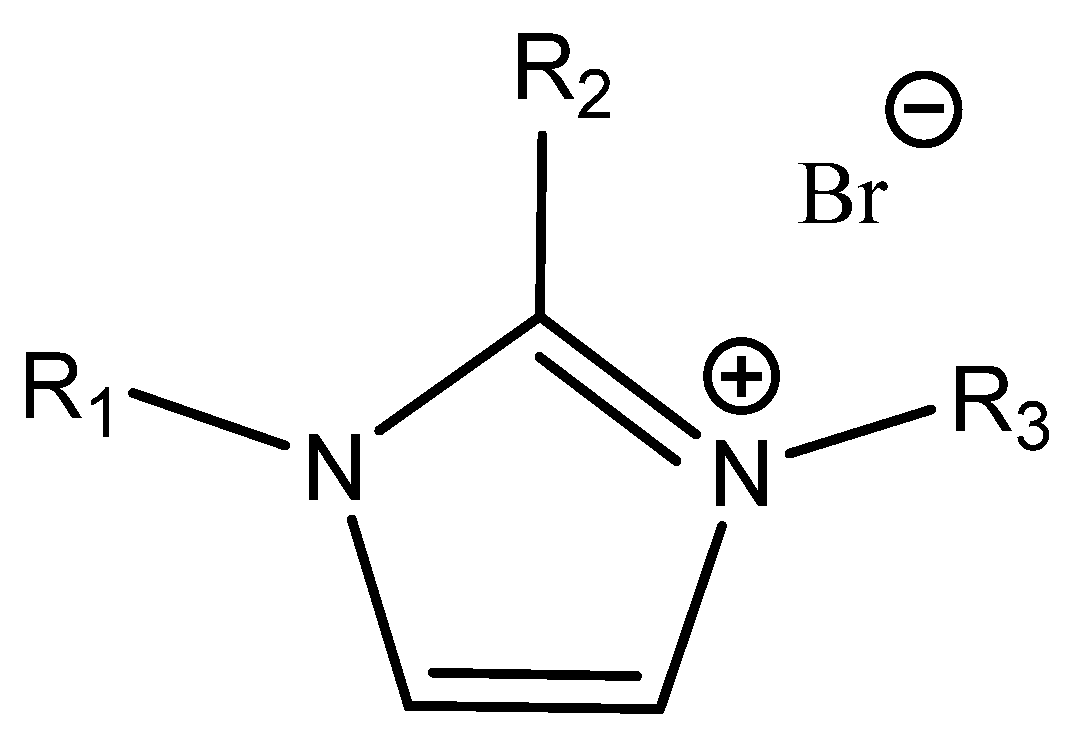
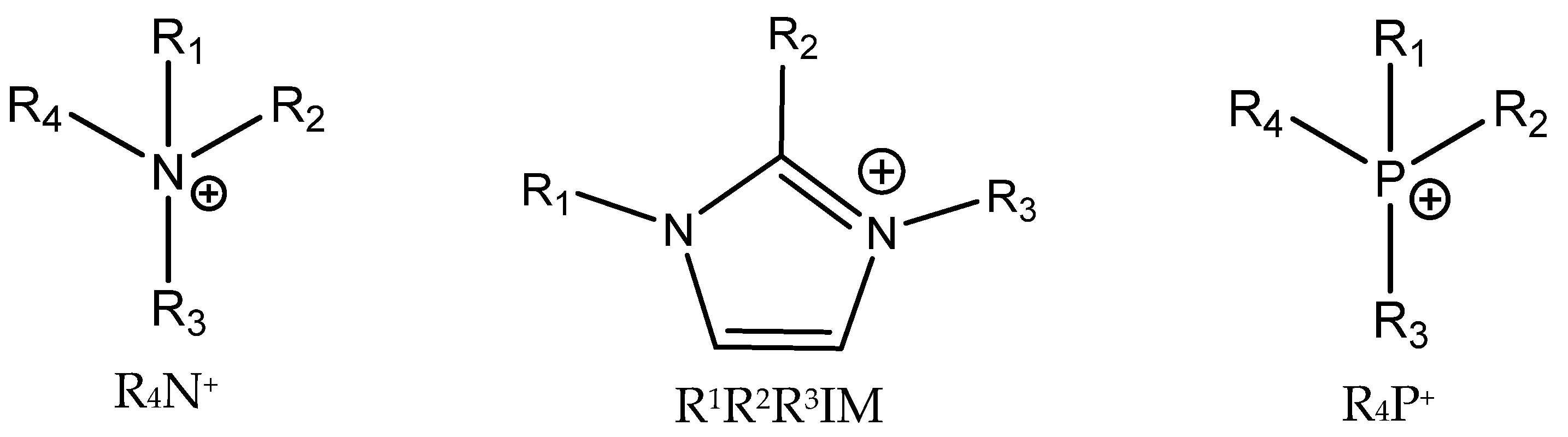
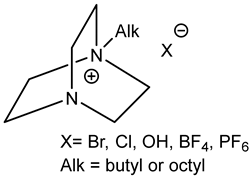
3.4. Onium Salts as Catalysts
Quaternary ammonium (most often tetrabutylammonium bromide, TBAB, and iodide (TBAI), phosphonium and sometimes even sulfonium salts [
55]) are common catalysts for the formation of cyclic carbonates from epoxides and CO
2 [
5,
6,
12] (
Figure 5).
The low-melting (below 100 °C) quaternary ammonium salts are called ionic liquids (ILs). In addition, ILs show good solvating ability including CO
2, variable polarity and negligible vapor pressure [
56]. ILs are considered to be sustainable (“green”) solvents because of their properties such as relatively high thermal stability and negligible vapor pressure, high chemical stability and simple separability, and the modularity of their properties changing the structure of anions and cations.
The first published article that mentioned the formation of cyclic carbonates via the cycloaddition of CO
2 in epoxides catalyzed solely by ILs was published by Deng and Peng [
57]. The authors studied different 1-butyl-3-methylimidazolium (BMIM) and
N-butylpyridinium (BPY) salts varying in anions (chloride and tetrafluoroborate (BF
4−, hexafluorophosphate (PF
6−)) and observed the highest activity in the case of BMIMBF
4 salt. The catalytic activity increase was in the order: BMIMPF
6 < BPYBF
4 < BMIMCl < BMIMBF
4. This observation is in agreement with the solubility of CO
2 in these Ils; the highest solubility of CO
2 was determined in BMIMBF
4 [
58].
Dyson et al. and Wang et al. studied the abilities of different ILs that differed in terms of the cations (alkylated imidazolium and tetraalkylammonium) and anions (halides) used [
59,
60]. Interestingly, cheap Bu
4NCl was found to be a very active catalyst compared with the much more expensive alkylated imidazolium halides. Based on the experimental results obtained at ambient pressure and 50 °C, they hypothesized that the balance between the nucleophilicity of anions and the acidity of hydrogens bound in the imidazolium ring of used IL is most important.
Yang et al. published an even higher catalytic activity of corresponding bromides when comparing them with tetrafluoroborates (
Table 9, Entries 10 and 11) [
29].
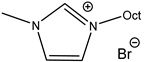
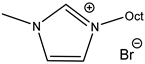
An increase in catalytical activity was observed with the increasing of the lipophilic alkyl chain length in RMIMXs when comparing 1-butyl-3-methylimidazolium and 1-octyl-3-methylimidazolium bromide [
29].
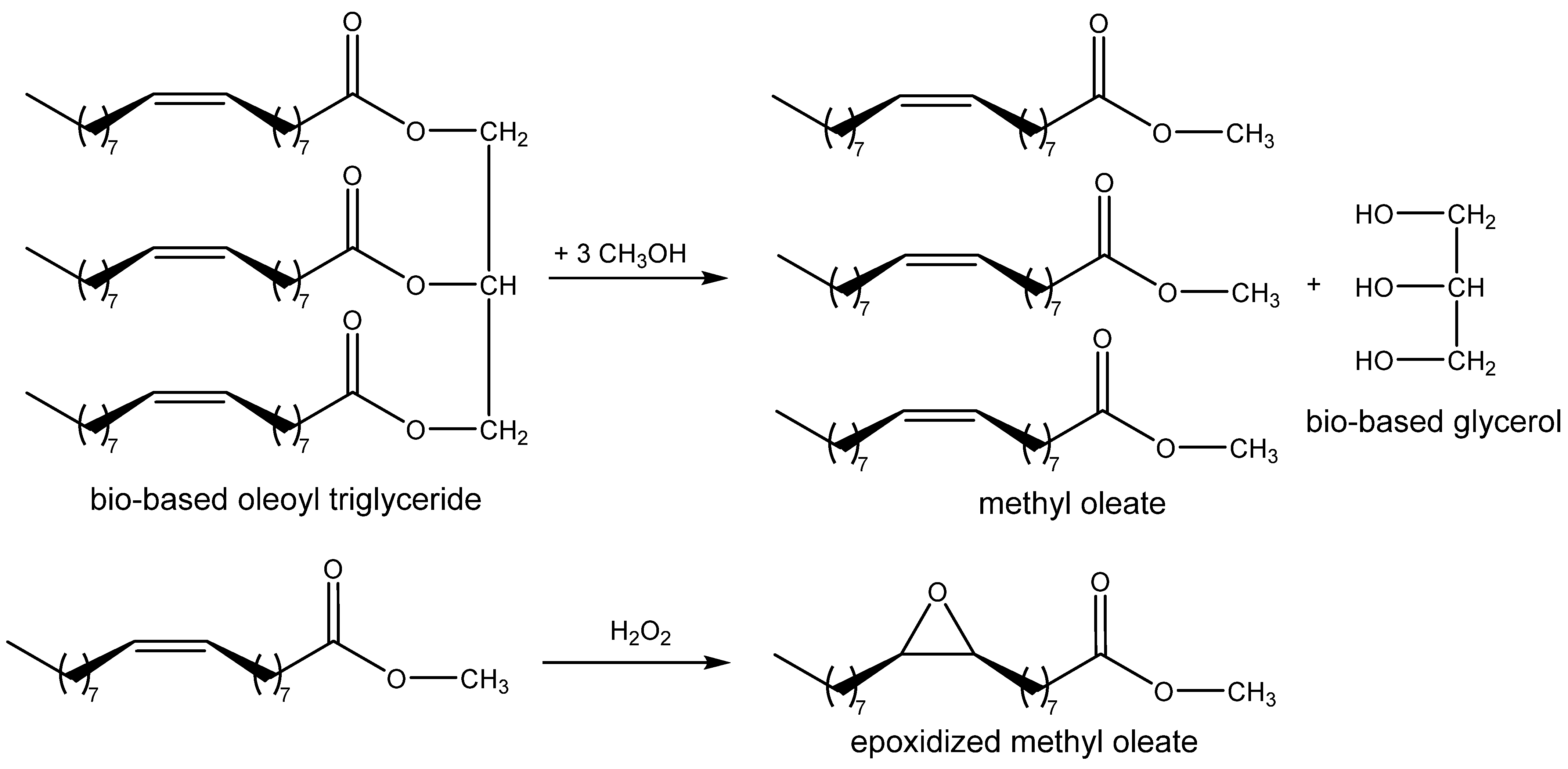
Based on this idea, Akhdar et al. successfully tested the carbonation of internal epoxide produced via the epoxidation of methyl oleate [
61,
62,
63]. The catalysts were prepared by means of the alkylation of
N-alkylimidazoles with oligoethylene iodide and modified by ion-exchange to the corresponding bromide.
N′-Oligoethylene-
N-butylimidazolium bromide was recognized as the most active catalyst enabling the carbonation of epoxidized methyl oleate in 96% yield even at 100 °C and 2 MPa of CO
2.
