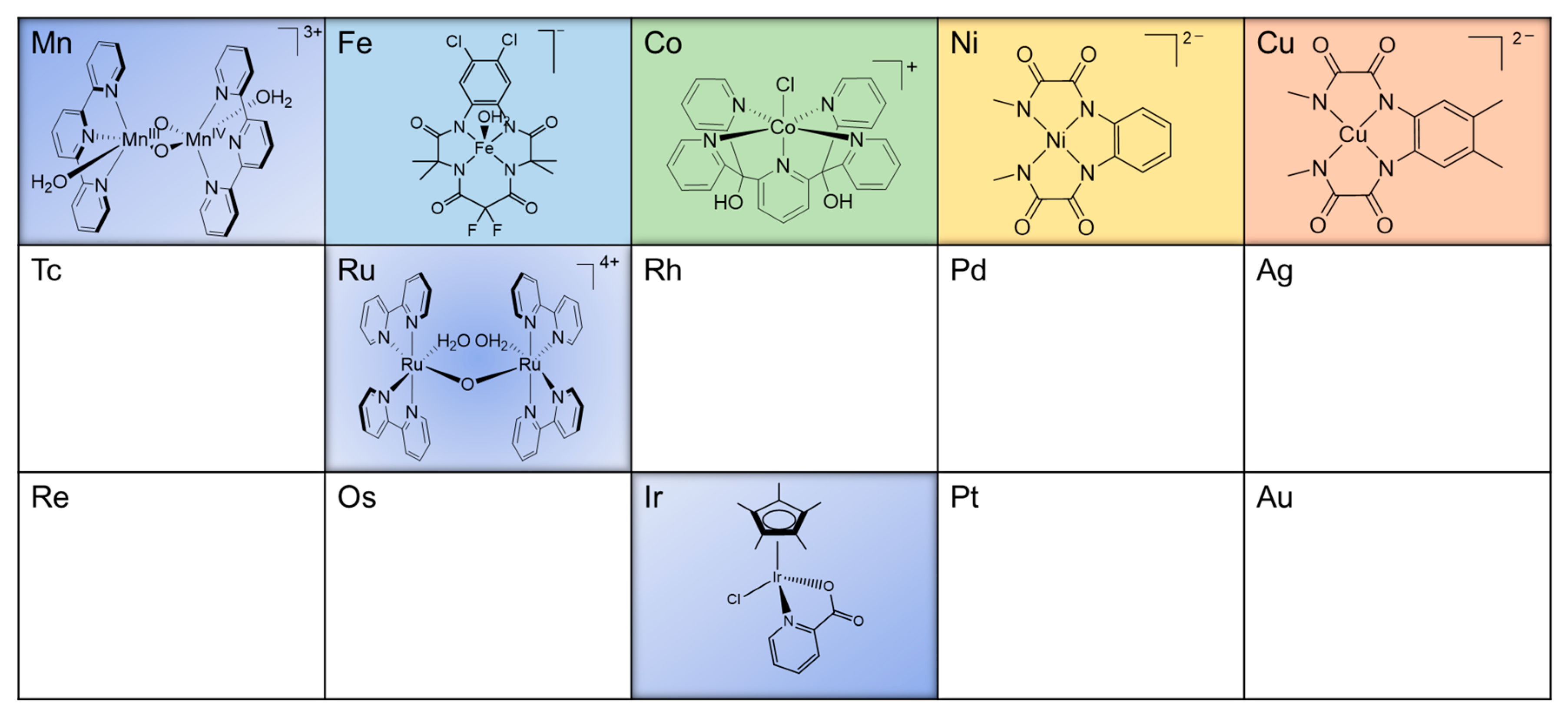
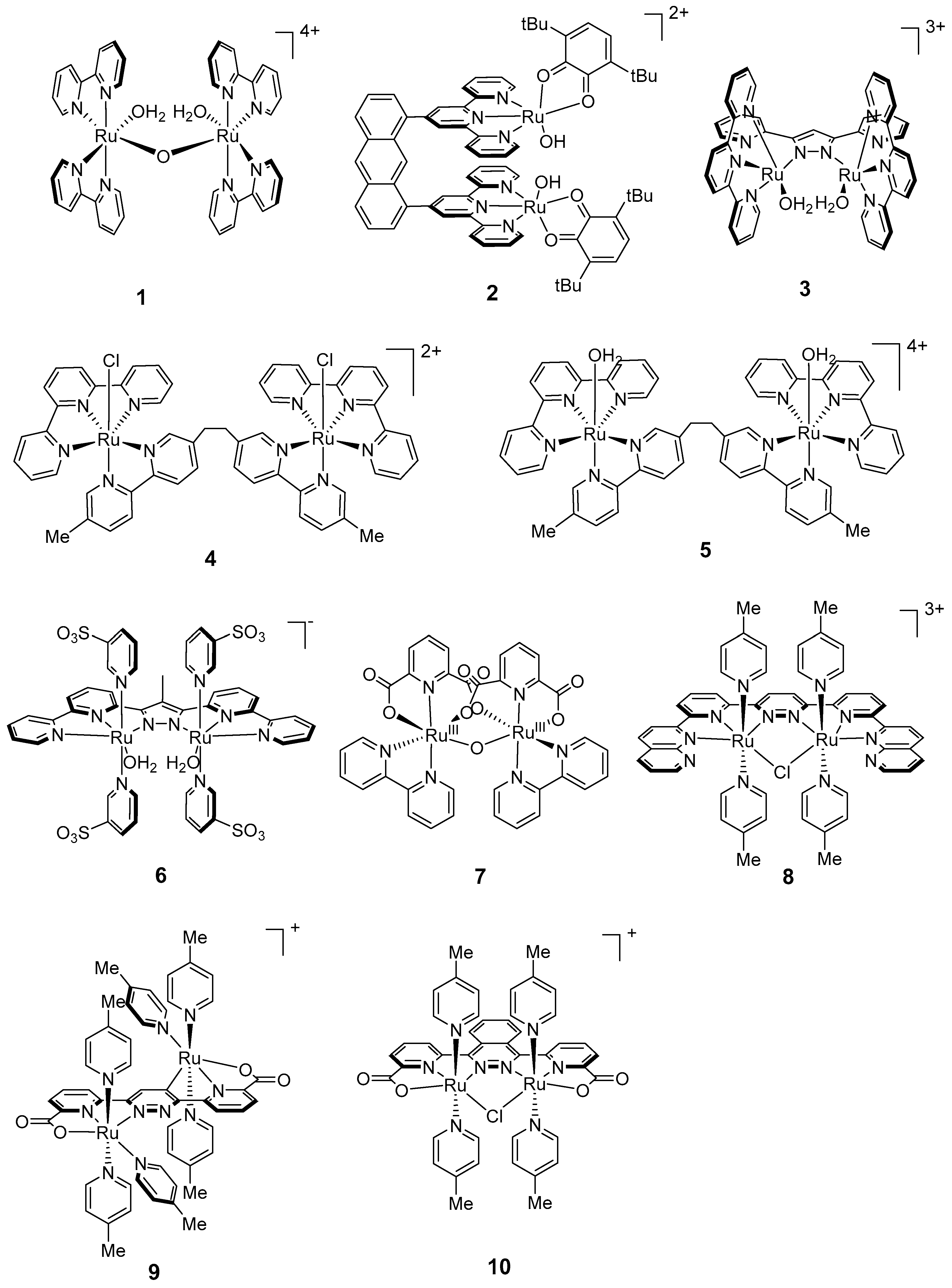
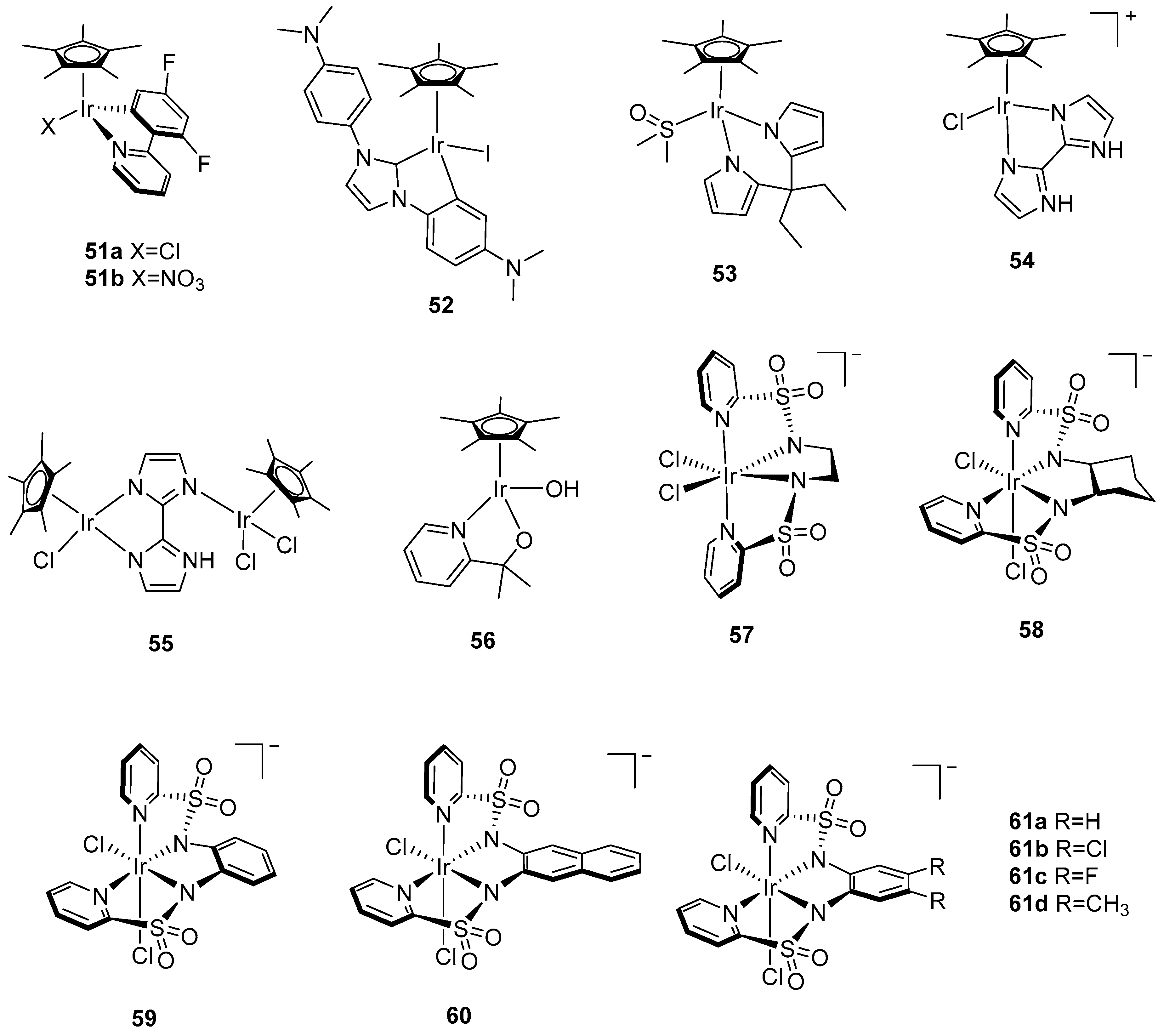
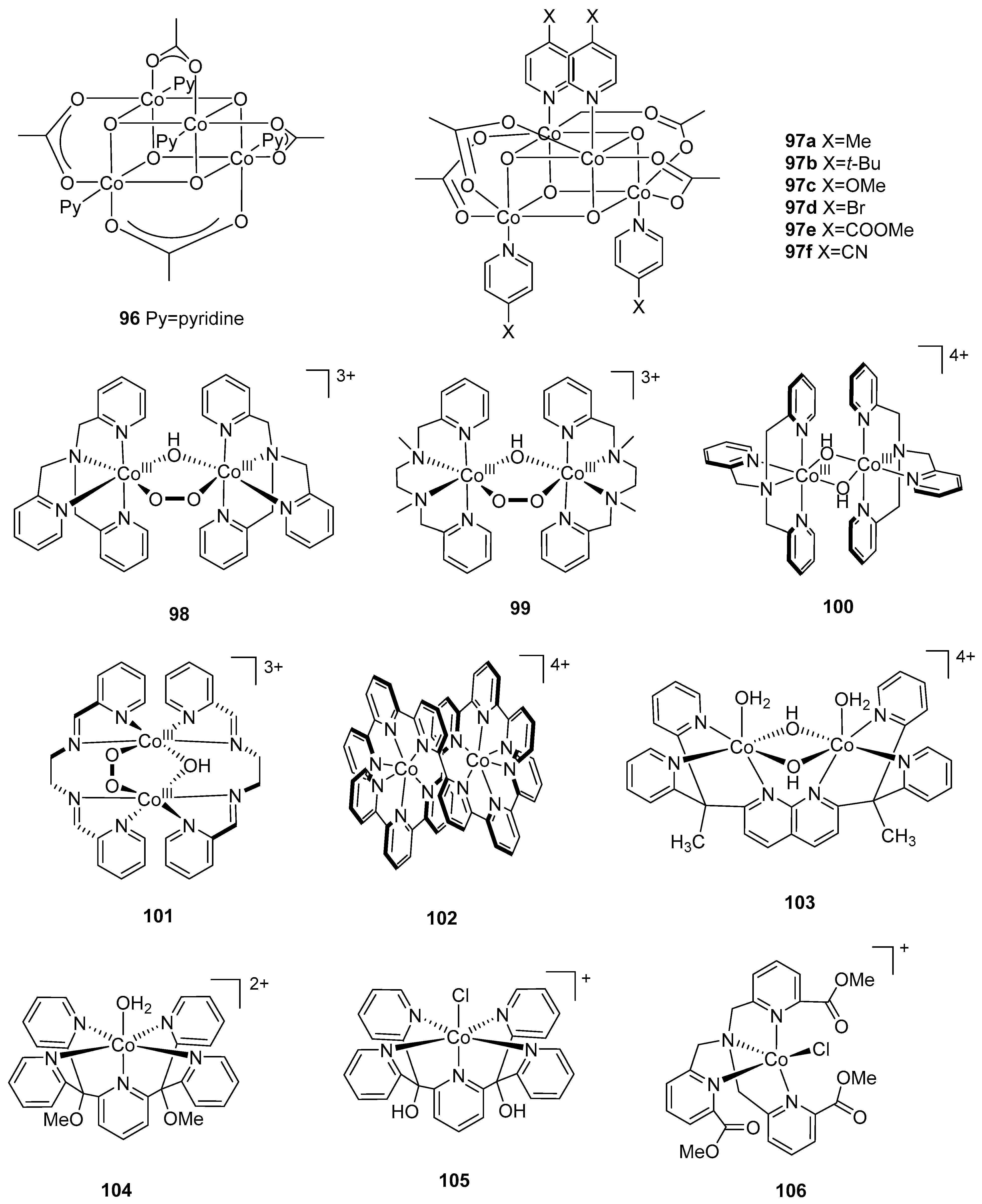
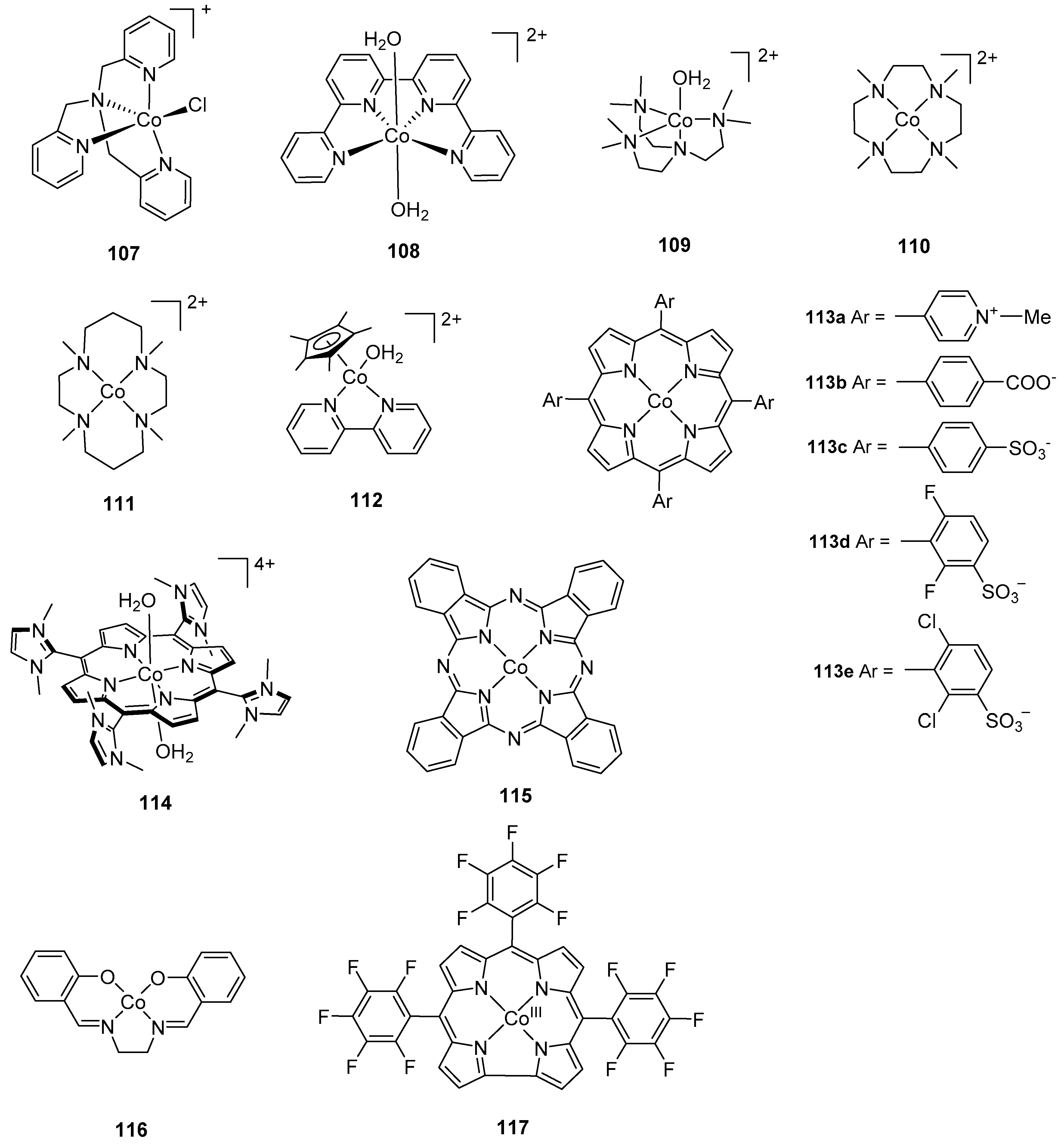
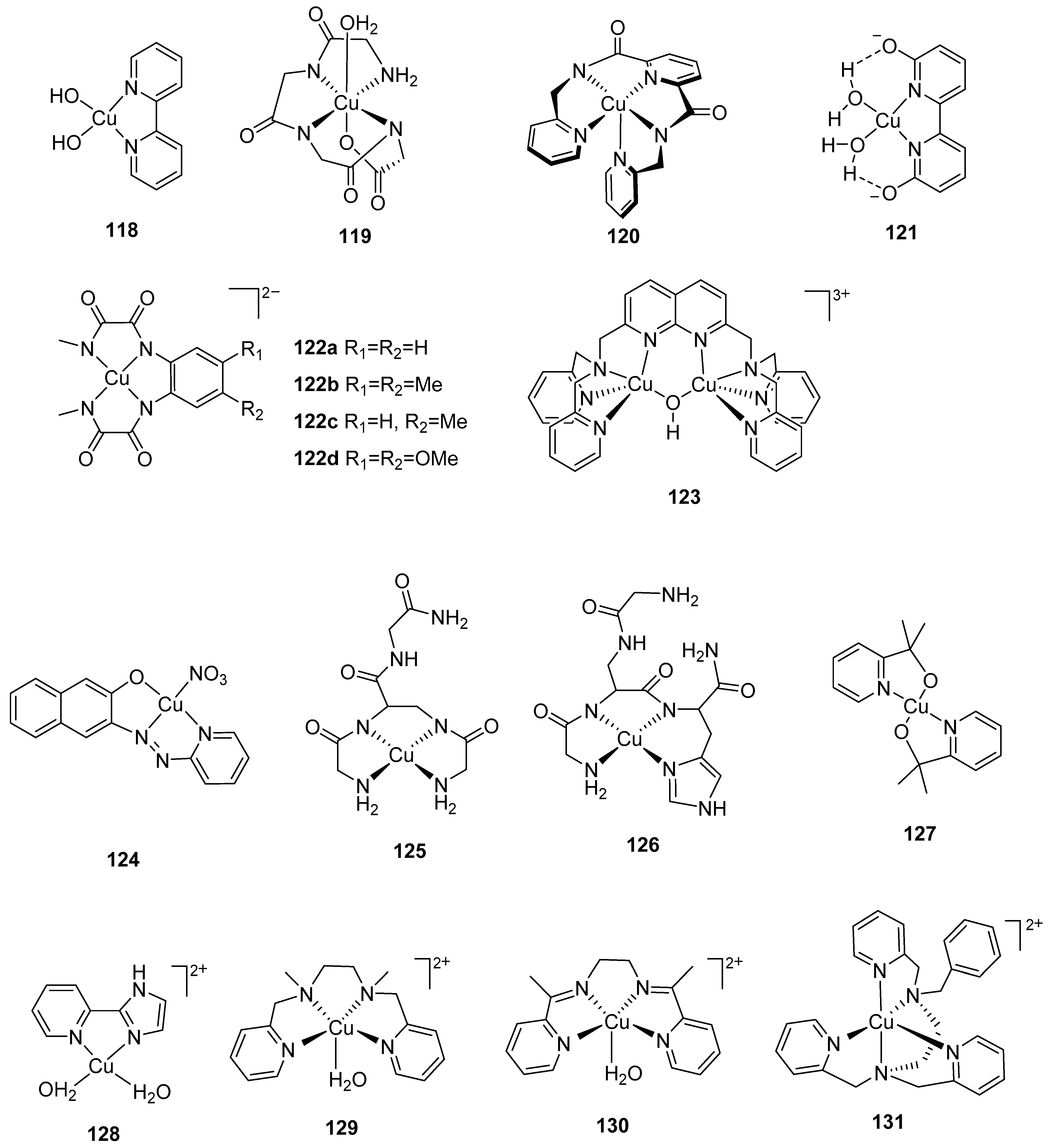
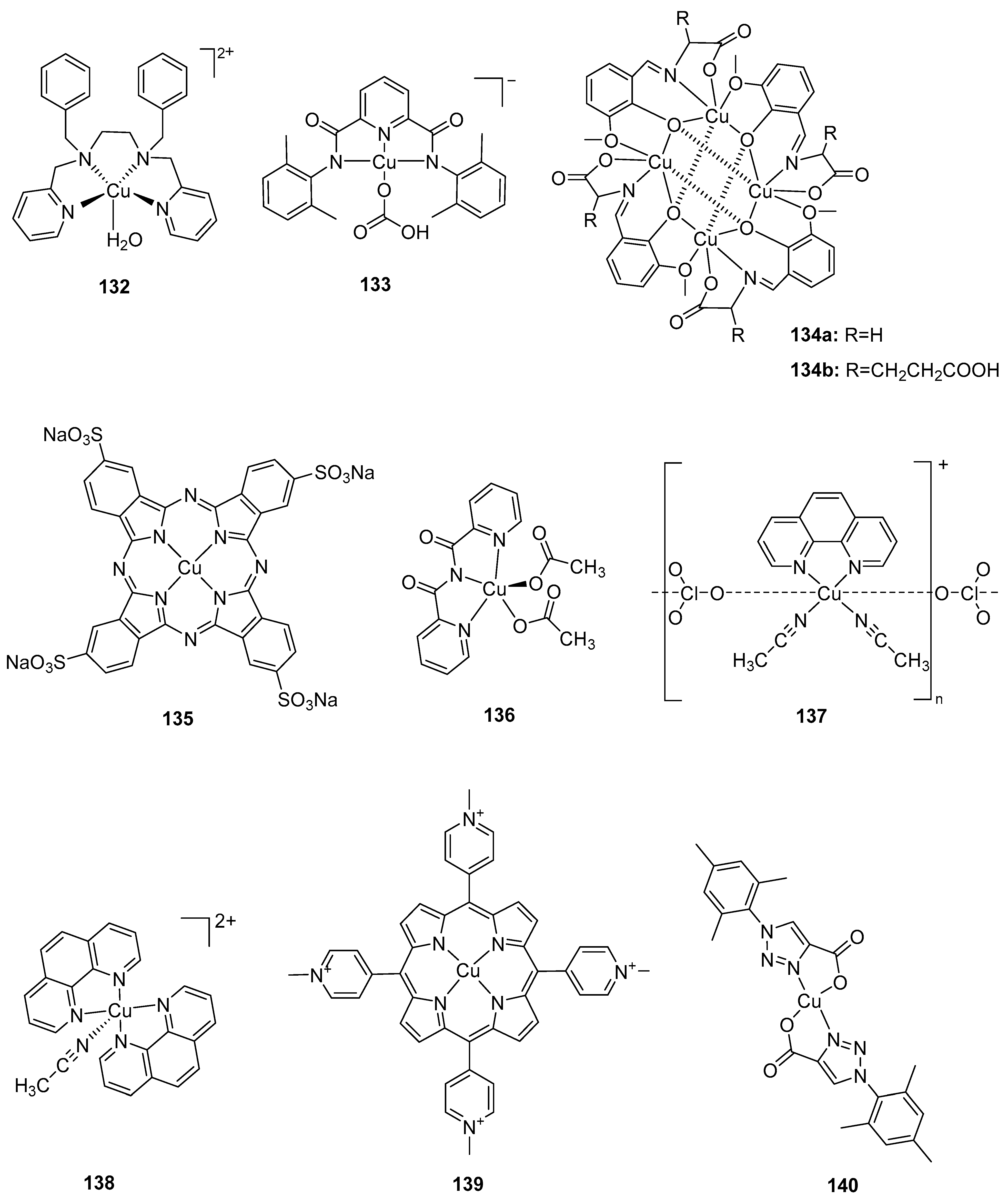
2.1. Dinuclear Ruthenium Catalysts
The ruthenium-based dinuclear molecular catalyst for water oxidation, known as the “blue dimer”, [(H
2O)Ru
III(bipy)
2(μ-O)Ru
III(bipy)
2(H
2O)]
4+ (
1) (
Figure 3), was first reported by Meyer and coworkers in 1982 [
49]. This complex is comprised of a Ru