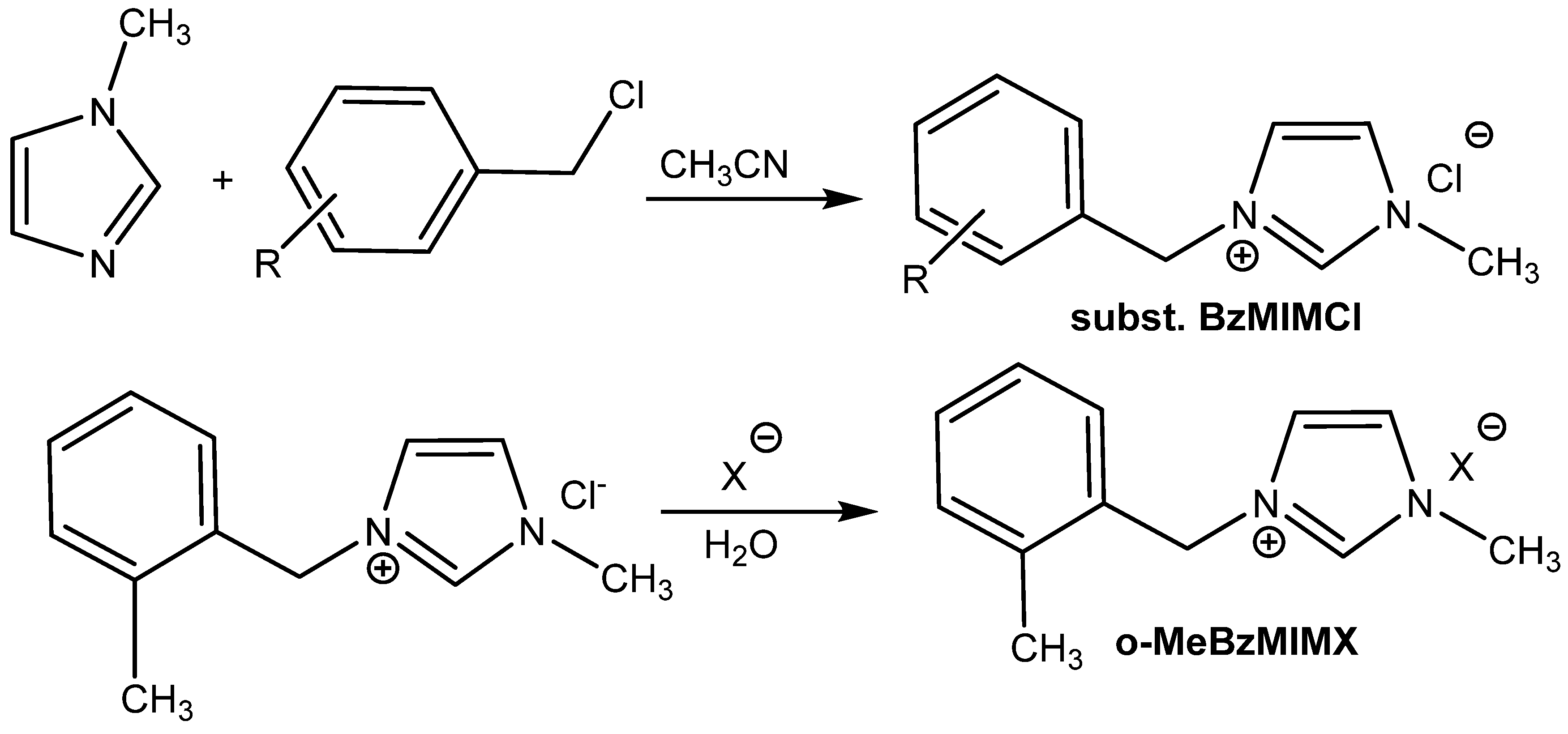
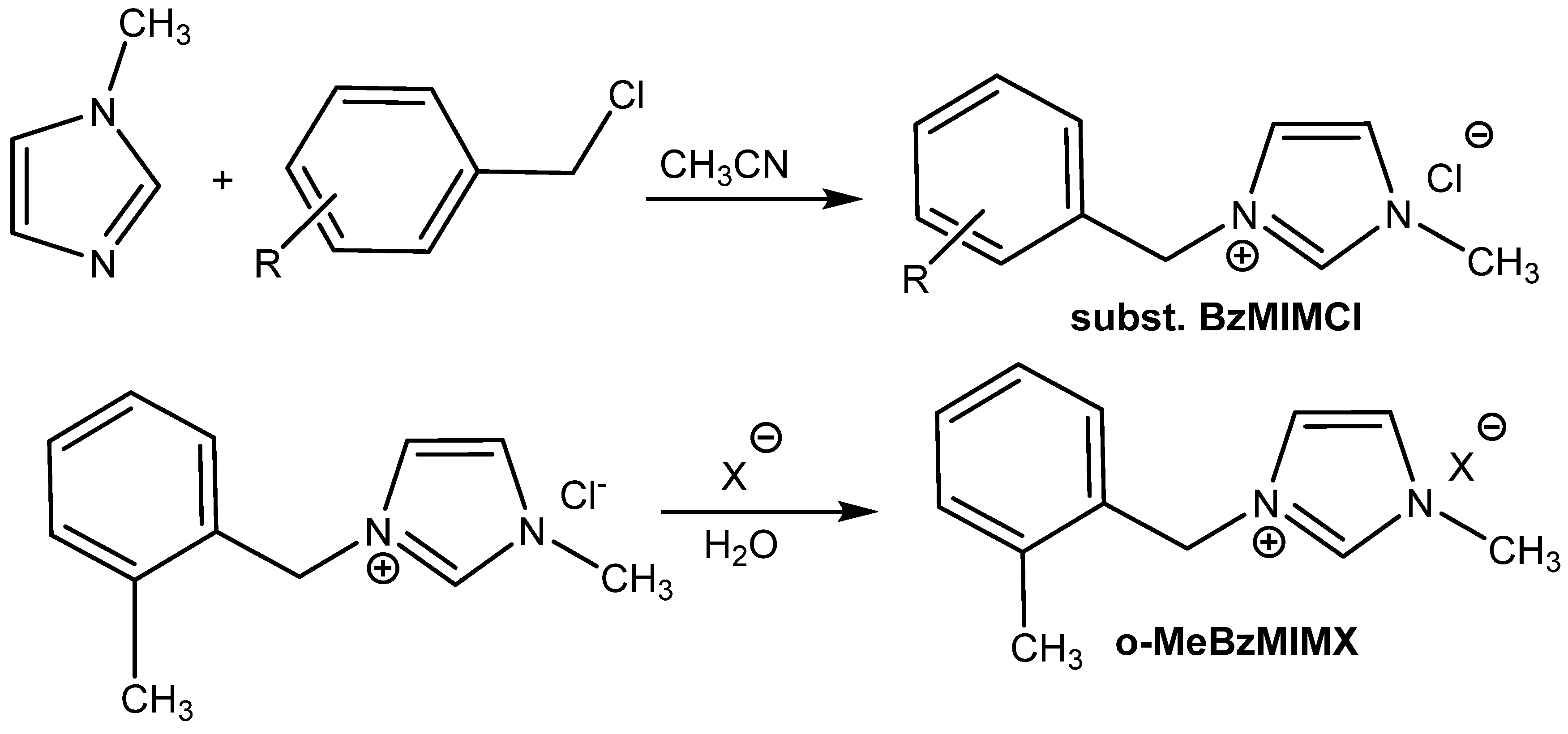
The catalytic activity of ILs cannot be known based on a comparison of the anion effects using one type of cation. In the case of
N-benzyl-
N′-methylimidazolium salts, the most active one is
N-(o-methyl)benzyl)-
N′-methylimidazolium chloride
o-MeBzMIMCl; the catalytic activity of the corresponding
o-MeBzMIMBF
4− and
o-MeBzMIMPF
6− salts is lower (
Table 9, Entries 14–16) [
60].
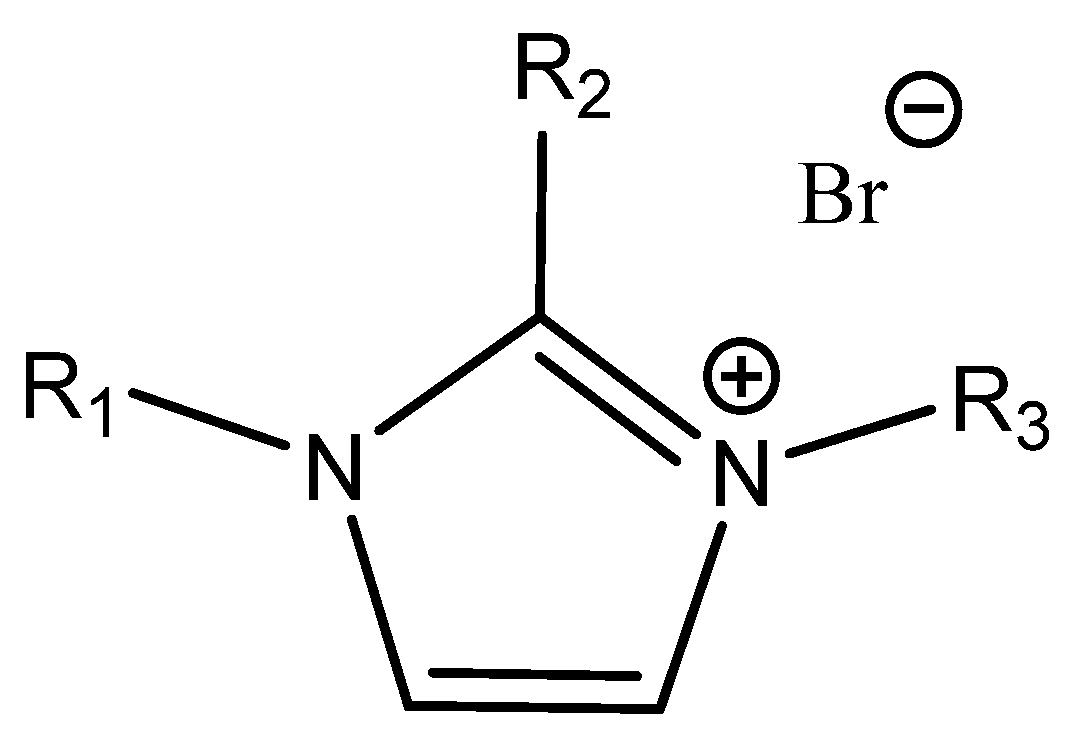
Anthofer et al. studied the relationship between the structure, affinity to CO
2 and catalytic activity of different low-melting
N,
N′-dialkylimidazoles in detail [
64] (
Figure 6).
As could be seen, the most active ILs were those that contained bulk lipophilic substituents (
N-octyl-
N′-pentafluorophenyl-,
N-butyl-
N′-pentafluorophenyl- or
N-methyl-
N′-octyl-imidazole). The observed catalytic activity correlates well with the measured sorption of CO
2 in the studied ILs under the reaction conditions of this study. The substitution of the 2-position also significantly reduced the activity of the tested ILs [
64].
The authors detected an interaction (hydrogen bond formation) between the acidic hydrogen of the used imidazole bromides (bound in position 2) and the oxygen atom of PO using FT-IR spectroscopy [
64]. The mentioned catalyst was very effective even for the carbonation of internal epoxides such as cyclohexene oxide (ChO).
As could be seen, the most active ILs were those that contained bulk lipophilic substituents (
N-octyl-
N′-pentafluorophenyl-,
N-butyl-
N′-pentafluorophenyl- or
N-methyl-
N′-octyl-imidazole). The observed catalytic activity correlates well with the measured sorption of CO
2 in the studied ILs under the reaction conditions of this study. The substitution of the 2-position also significantly reduced the activity of the tested ILs [
64] (
Table 10).
The authors detected an interaction (hydrogen bond formation) between the acidic hydrogen of the used imidazole bromides (bound in position 2) and the oxygen atom of PO using FT-IR spectroscopy [
64]. The mentioned catalyst was very effective even for the carbonation of internal epoxides such as ChO (
Table 11).
As was published by Yang et al., in the case of butylated DABCO (BuDABCO), the corresponding bromides, chlorides and hydroxides were recognized as the most effective cycloaddition catalysts [
29] (
Table 12, Entries 1–4). In contrast, non-nucleophilic anions such as NTf
2−, PF
6− or BF
4− caused the loss of the catalytical activity of the studied BuDABCO salts.
Surprisingly, when ILs containing activated CO
2 in their structures in
N,
N′-di(alkyl)imidazolium-2-carboxylates were tested as CO
2 cycloaddition catalysts by Kayaki et al. [
65], the observed activity was quite low and a high CO
2 pressure was required to obtain satisfactory conversion (
Table 13). The hydrogen in position 2 on the imidazolium ring is, in all probability, important as an HBD for epoxide ring activation and substitution with -COO
− causes a decrease in catalytic activity (
Table 13).
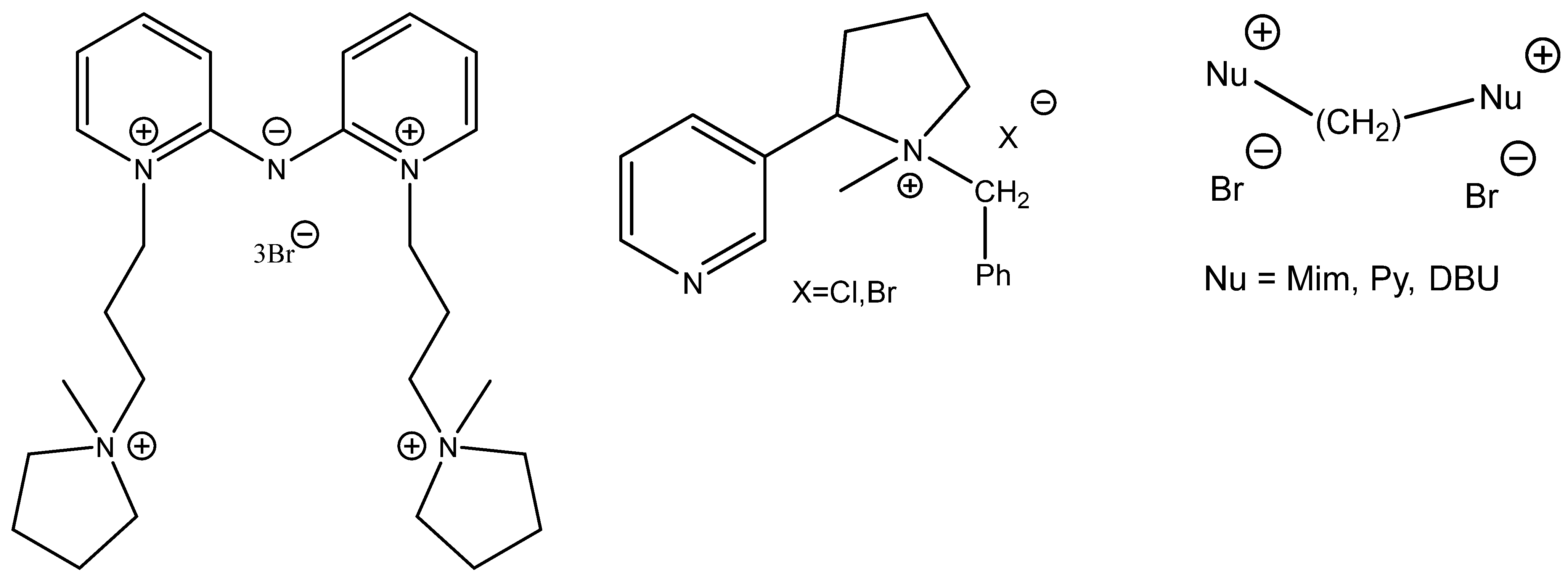
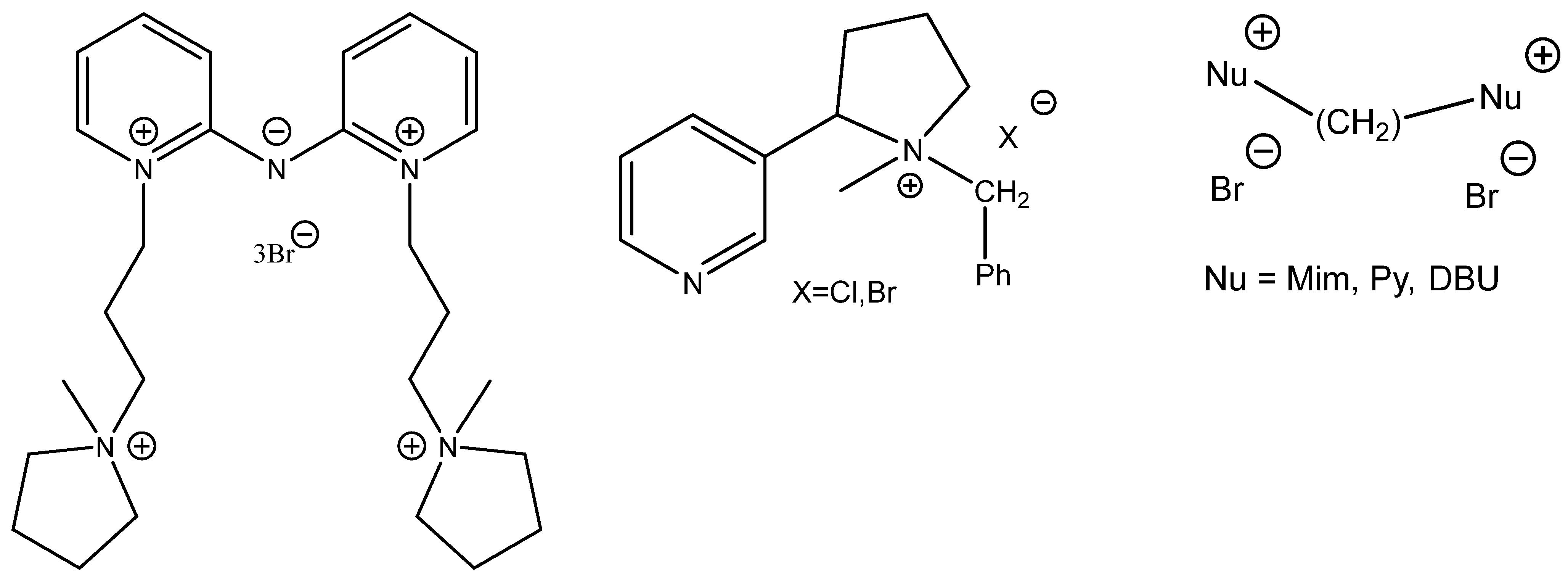
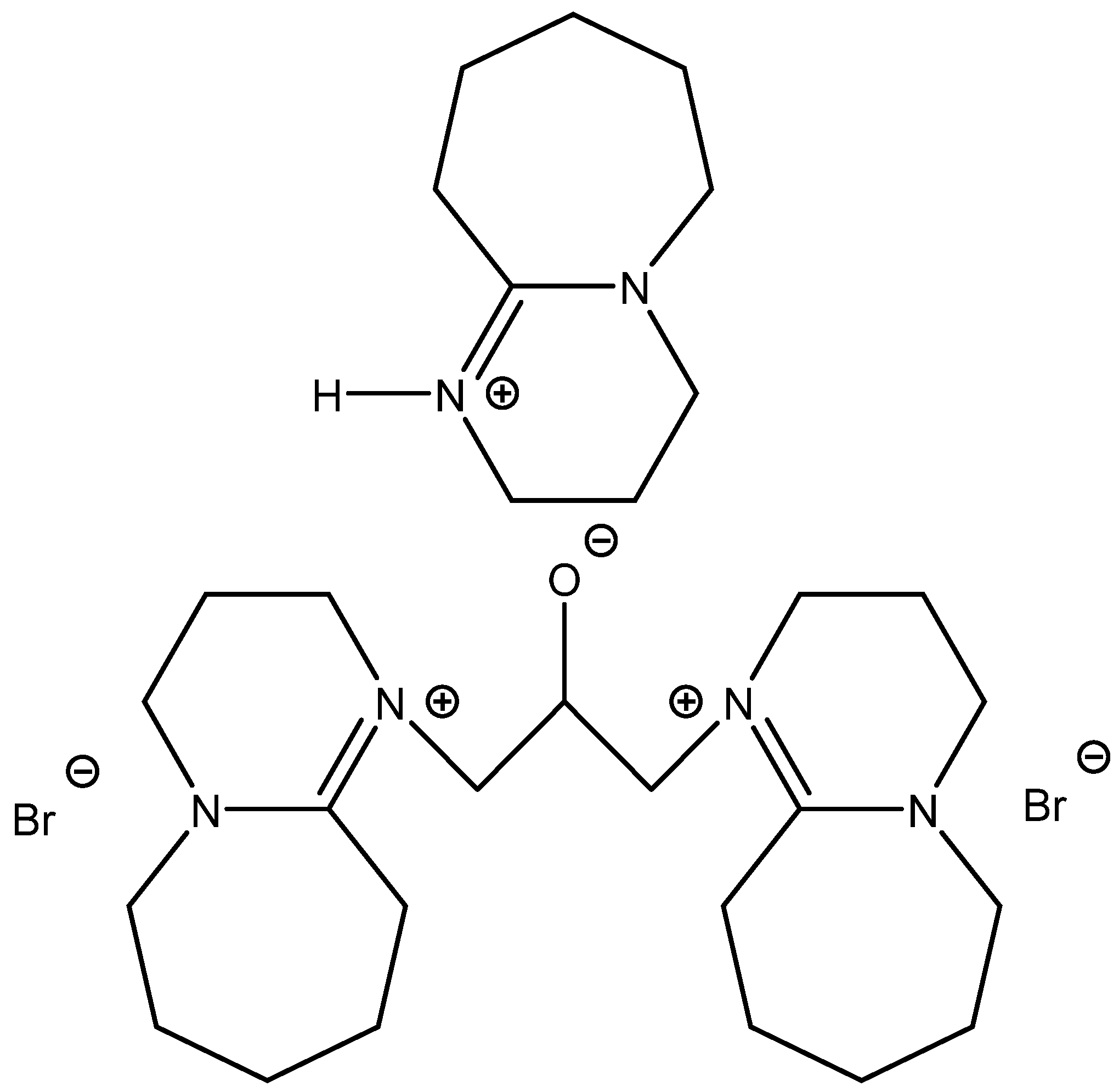
Interestingly, some of the attempts to boost the catalytic activity of onium salts constructing di- or tricationic ILs (
Scheme 7) often fall flat (
Figure 7) [
66,
67,
68].
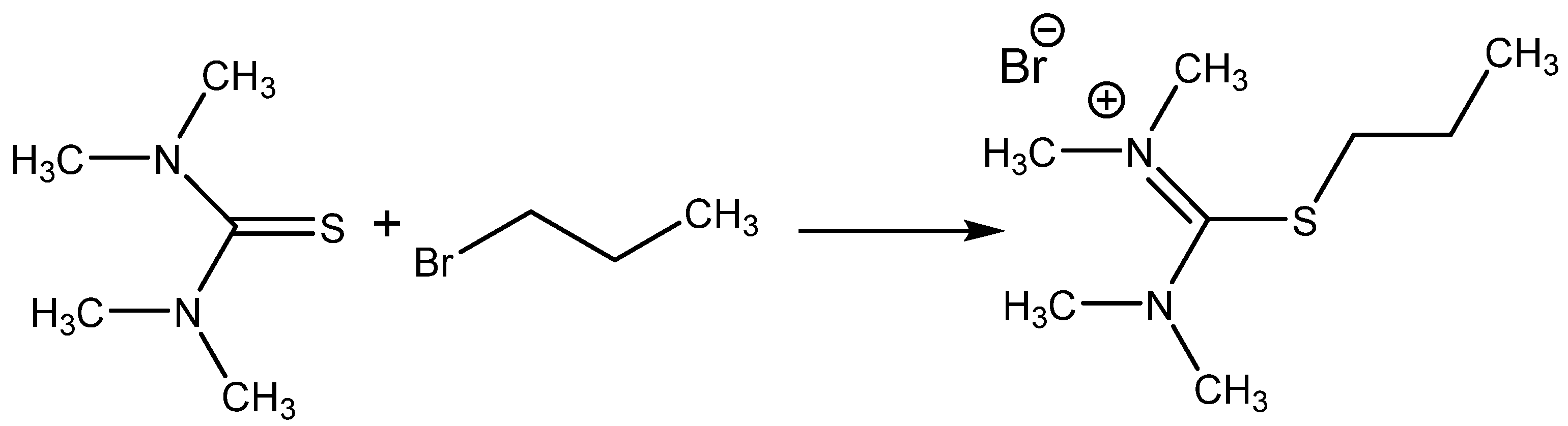
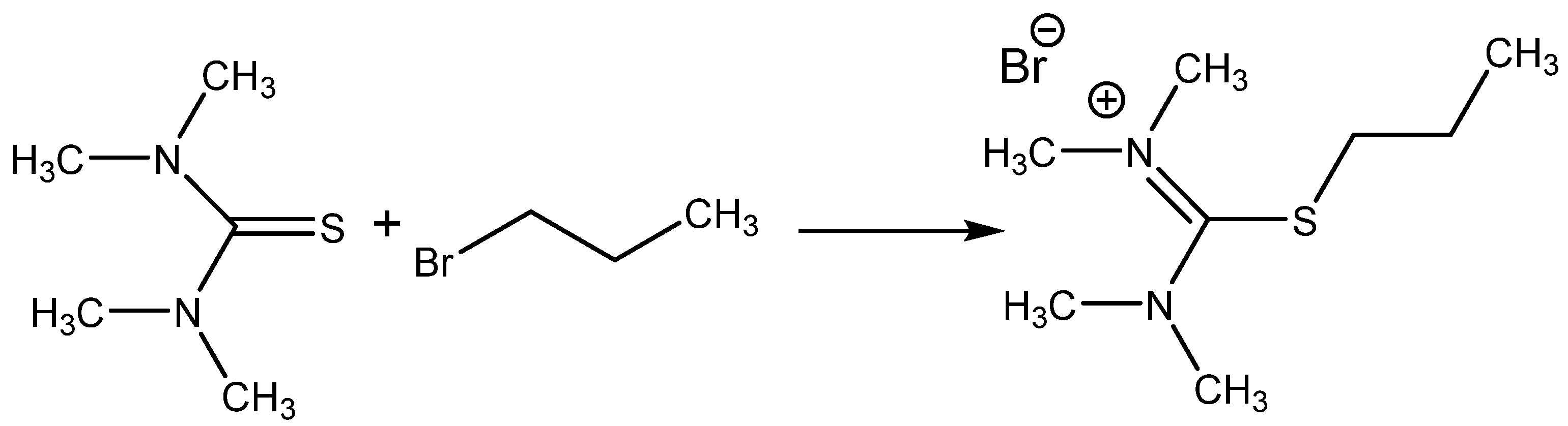
Isothiouronium salts (
Scheme 8) were also chosen for the testing of catalytic activity for CO
2 addition, providing encouraging results (over 90% yield with selectivity over 99%) at 2 MPa pressure of CO
2 and 140 °C after 2 h of action using 1 molar % of catalyst. The corresponding thiourea was practically nonactive [
55].
Apart from ammonium and sulfonium salts, the methyl-trioctylphosphonium-based ILs with organic anions were studied as cycloaddition catalysts. Their catalytic activity was remarkable even for cycloaddition reaction of less reactive styrene oxide (SO) with CO
2 at ambient pressure [
46].
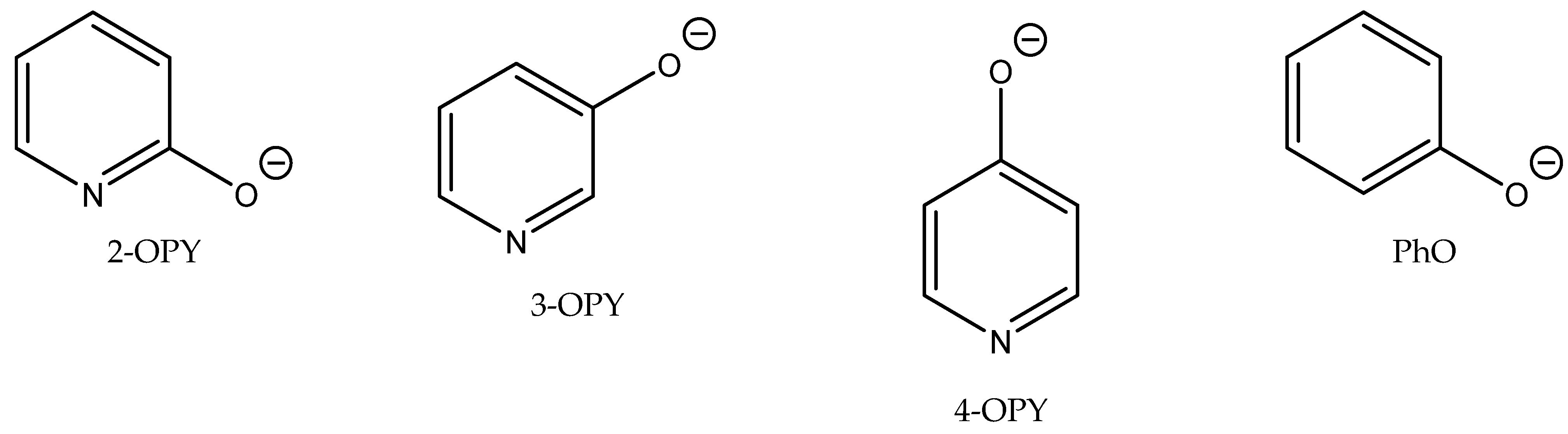
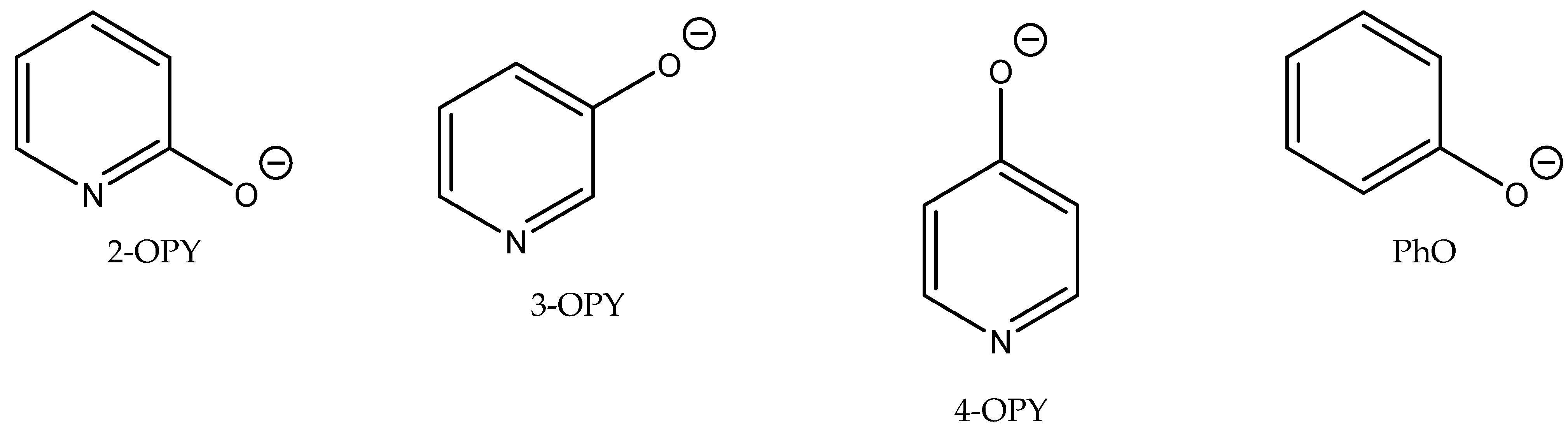
Wilhelm et al. compared the action of different aromatic or heterocyclic alcoholates (phenolates or anions of hydroxypyridine regioisomers,
Figure 8) used as anions in combination with tetrabutylphosphonium, tetrabutylammonium and
N-ethyl-DBU-based cations [
69] (
Table 14). The authors discovered the cooperative effect of the alcoholate anion of 2-hydroxypyridine with the tetrabutylphosphonium cation in the case of a reaction of CO
2 with epichlorohydrin. The catalytic activity, however, of other ILs containing Bu
4N
+ or Et-DBU
+ cations was slender (
Table 14).