III dimer, where the metal ions are linked by a μ-oxo bridge. Each Ru
III ion is coordinated by two bipyridine (bipy) ligands that provide an open coordination site to accommodate a water ligand. The blue dimer displayed a turnover number (TON) and turnover frequency (TOF) of 13.2 and 0.0042 s
−1, respectively, for the chemical oxidation of water using [Ce(NO
3)
6][(NH
4)
2] (CAN) as a sacrificial oxidant (
Table 1) [
61,
62]. CAN provides a sufficient oxidation potential to oxidize the investigated catalysts at approximately 1.75 V vs. NHE (pH 0.9) [
60,
63]. The mechanism of water oxidation by complex
1 was investigated by kinetic measurements in combination with isotope replacement studies to determine the kinetic isotope effect, which suggested that the reaction predominantly proceeded through a water nucleophilic attack (WNA) mechanism with the formation of a high-valent intermediate, [(O)Ru
V(μ-O)Ru
V(O)]
4+, where uncoordinated water molecules attack the Ru
V=O group of the intermediate. However, the results were unable to exclude the possibility of intramolecular or bimolecular pathways in the reaction [
64,
65,
66,
67]. The moderately low catalytic performance of complex
1 was attributed to the anation of an active intermediate, in which the [(O)Ru
V(μ-O)Ru
V(O)]
4+ cation coordinated an anion forming the species, [(bipy)
2(H
2O)Ru
IV(μ-O)Ru
III(X)(bipy)
2]
4+ (where, X = ClO
4−, CF
3SO
3− and NO
3−), which resulted in the deactivation of the system [
68].
Studies of the blue dimer were followed by the synthesis and characterization of a series of dinuclear ruthenium complexes with a variety of ligands bridging the ruthenium ions, such as pyrazole [
69,
70], pyridazine [
71,
72], and phthalazine [
73]. Tanaka et al. (2001) reported a dinuclear complex, [Ru
II2(OH)
2(3,6-
tBu
2qui)
2(btpyan)]
2+ (
2) (where 3,6-
tBu
2qui = 3,6-di-tert-butyl-1,2-benzoquinone), that featured a novel ligand, 1,8-bis(2,2′:6′,2′′-terpyridyl)anthracene (btpyan), to bridge the two ruthenium ions [
74]. Complex
2 was capable of electrochemical water oxidation with a TON of 21 in the presence of water in 1,1,1-triflurorethanol and displayed a TON of 33,500 on the surface of an indium tin oxide (ITO) electrode [
74]. Subsequently, Llobet and coworkers (2004) demonstrated that the complex [(H
2O)Ru
II(terpy)
2(μ-bpp)Ru
II(terpy)
2(H
2O)]
3+ (
3) (where, terpy = 2,2′:6′,2′′-terpyridine) with the bridging ligand, 3,5-bis(2-pyridyl)pyrazolate (bpp
−) displayed improved catalytic activity with a TON of 512 and a TOF of 0.014 s
−1 for chemical water oxidation using CAN as a sacrificial oxidant [
69]. The presence of the btpyan and bpp
− bridging ligands in the complexes
2 and
3, respectively, offered alternatives to the previous strategy of μ-oxo-bridged dinuclear ruthenium complexes [
4], which is unique as there are very few dinuclear ruthenium complexes that have been shown to be catalytically active in the absence of bridging μ-oxo ions.