The authors suggested the mechanism of this reaction based on activation of the epoxide ring with the tetrabutylphosphonium cation as the Lewis acid with the simultaneous activation of CO
2 and phenolate [
69]. The most active [Bu
4P] 2-OPY was tested at ambient pressure for the carbonation of different terminal epoxides with satisfactory results, using 50 molar % quantity of [Bu
4P] 2-OPY to an appropriate epoxide (
Table 15).
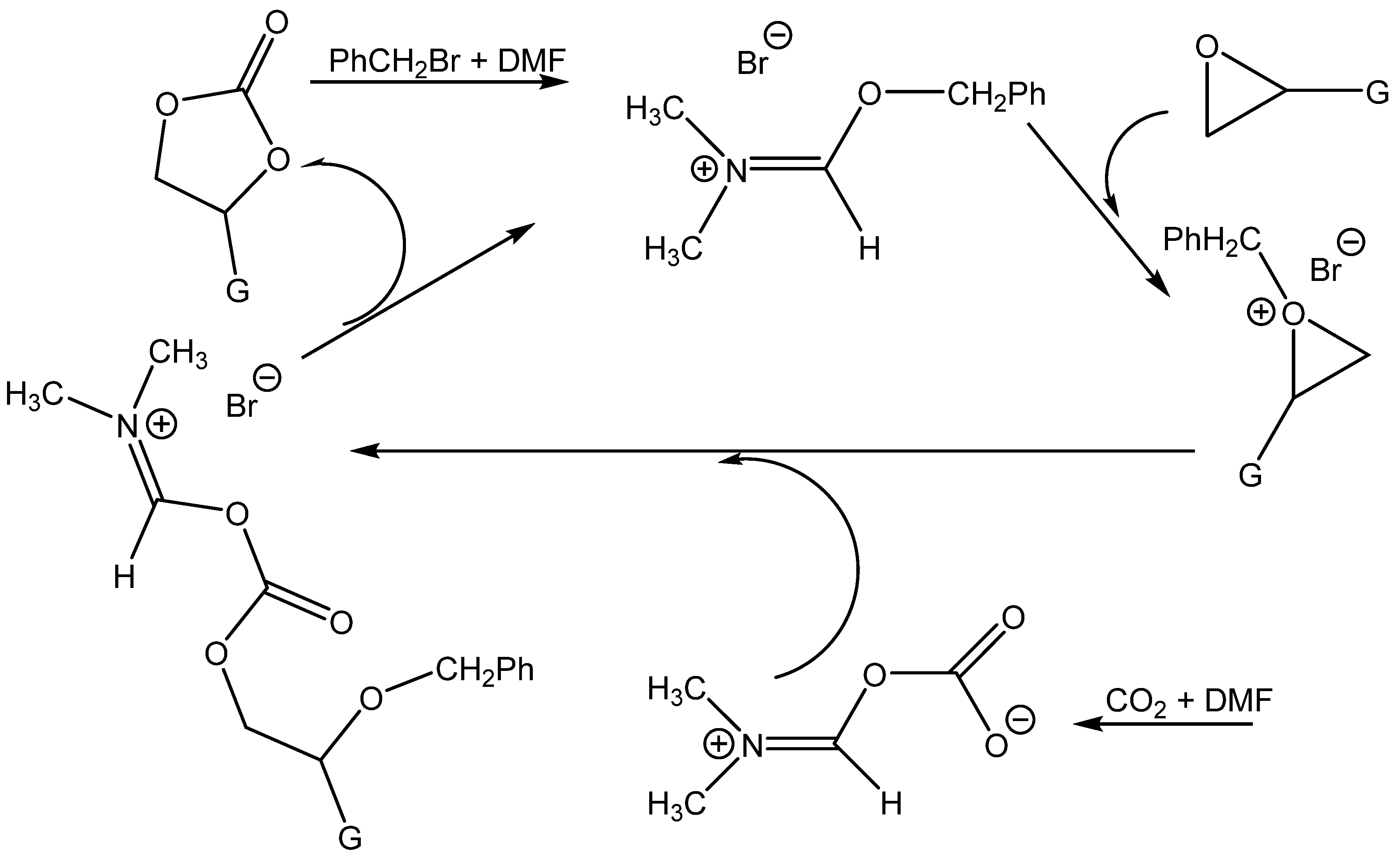
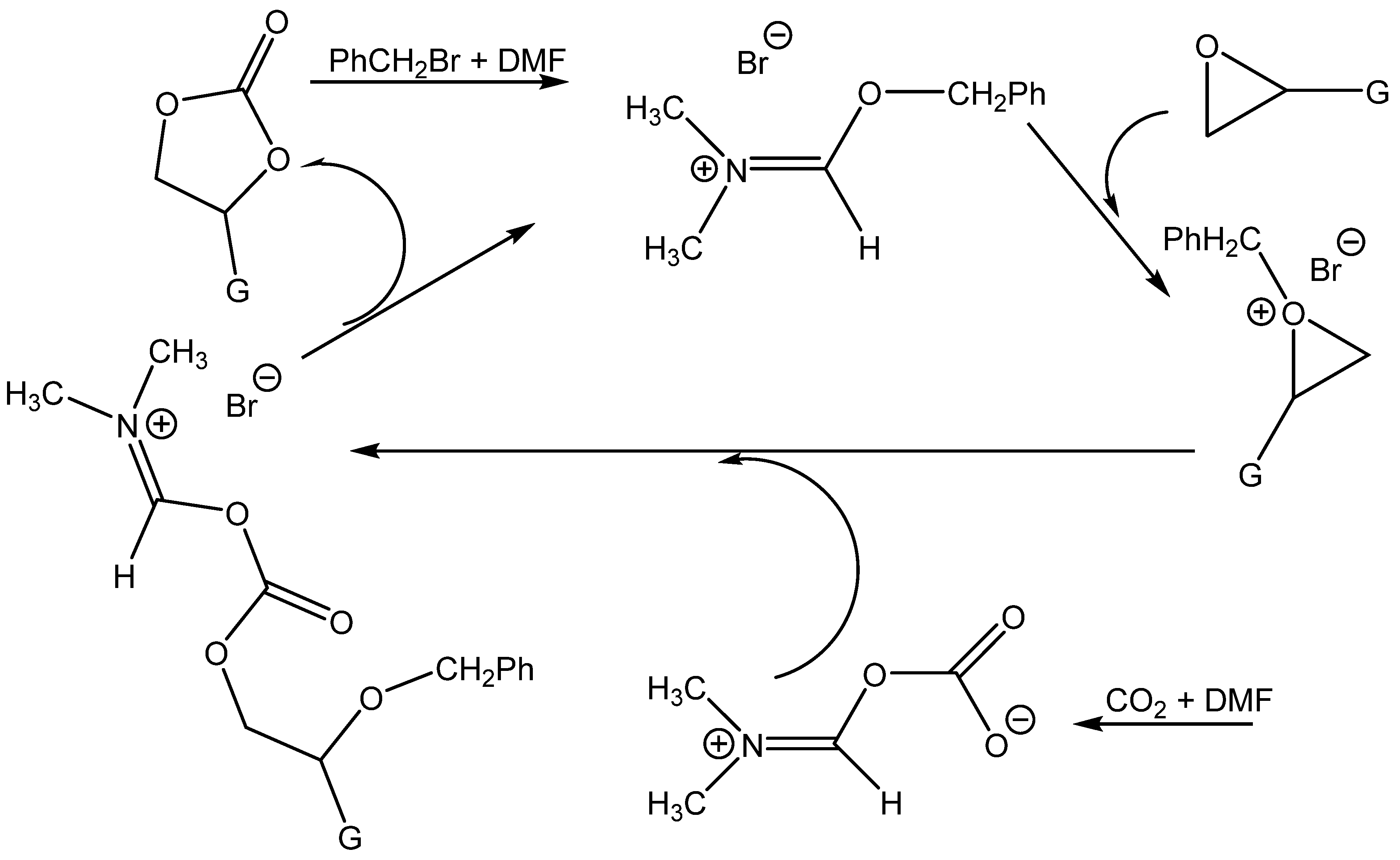
Wang et al. prepared ammonium salts in situ by alkylating tertiary amides (
N,
N-dimethylformamide (DMF),
N,
N-dimethylacetamide (DMAc),
N-formylmorpholine,
N-methylpyrrolidone, tetramethylurea and
N-formylpiperidine) with benzyl halogenides. The prepared ammonium salts enabled the formation of cyclic carbonates even at an ambient pressure, especially those prepared from DMF using benzylbromide [
70] (
Table 16).
Wang et al. attributed the high activity of the DMF + BnBr mixture in particular to the activation of the oxirane ring by benzyl cations and the contemporary nucleophilic activation of CO
2 by DMF [
70] (
Scheme 9).
Similarly, the effectiveness tertiary amines described earlier as active catalysts (see
Section 3.1) was satisfactorily proven for the reaction of epoxides with CO
2 at an ambient pressure after in situ quaternization via benzylation [
71] (
Table 16).
For a comparison of the action with the ammonium salts formed using arylmethylbromide derivatives, Bu
4NBr was employed as the bromide source using the same reaction conditions as the model reaction (
Table 16, Entry 15).
As could be seen, the yield was lower than that using benzyl bromide as the bromide anion source, which was presumably due to the electrostatic interaction between the bromide anion and the ammonium center decreasing with the bulkiness of the cation. The authors stated, based on above-mentioned results, that the nucleophilicity of the bromide anion is weaker for Bu4NBr than for the salts (Bn-DBU+.Br−).
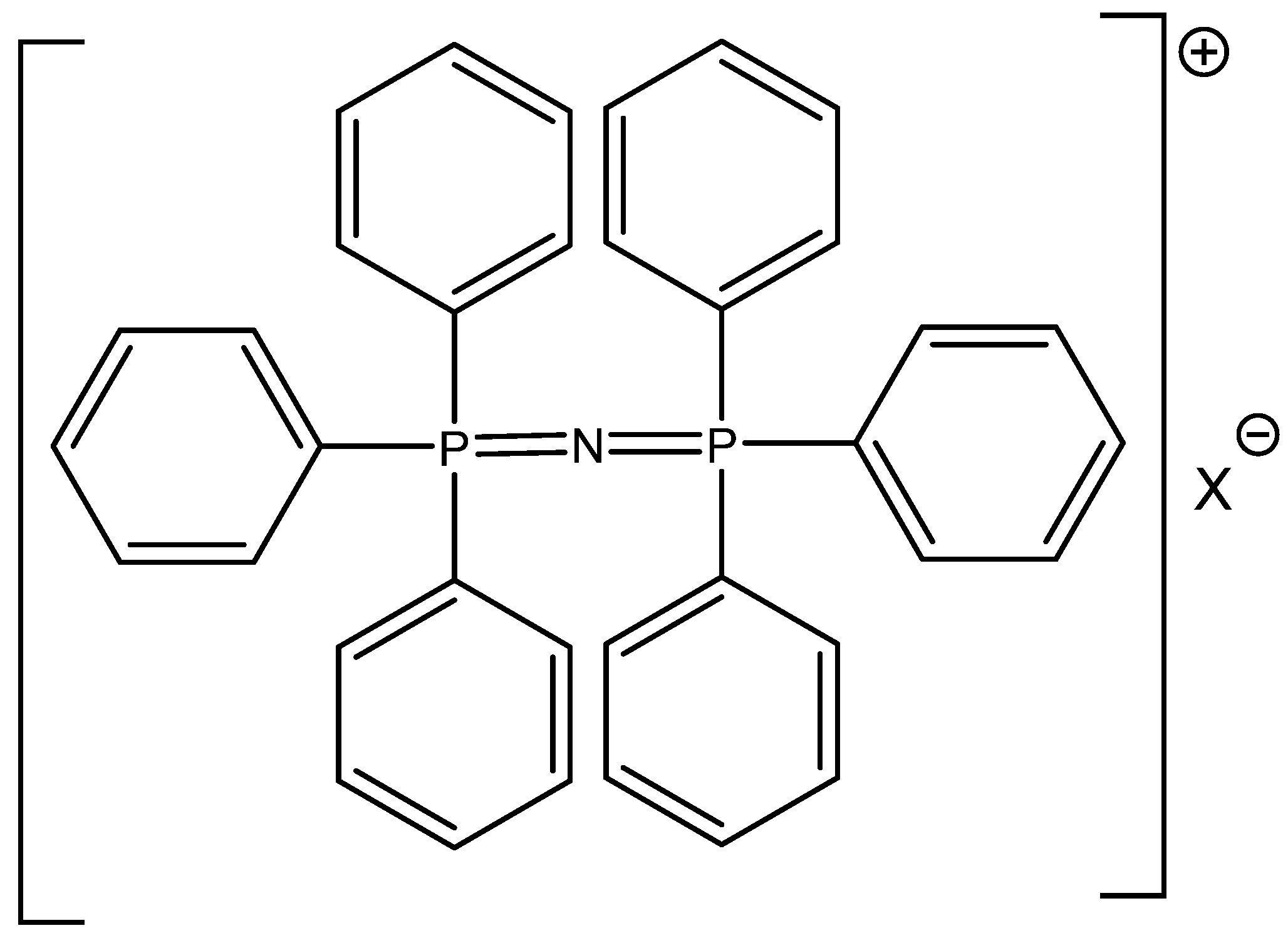
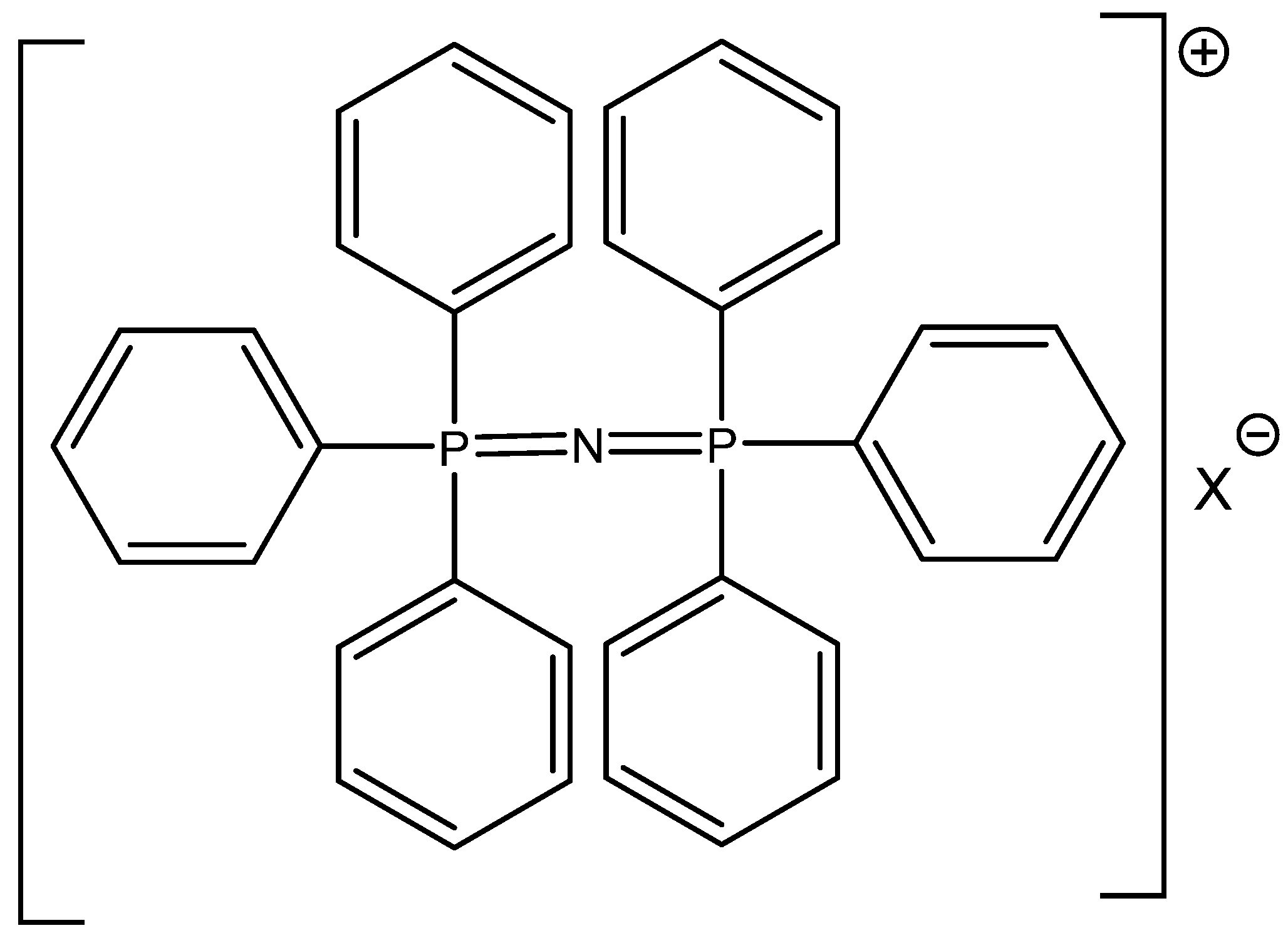
The successful utilization of tetrabutylammonium halides, especially bromide and iodide, in CO
2 cycloaddition reactions was reported by Calo [
72] (
Table 17). The higher reactivity of Bu
4NBr/Bu
4NI in comparison with RMIMBr or RPYBr salts was explained by less coordination of halide with the bulkier Bu
4N
+ cation [
72]. In addition, the catalytic activity of cheap and commercially available Bu
4NXs is high and quite comparable with much more expensive PPNXs salts (
Figure 9).
3.5. Two Component Catalysts Containing HDBs and Onium Salts
The cheap and easily available quaternary ammonium halides TBAB and TBAI are often combined with different HDBs with the aim of boosting catalytic activity for the insertion of CO2 in the oxirane ring.
It was observed that even the addition of glycidol to the Bu
4NX significantly increased the yield of PO compared with Bu
4NX used alone [
74] (
Table 18, Entries 4, 7–9).
Some mixtures of onium salts with HBDs produce low-melting eutectic solvents (DESs) that readily dissolve both CO
2 and epoxide, enabling cycloaddition even at ambient pressure and low temperature [
75]. DES is defined as a mixture of two or more compounds that are typically solid at room temperature, but when combined at a particular molar ratio, changes into liquid at room temperature [
76].
They not only possessed comparable physicochemical properties to traditional ILs (designability, non-volatility and high thermal stability), but also had advantages such as low cost and a simple preparation process (mixing and melting) without the need for purification.
DESs prepared via the mixing of tertiary amines hydrogen halides (R
3N.HX) and ethylene diamine or different aminoethanols were compared in the carbonation of SO at an ambient pressure, obtaining intriguing yields and selectivities of SC even in the case of DES prepared from hydrobromide of cheap triethyl amine and diethanolamine [
75] (
Table 18, Entries 10–13). DBU hydrobromide mixed with diethanolamine at a molar ratio of 2:1 was recognized as the most catalytically active. Testing this most effective DES, high yields of different carbonates were determined by GC-MS even at an ambient pressure and room temperature of CO
2 after 48 h using 20 molar % of this DES. Testing the carbonation of internal ChO, the yield of CC was 43%. Applying a mixture of 15% CO
2 with nitrogen (simulated flue gas) drops, however, the yield decreased from 92% (100% CO
2 at an ambient pressure after 48 h at room temperature) to 10% (using 15%CO
2 in nitrogen under the same reaction conditions,
Table 18, Entries 14–21).
Comparing the catalytic activity of different DESs with various protic ILs, the DES (2 DBU.HBr + 1 DEA) is much more active than PIL DBU hydroiodide (DBU.HI) alone. The observed high catalytic activity was explained by the synergistic action of DEA (as HBD) and DBU.HBr as a source of highly nucleophilic naked bromide [
75] (
Table 18, Entries 16–21 and 24).
Similarly, high activity was observed using DES prepared from Bu
4PBr with 2-aminophenol for the carbonating of terminal epoxides. The carbonation of internal epoxide CO to CC was, however, very slow [
79] (
Table 18, Entry 28).
Pentaerythritol as an aliphatic polyol-based HDB was recognized as effective for the carbonation of PO at elevated pressure [
80]. Although completely inactive used alone or with KI, in a mixture with Bu
4N
+ bromide or iodide, it is very active, obtaining a 97% yield of PC at 70 °C after 22 h of CO
2 (0.4 MPa) action (
Table 19).
Choline iodide together with
N-hydroxysuccinimide forms DES, enabling the high-yield carbonation of PO to SC at 30–80 °C and 1 MPa pressure of CO
2. Instead of choline iodide, Bu
4NX in a mixture with
N-hydroxysuccinimide is applicable [
81]. Using a 2 MPa pressure of CO
2, a high yield of CC from internal ChO was obtained at 70 °C after 10 h of reaction (
Table 18, Entry 29).
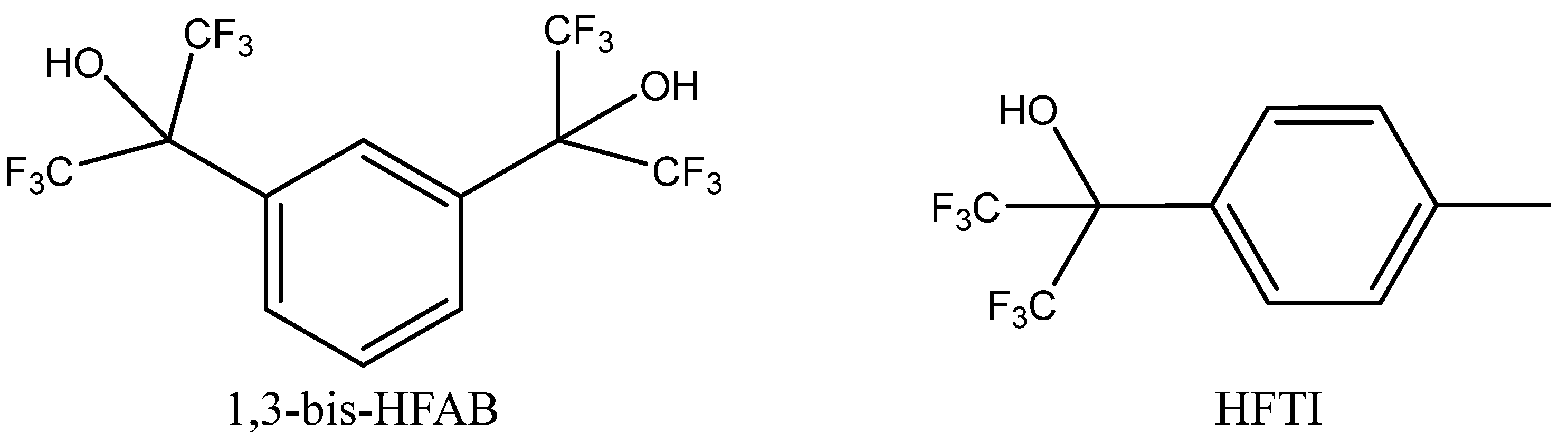
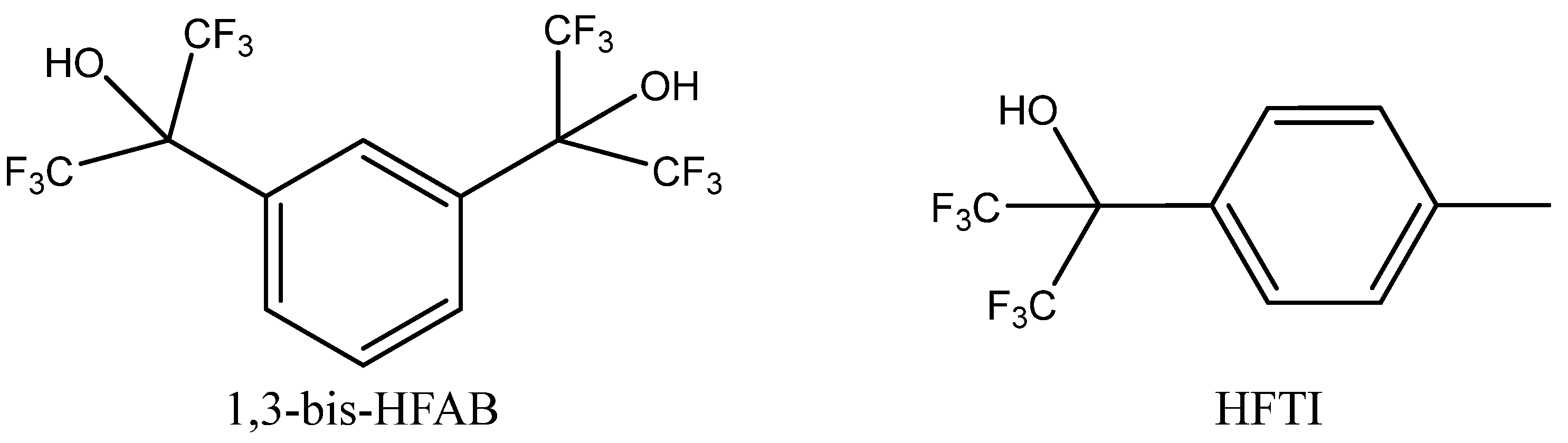
A broad set of aliphatic and aromatic alcohols in terms of their role as potential HDBs was studied by Alves et al. [
82] in the co-action of Bu
4NBr using pressurized CO
2 (2 MPa) for PO carbonation. The authors observed that the most active HDBs were low-polar polyfluorinated secondary alcohols such as tertiary alcohols HFTI or 1,3-bis-HFAB (
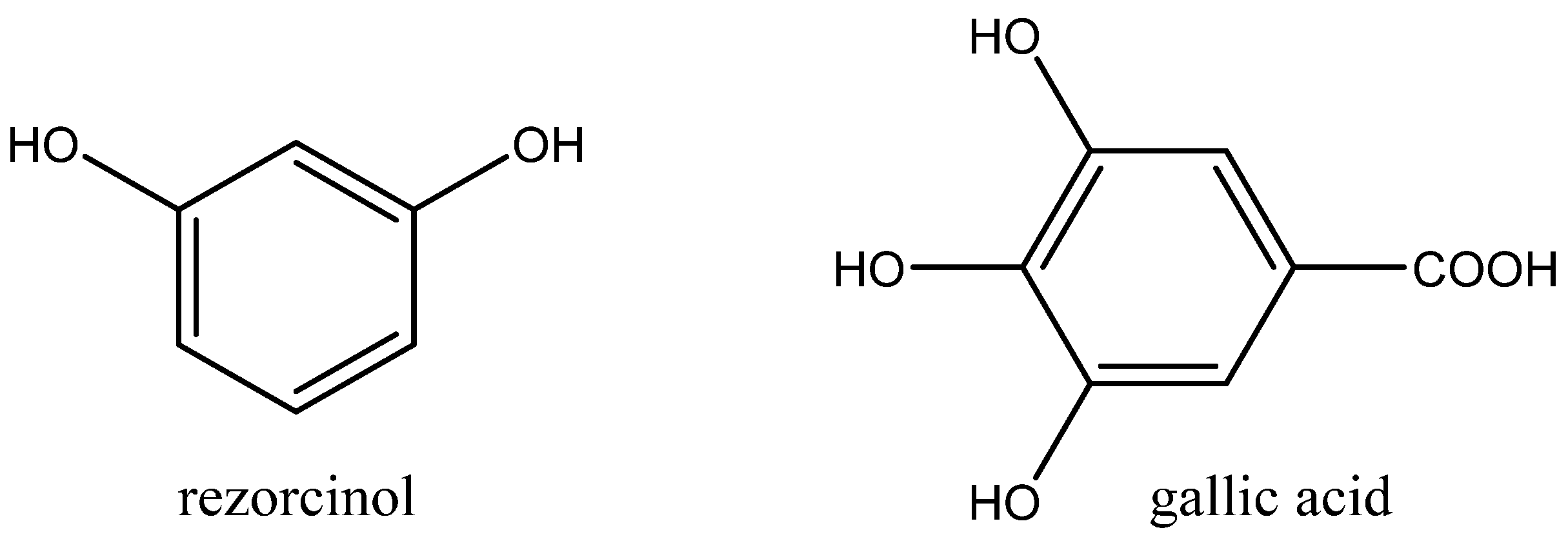
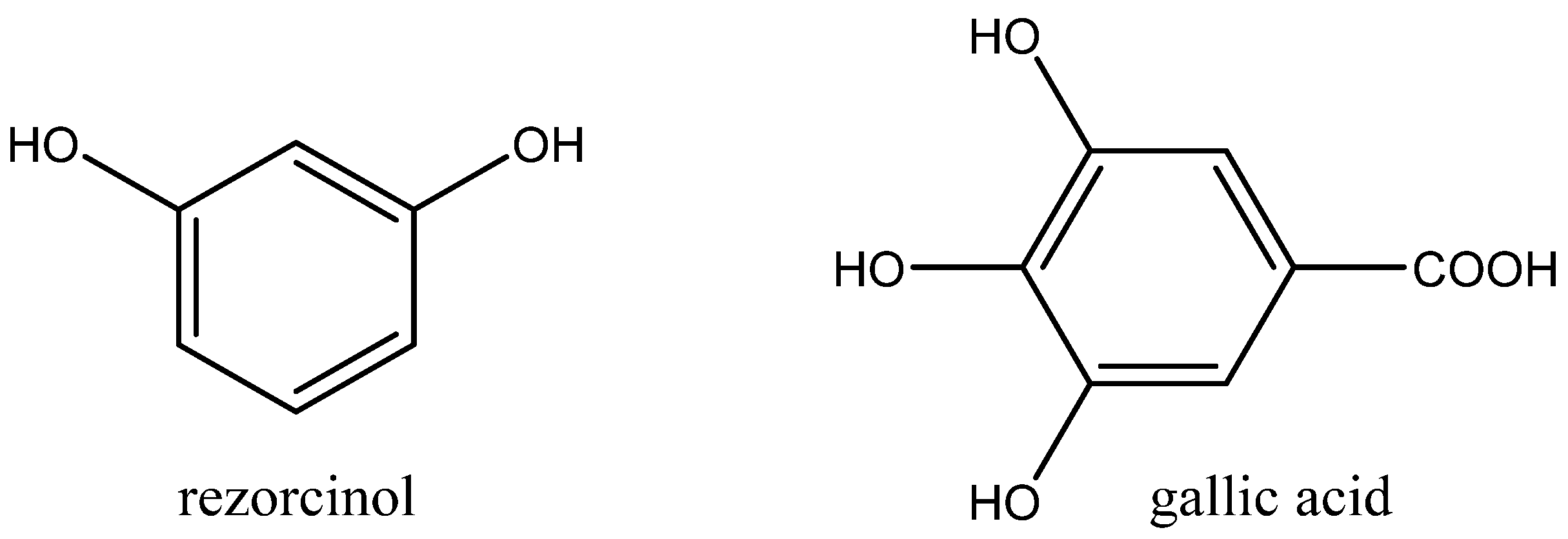
Figure 10).