This was followed by the development of two dinuclear ruthenium complexes [{Ru
II(terpy)Cl}
2(μ-L)]
2+ (
4) and [{Ru
II(terpy)(H
2O)}
2(μ-L)]
4+ (
5) (where, L = bis[5-(5′-methyl-2,2′-bipyridinyl)]ethane) by Sakai and coworkers (2009) [
75] that displayed activity for chemical water oxidation with TONs of 75 and 106, respectively, using CAN as an oxidant. It was found that the activities of complexes
4 and
5 were much higher than that of the μ-O bridged complex
1. More importantly, an induction time of 2–3 h was observed for complex
4. However, the initial rate of O
2 formation for complex
5, the aqua species, did not show an induction period for oxygen formation, implying that the aqua (instead of the chloro) species was active during the catalysis of the water oxidation reaction [
75]. Subsequently, Llobet and coworkers (2014) developed a powerful and oxidatively rugged complex, [{Ru
II(py-SO
3)
2(H
2O)}
2(μ-Mebbp)]
− (
6) (where, HMebbp = 2,4-bis(bipyridin)-3-methyl-pyrazole and py-SO
3 = pyridine-3-sulfonate), with a pyrazolate-based equatorial ligand that featured a TON of 22.6 and a TOF of 0.068 s
−1 for chemical water oxidation using CAN as an oxidant [
70]. In complex
6, Mebbp
− is a bis(tridentate) monoanionic ligand that was designed to act as a bridging scaffold that placed the two ruthenium ions in close proximity. The Mebbp
− ligand appeared to induce subtle geometric variations on the relative disposition of the active Ru–OH
X groups that regulated the O–O bond formation pathway and influenced the mechanism toward WNA. This was in contrast to the intermolecular bimolecular (I2M) mechanism that was suggested for analogous complexes with 3,5-bis(2-pyridyl)pyrazolato dinucleating, Hbpp
−, ligand due to the strategic disposition and encumbrance of the terpy ligands [
57,
69,
76,
77]. This study demonstrated that subtle variations in ligand design could be used to regulate the O−O bond formation pathway of the water oxidation reaction. Additionally, the tridentate dianionic meridional pyridyl-2,6-dicarboxylato (pdc
2−) ligand was used to generate a dinuclear ruthenium complex
7. Although complex
7 was not a catalyst for water oxidation, it was shown to act as a precursor for a ruthenium-aqua mononuclear complex, [Ru
II(pdc-κ
3-N
1O
2)(bipy)(H
2O)] (
31) (where, pdc = pyridyl-2,6-dicarboxylato and bipy = 2,2′-bipyridine), that was an active water oxidation catalyst with low overpotential of 240 mV at pH 1 and a TOF of 0.2 s
−1 [
78].
Further improvements in the design of dinuclear ruthenium catalysts included the incorporation of a rigid polypyridyl equatorial ligand in [Ru
II2(μ-L)(μ-Cl)(pic)
4]
3+ (
8) (where L = 6-di-(6′-[1″,8″-naphthyrid-2″-yl]-pyridin-2′-yl)pyrazine and pic = 4-picoline), which improved the catalytic performance for chemical water oxidation using CAN as a sacrificial oxidant at pH 1 with a TON and TOF of 538 [
50,
79] and 0.046 s
−1, respectively [
50,
71]. During this time, biophysical studies of PSII indicated that the presence of negatively-charged carboxylate ligands in the vicinity of the OEC likely improve the stability of the high-valent manganese intermediates by lowering the oxidation potential of the catalytic Mn
4Ca-oxo cluster [
45,
50,
72,
80]. This led to the incorporation of carboxylate ligands in the design of dinuclear ruthenium complexes [
72,
73]. While the dinuclear ruthenium complexes containing neutral ligands displayed high oxidation potentials, which required the use of strong chemical oxidants, such as Ce
IV for catalytic water oxidation, it was thought that the redox potentials of these complexes could be decreased by ligand modification. The presence of negatively-charged ligands could lower the oxidation potential of the complexes and stabilize the higher oxidation states of the metal ions [
72]. In principle, this could present the possibility of driving the water oxidation reaction by a mild oxidant. Using this strategy, Sun and coworkers prepared a dinuclear ruthenium catalyst with a negatively charged dicarboxylate ligand. The complex [Ru(pic)
3(μ-cppd)Ru(pic)
3]
+ (
9) (where, H
2cppd = 3,6-bis-(6′-carboxypyrid-2′-yl)-pyridazine) yielded a TON and TOF of 4700 and 0.28 s
−1, respectively, for chemical water oxidation using CAN as an oxidant [
50,
73]. Moreover, the complex [Ru(pic)
2(μ-Cl)(μ-cpptz)Ru(pic)
2]
+ (
10) (where, H
2cpptz = 1,4-bis(6′-COOH-pyrid-2′-yl)phthalazine) displayed improved catalytic activity under identical conditions with a TON of 10,400 and a TOF of 1.2 s
−1 [
73]. Both complex
9 and
10 provided direct evidence of the benefit of introducing carboxylate functionalities in the equatorial ligand framework of dinuclear ruthenium catalysts. In 2021, Meyerstein and coworkers reported a dinuclear ruthenium carbonate complex, Na
3[Ru
2(µ-CO
3)
4], that is electrochemically active for water oxidation with a TOF of 0.10 s
−1 under pH-neutral conditions and 1.48 s
−1 in bicarbonate media (pH 8.3) [
81].