Aromatic polyols such as pyrocatechol, pyrogallol and gallic acid were less catalytically active. Aliphatic alcohols exhibited low cooperative activity in the case of the tested Bu4NBr, which was practically comparable with the catalytic effect of sole Bu4NBr. Interestingly, some of tested alcohols even exhibited inhibition effects.
The high catalytical activity of RMIMs/phenols-based DESs was published by Liu et al. even at an ambient pressure of CO
2 and at room temperature for SO [
83]. Especially
N-ethyl-
N′-methylimidazolium iodide (EMIMI) was recognized as a very suitable part of DES in co-action with phenols substituted with electron-donating groups such as –NH
2, –C(CH
3)
3 and –Cl, –OH. The most effective DES contained EMIMI (2 mol) and resorcinol (1 mol). The authors explained its high catalytic activity as multifunctional HBD-based activation by acidic hydrogen bound in position 2 of EMIM salt together with hydrogen from the –OH group of resorcinol (
Figure 11) and the subsequent action of iodide as a nucleophile. Interestingly, comparing the activity of (2 EMIMI + 1 resorcinol) DES with the much cheaper (2 Bu
4NI + 1 resorcinol) binary system for SO carbonation, the obtained yields of PEC were very similar [
83]. SO was the single epoxide studied, however, in this article. Another catalytically very effective DESs containing mixture of choline chloride and malic acid or choline iodide and glycerol published Vagnoni et al. [
84].
Additional very effective DESs were obtained as catalysts by mixing 2-hydroxymethylpyridine or 2,6-hydroxymethylpyridine with Bu
4NI [
40]. These DESs were able to catalyze the carbonation of EPIC to chloromethyl-ethylenecarbonate (CMEC) even at room temperature and ambient CO
2 pressure. The carbonation of internal epoxide ChO was very slow, however, under ambient conditions even after 20 h using 8 molar % of catalyst [
40].
Gallic acid (
Figure 11), as a green, biobased and biodegradable HDB, was discovered by Sopena et al. as a more effective alternative of resorcinol in a binary Bu
4NI + gallic acid catalytic system dissolved in 2-butanone [
85]. Even internal epoxide was carbonated with a high yield at 80 °C and 1 MPa pressure of CO
2 after 18 h [
85].
Polycarboxylic acids such as citric acid were effectively applied as the HDB part of DES together with choline iodide [
86]. The other tested carboxylic acids were less active HDBs compared with citric acid. Additionally, it was observed that the molar ratio of the used HBA and HDB is crucial. For DES obtained by the melting of choline iodide, citric acid at a molar ratio of 2:1 (excess of iodide source) is highly active. Changing the molar ratios significantly decreased the reaction yield (but not selectivity). ChO tested as an internal epoxide at 70 °C and 0.5 MPa of CO
2 produced only 36% CC after 6 h [
86].
The attempts to substitute ILs-based iodides or bromides as key parts of DESs were described by Wang et al. [
87]. Applying boric and glutaric acids, together with BMIMCl, the authors described significant catalytic activity even without the presence of bromide or iodide ions for the carbonation of terminal epoxides at 0.8 MPa of CO
2 and 70 °C. [
87]. The carbonation of internal ChO was below 40% after 7 h of CO
2 action.
The most active HDB described until this time for the catalysis of epoxides’ carbonation is ascorbic acid in co-action with Bu
4NI [
15] (
Table 20). This mixture was effective even for the carbonation of internal epoxides, even at an elevated temperature (100 °C) and 2 MPa CO
2 pressure [
15] (
Table 21).
Encouraged by the robustness of this catalytic system, Elia et al. tested the Bu
4NI/ascorbic acid system for the cycloaddition of CO
2 in epoxidized fatty acid esters [
91]. Cyclic carbonates based on fatty acid esters seemed to be potential plasticizers for polyvinyl chloride instead of harmful phtalates, for example [
92].
As can be seen in
Table 21, the most effective catalytic mixture found contains Bu
4NCl/ascorbic acid. Bu
4NCl is superior because overly nucleophilic Bu
4NI causes undesirable Meinwald rearrangement producing ketones instead of cyclic carbonates, probably due to the sterical hindrance in the case of epoxidized oleic acid methyl ester [
91]. In the case of epoxidized polyunsaturated fatty acid esters, allylic alcohols are produced as by-products using Bu
4NI [
91].
3.6. Application of Bifunctional (or Multifunctional) Onium Salts
The functionalization of ILs involves an increase in catalytic activity owing the synergistic effect between the bound functional groups (nucleophilic iodide or bromide anions together with –NH
2, –COOH or –OH groups in the role of HBDs). The reached synergism enables a decrease in the quantity of the multifunctional catalyst and simpler separation in an optimal case [
93] (
Table 22,
Figure 12). On the other hand, multifunctional ILs are more difficult to prepare and more expensive due to this reason.
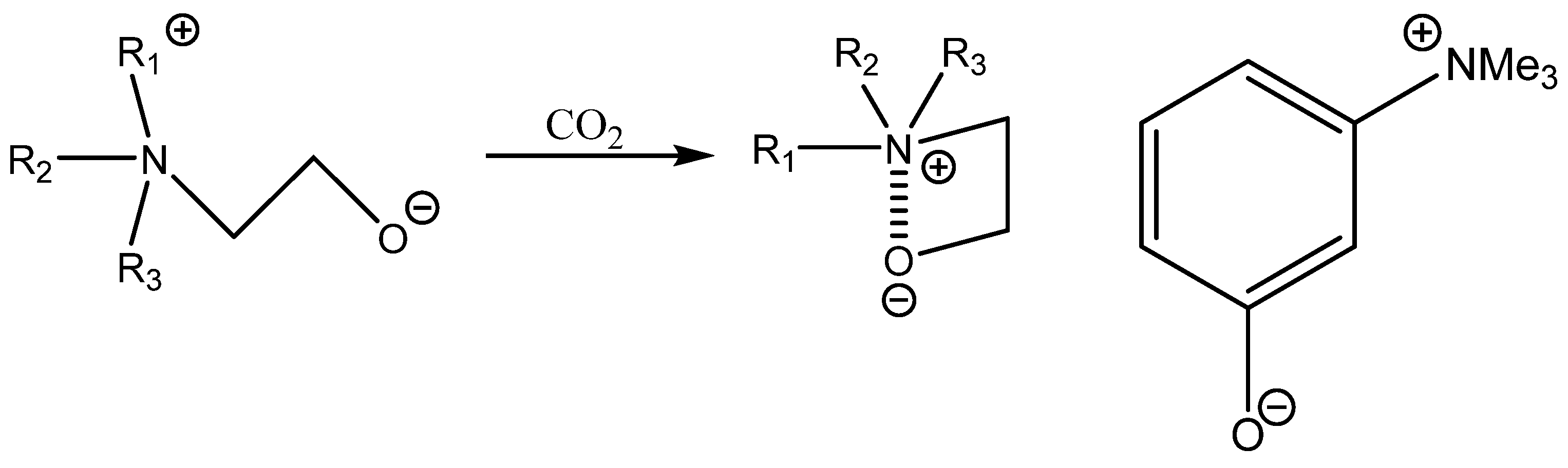
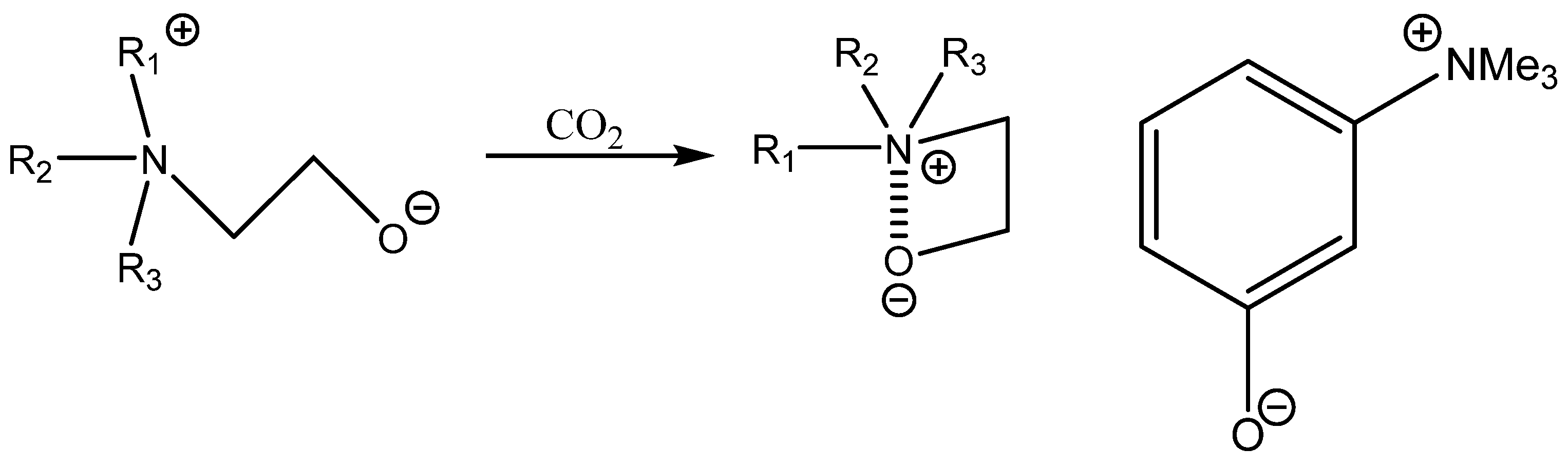
Bifunctional catalysis based on aromatic OH-functionalized onium salt was described by Tsutsumi et al. [
94]. This research work demonstrates that it still possible to discover a very active and quite cost-effective bifunctional catalyst such as m-trimethylammonium phenolate, which is much more effective than more structurally complicated ones [
94]. This catalyst was only tested, however, for the carbonation of terminal epoxides and still requires higher pressure of CO
2 at an elevated temperature [
94] (
Scheme 10,
Table 23).
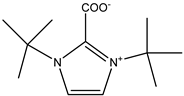
Meng et al. published very promising results obtained by testing a series of OH-functionalized DBU-based ILs (
Figure 13) [
95]. They discovered not only a recyclable organocatalyst with excellent activity for carbonation of EPIC at 30 °C and an ambient pressure of CO
2, but also a compound suitable for the utilization of CO
2 from a simulated flue gas (
Scheme 11). Its high activity is explained by the cooperative activation of the epoxide ring by protonated DBU in the role of HDB and the activation of CO
2 via carbonate formation utilizing alcoholate on a bridge-functionalized bis-DBU anion.
Another described group of catalytically active bifunctional catalysts consists of 1-alkyl-2-hydroxyalkyl pyrazolium salts [
96]. The most active one from the broad group of tested derivatives was 1-ethyl-2-(2-hydroxy)ethylpyrazolium bromide (
Figure 14). It was demonstrated that using 1 MPa pressure of CO
2 at 110 °C, even internal epoxide ChO was carbonated to CC with a considerable yield of over 60% after 4 h of action [
96].
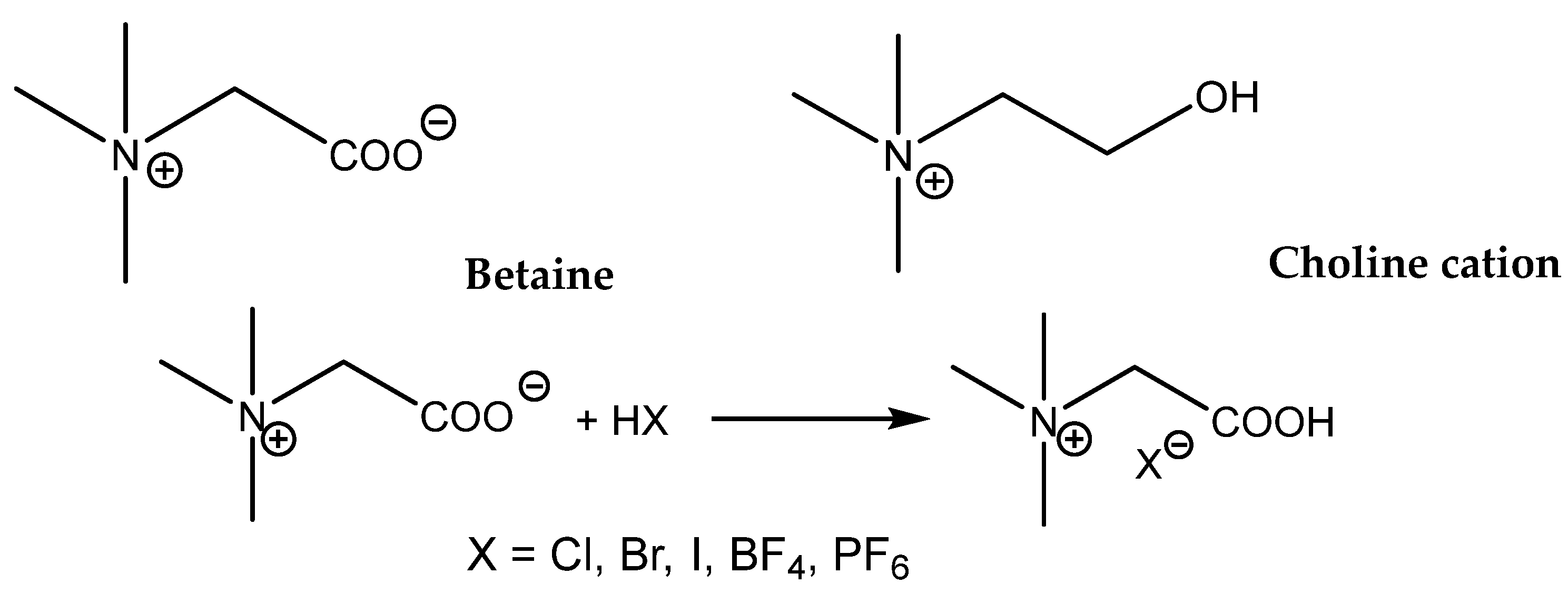
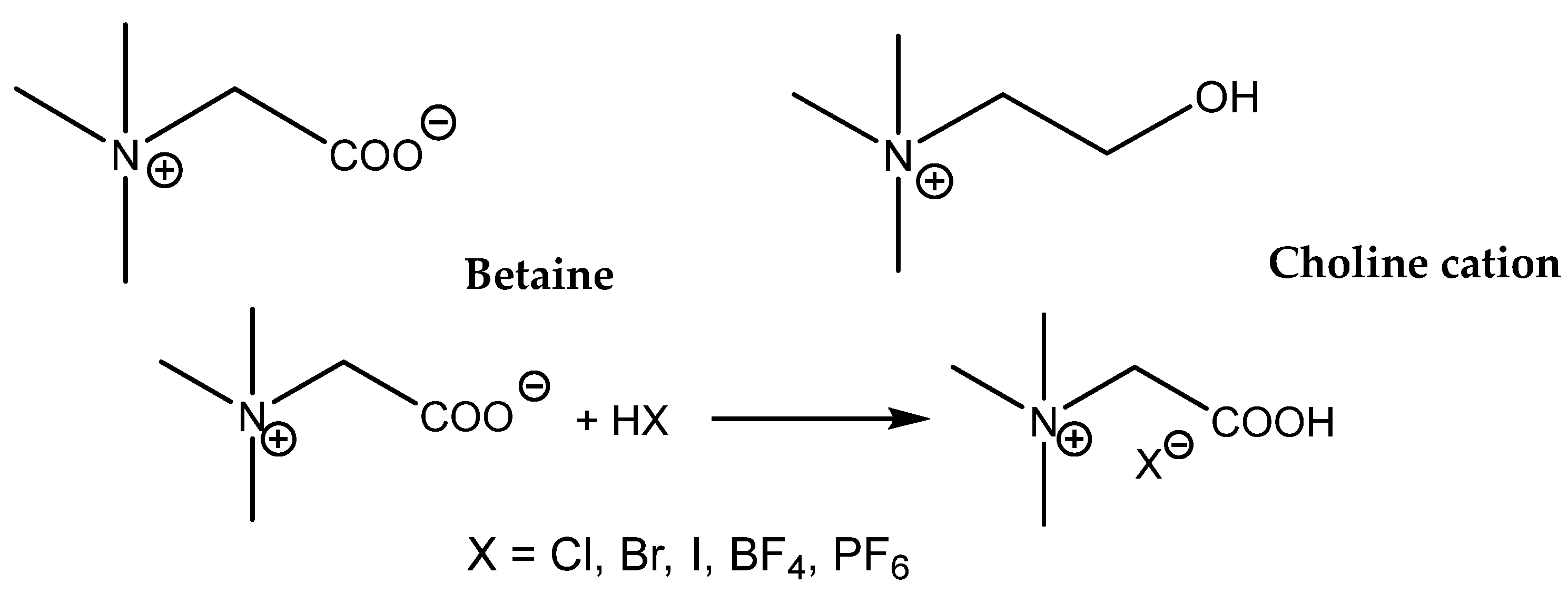
Zhou et al. compared the catalytic activity of quaternized aminoacid glycine (betaine) and quaternized aminoethanol salts (choline salts,
Scheme 12) [
97].
The main difference in the structures of these onium salts is the presence of a more acidic carboxylic group (a stronger HBD) in the betaine structure compared with the choline hydroxyl group. In addition, the effects of different anions in the case of protonated betaine were compared, and it was observed that the most active was the corresponding iodide salt. The catalytic activity of different betaine and choline salts decreased in the range: betaine.HI > betaine.HCl > choline.HCl > betaine.HBr > betaine.BF
4−. The authors interpret the low betaine.HBr activity as the effect of its low solubility in the reaction mixture. The tested choline.HCl possesses an activity comparable with Bu
4NBr, which supports the positive effect of the hydroxyl group in the activation of the oxirane ring of PO. This positive effect could be both based on HBD action and/or the activation of CO
2 on account of carbonate formation. The considerable HDB effect of the carboxylic group of protonated betaines exceeds, however, the effect of the hydroxyl group in choline. The reaction conditions for effective carbonation even of terminal epoxides are, however, harsh (8 MPa CO
2, 140 °C) [
97]. Due to the above-mentioned reasons, the research that focused on ILs functionalized with the carboxylic group(s) provided fruitful results.
Xiao et al. suggested that the influence of suitable acidity, even with the flexibility of the bound -(CH
2)
n-COOH chain in the IL structure, is crucial for the carbonation of epoxides due to the cooperation function of the ring-opening of oxiranes [
98]. When 1-(2-carboxyethyl)-3-methylimidazolium bromide was used as bifunctional IL, the PC from PO was obtained with ca. 96% yield using pressurized CO
2 (1.5 MPa) at 110 °C after 2 h. The IL showed high thermal stability and could be recycled with a slight loss in activity, while the selectivity of the cyclic carbonates remained at over 98%. The catalytic activity of the described functionalized IL-based carboxylic acids is still not unique enough, however, and these types of catalysts still require elevated pressure of CO
2 to maintain a high conversion of epoxides to cyclic carbonates. In addition, the carbonation of internal epoxide is still quite slow even at the above-mentioned high pressure and elevated temperature [
98].
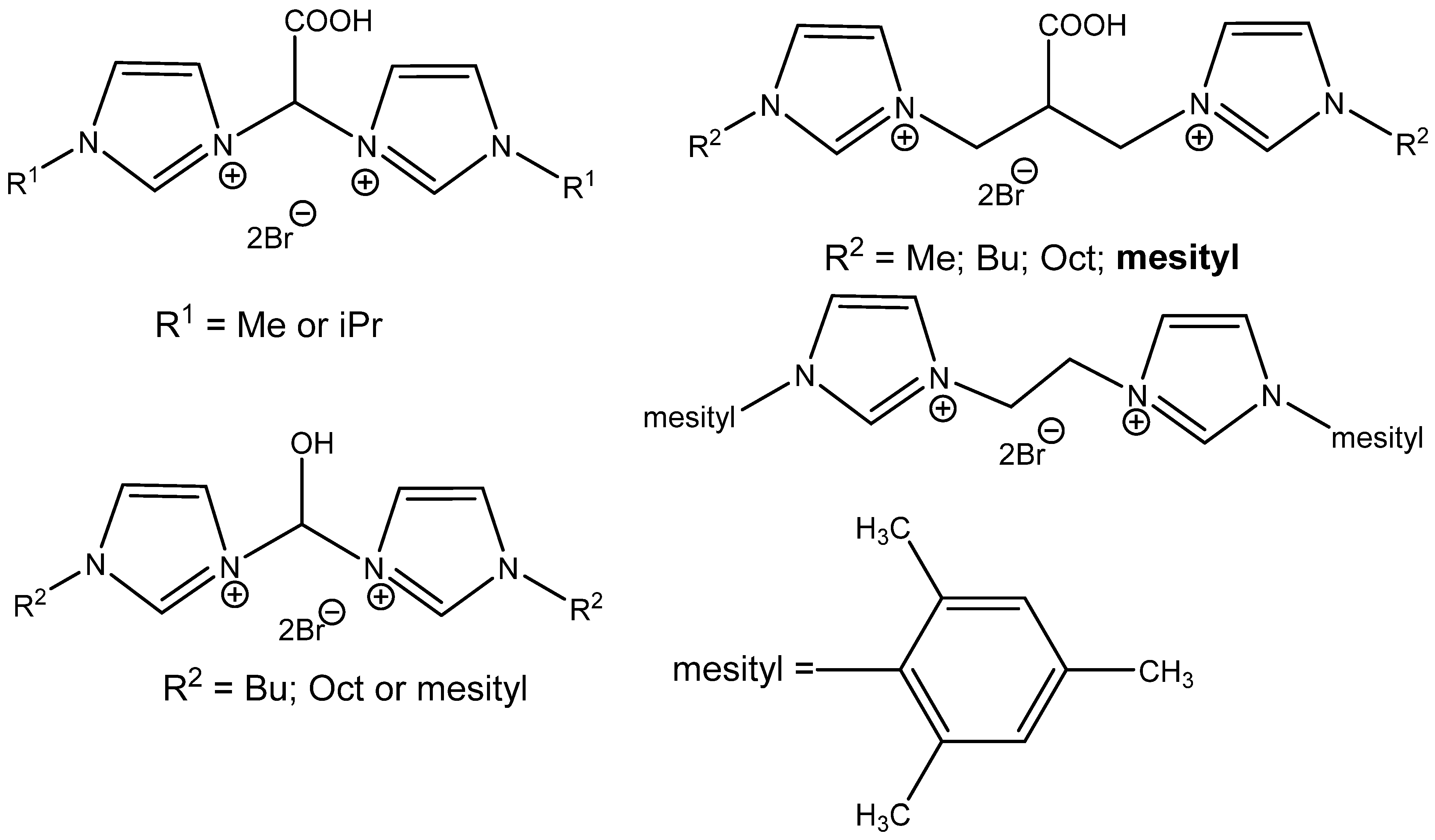
The construction of bridge-functionalized bisimidazolium-based ILs improves the catalytic activity of acidic ILs, as was discovered by Kuhn et al. [
99]. The most active catalyst is the most branched one, bis(imidazoyl)isobutyric acid derivative,
N-arylated with hydrophobic mesitylene (
Figure 15). It is well recyclable and active even using 0.4 MPa CO
2 at 70 °C. It is ineffective, however, in the case of internal epoxides’ carbonation [
99].
An additional direction of research related to the significant increasing of catalytic activity was discovered by Han et al. [
100] and Wang et al. [
101]. The Han and Wang research groups recognized the crucial role of ion pairs produced by a combination of acidic ILs with guanidinium cations. Using the same acidic ILs, neutralized by alkylated guanidines, enables an increase in activity, probably due to the distinctive activation of reacting CO
2. This catalytic system is active at 0.1 MPa CO
2 and 30 °C for the carbonation of EPIC, but fails even in the case of PO (
Table 24). Additionally, the used ion pairs are simply separable from the produced cyclic carbonates by means of extraction with ethyl acetate, enabling simple recycling without a significant drop in conversion.
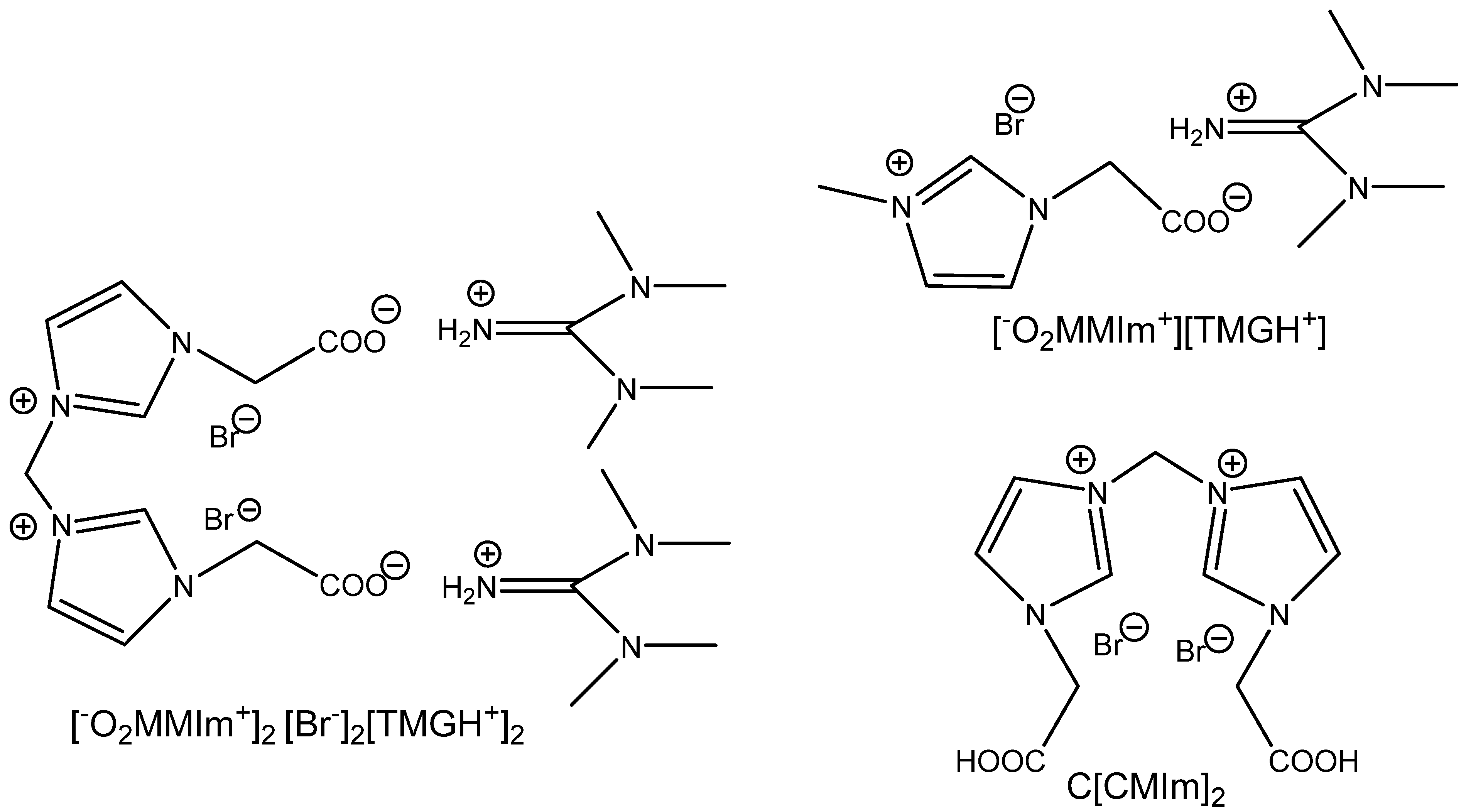
Bridged methylene(bis)imidazolium salts substituted on both
N′-nitrogens by carboxymethyl groups are more active after neutralization with tetramethylguanidine [
101] (
Figure 16,
Table 24). Even this catalytic system is active at 0.1 MPa CO
2 and 50 °C but the carbonation of internal ChO seems to be sluggish using the above-mentioned reaction conditions. Additionally, the used catalyst is simply separable from the produced cyclic carbonates and enables simple recycling without a significant drop in conversion.
The reverse activation of dual amino-functionalized ILs neutralized with acidic aminoacids, such as glutamic or aspartic acids, is possible, as was documented by Yue et al. [
102]. This ion-pair-based catalytic system produces, however, high yields at 0.5 MPa and a temperature of 105 °C after 13 h of CO
2 action. It is recyclable without loss of activity and works well in the case of terminal epoxides [
102] (
Figure 17).
A very attractive alternative approach was published by Kumar et al. [
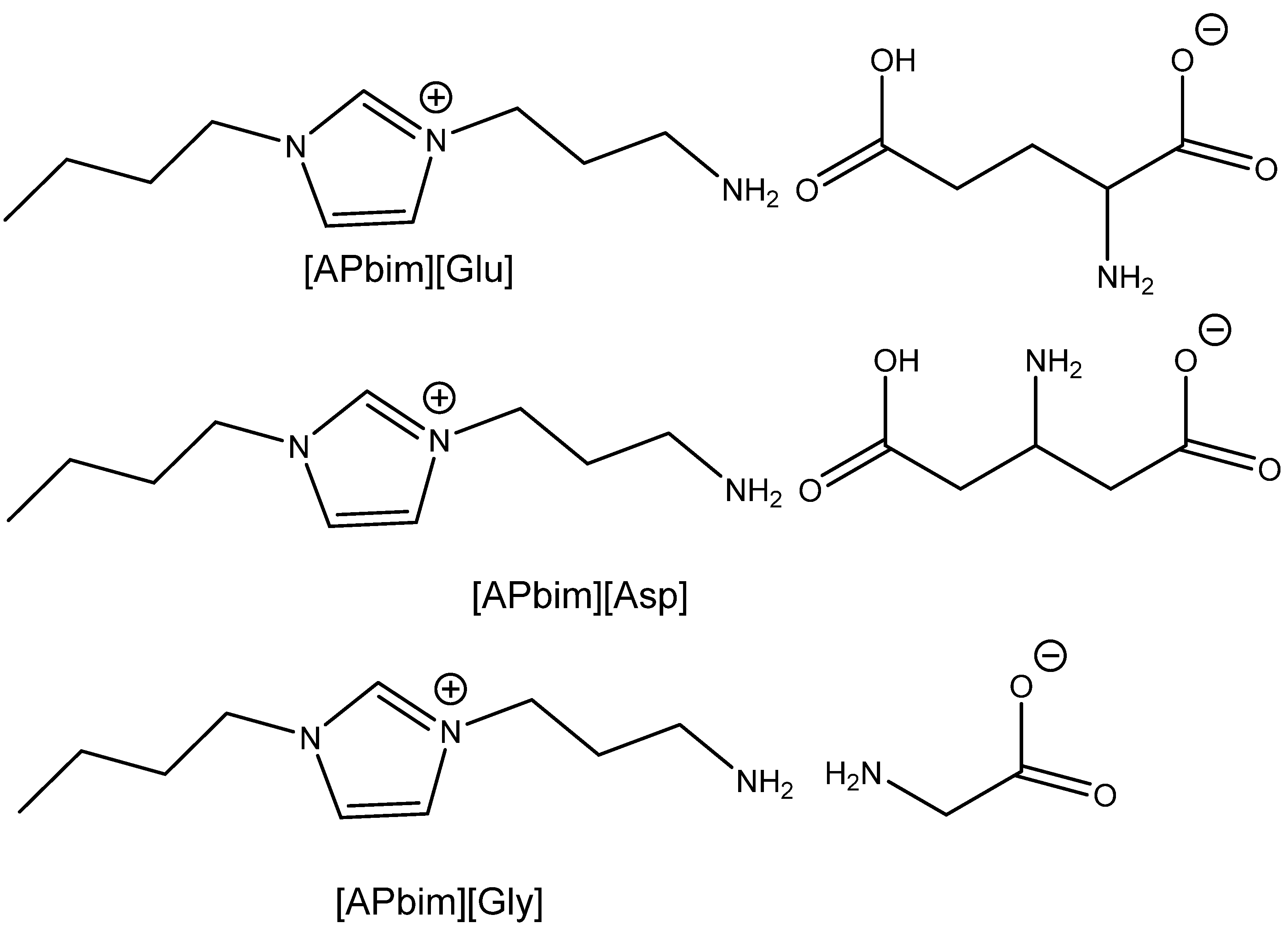
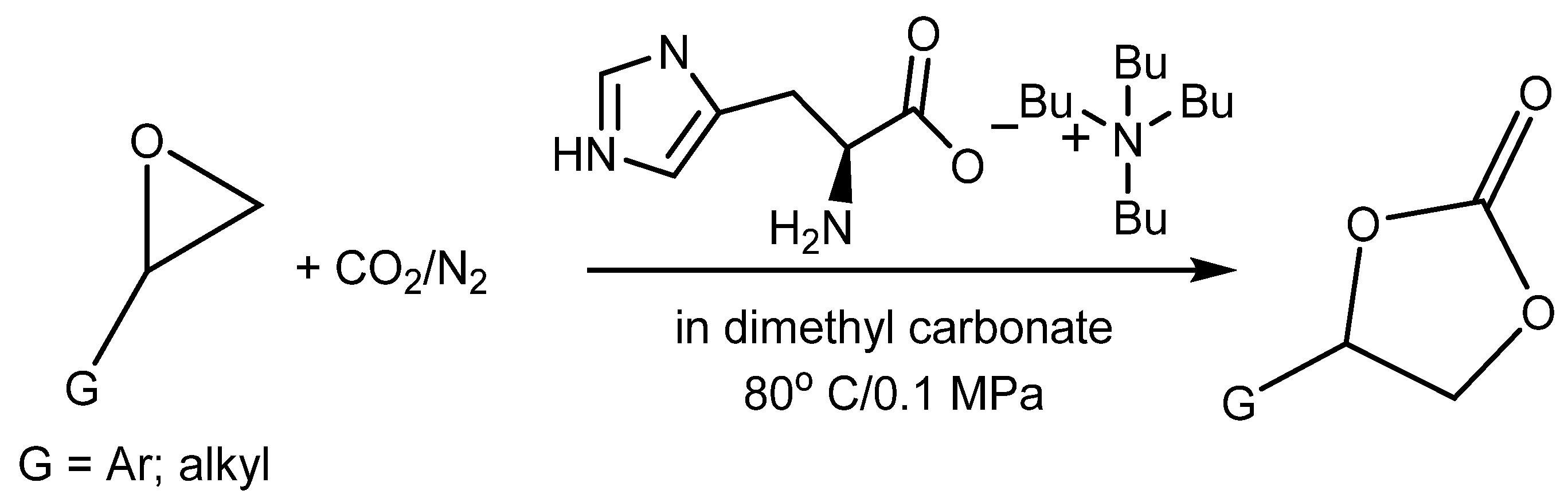
103]. Their research was focused on the utilization of CO
2 from model flue gas (5–15% CO
2 in N
2 stream at atmospheric pressure) at 80 °C using task specific AA-based ILs (
Scheme 13). The authors verified that tetrabutylammonium salt with histidine dissolved in dialkyl carbonate enables the capture and usage of CO
2 for the carbonation of terminal epoxides at the above-mentioned reaction conditions, with a high yield. This research work is one of the very infrequent examples of the direct capture and subsequent utilization of CO
2 from (model) flue gas. The authors documented that this catalyst is recyclable with no drop in efficiency after six recycling steps. This catalytic system was proved, however, only on terminal epoxides [
103].
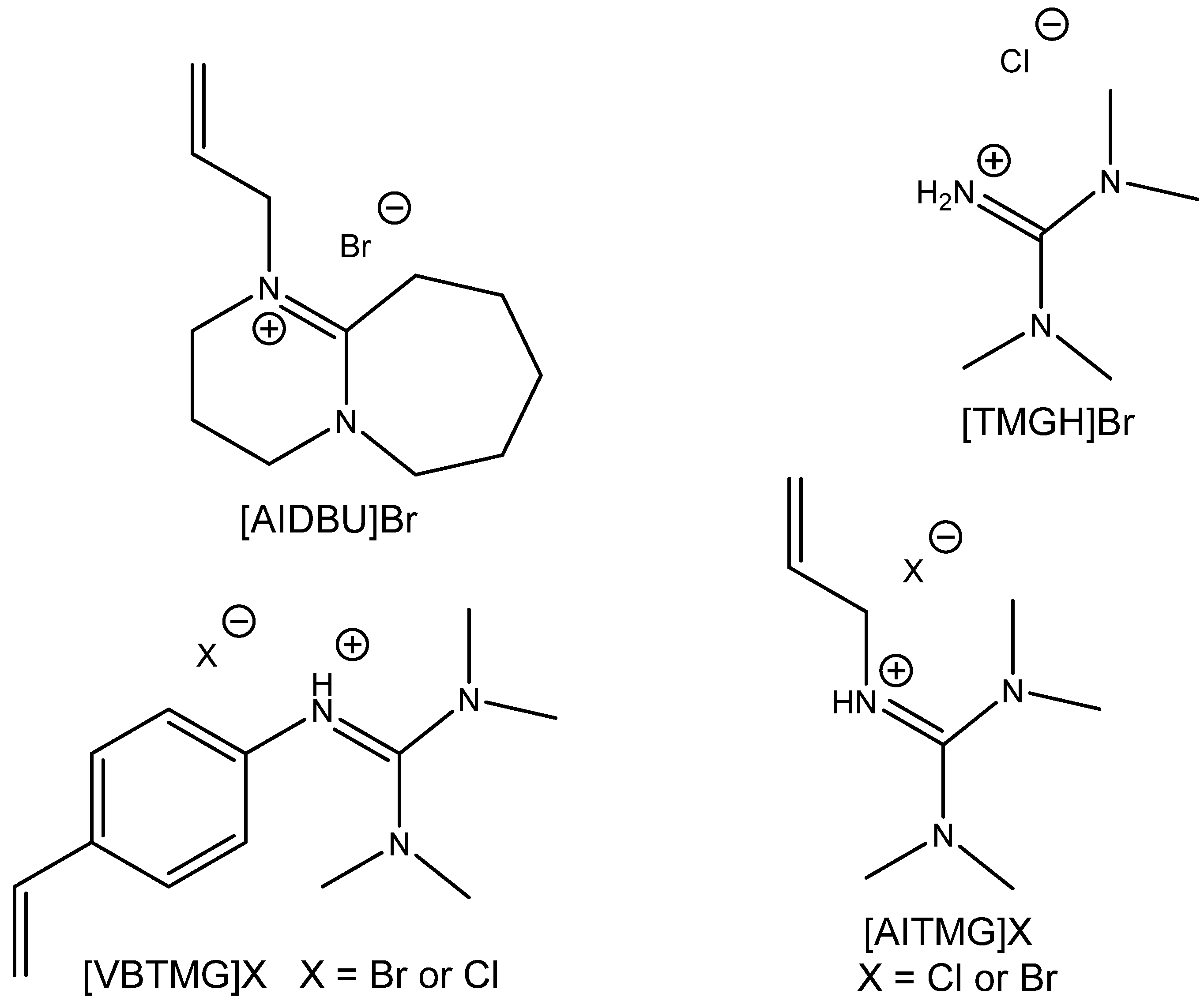
A different approach was used for the preparation of highly catalytically active bifunctional ILs using allylation by Hui et al. [
104] (
Figure 18). They discovered tetramethylguanidine-based allylated IL with superior activity for the capture and utilization of CO
2 from simulated flue gas at 120 °C, ambient pressure and solventless conditions (
Table 25). This catalyst is effective even for carbonation of ChO and reusable with low loss of activity after five recycling steps [
104]. This type of catalyst seems to be very attractive even for the carbonation of other internal epoxides including eventually epoxidized fatty acid esters.



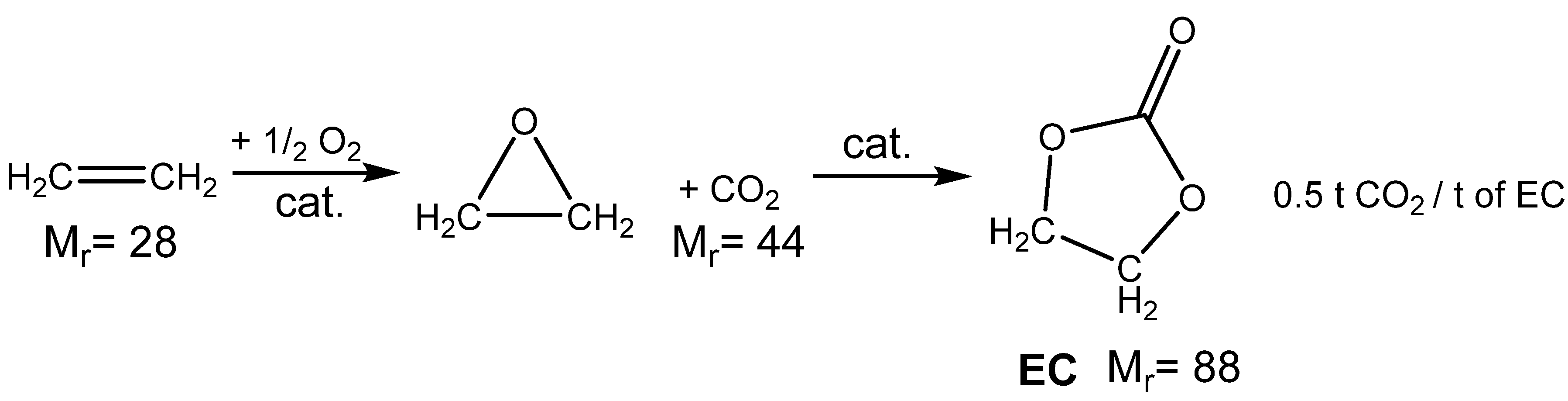




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



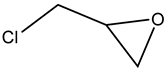

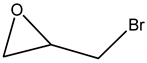
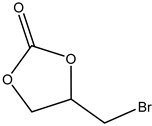

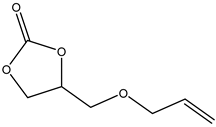



 .
.