Figure 3.
Chemical structures of recent dinuclear ruthenium catalysts
1–10 for water oxidation: [(H
2O)Ru
III(bipy)
2(μ-O)Ru
III(bipy)
2(H
2O)]
4+ (
1, bipy = 2,2′-bipyridine) [
49]; [Ru
II2(OH)
2(3,6-
tBu
2qui)
2(btpyan)]
2+ (
2, btpyan = 1,8-bis(2,2′:6′,2″-terpyridyl)anthracene, 3,6-
tBu
2qui = 3,6-di-tert-butyl-1,2-benzoquinone) [
74]; [(H
2O)Ru
II(terpy)
2(μ-bpp)Ru
II(terpy)
2(H
2O)]
3+ (
3, bpp = 3,5-bis(2-pyridyl)pyrazolate, terpy = 2,2′:6′,2″-terpyridine) [
69]; [{Ru
II(terpy)Cl}
2(μ-L)]
2+ (
4, L = bis[5-(5′-methyl-2,2′-bipyridinyl)]ethane) [
75]; [{Ru
II(terpy)(H
2O)}
2(μ-L)]
4+ (
5, L = bis[5-(5′-methyl-2,2′-bipyridinyl)]ethane) [
75]; [{Ru
II(py-SO
3)
2(H
2O)}
2(μ-Mebbp)]
− (
6, Mebbp = 2,4-bis(bipyridin)-3-methyl-pyrazole) [
70]; [{Ru
III(pdc-
κ3-N
1O
2)(bipy)}
2(μ-O)] (
7, pdc = 2,6-pyridinedicarboxylato, bipy= 2,2′-bipyridine) [
78]; [Ru
II2(μ-L)(μ-Cl)(pic)
4]
3+ (
8, L = 6-di-(6′-[1″,8″-naphthyrid-2″-yl]-pyridin-2′-yl)pyrazine, pic = 4-picoline) [
71]; [Ru(pic)
3(μ-cppd)Ru(pic)
3]
+ (
9, ccpd = 3,6-bis-(6′-carboxypyrid-2′-yl)-pyridazine) [
72]; [Ru(pic)
2(μ-Cl)(μ-cpptz)Ru(pic)
2]
+ (
10, cpptz = 1,4-bis-(6′-carboxypyrid-2′-yl)-phthalazine) [
73].
Figure 3.
Chemical structures of recent dinuclear ruthenium catalysts
1–10 for water oxidation: [(H
2O)Ru
III(bipy)
2(μ-O)Ru
III(bipy)
2(H
2O)]
4+ (
1, bipy = 2,2′-bipyridine) [
49]; [Ru
II2(OH)
2(3,6-
tBu
2qui)
2(btpyan)]
2+ (
2, btpyan = 1,8-bis(2,2′:6′,2″-terpyridyl)anthracene, 3,6-
tBu
2qui = 3,6-di-tert-butyl-1,2-benzoquinone) [
74]; [(H
2O)Ru
II(terpy)
2(μ-bpp)Ru
II(terpy)
2(H
2O)]
3+ (
3, bpp = 3,5-bis(2-pyridyl)pyrazolate, terpy = 2,2′:6′,2″-terpyridine) [
69]; [{Ru
II(terpy)Cl}
2(μ-L)]
2+ (
4, L = bis[5-(5′-methyl-2,2′-bipyridinyl)]ethane) [
75]; [{Ru
II(terpy)(H
2O)}
2(μ-L)]
4+ (
5, L = bis[5-(5′-methyl-2,2′-bipyridinyl)]ethane) [
75]; [{Ru
II(py-SO
3)
2(H
2O)}
2(μ-Mebbp)]
− (
6, Mebbp = 2,4-bis(bipyridin)-3-methyl-pyrazole) [
70]; [{Ru
III(pdc-
κ3-N
1O
2)(bipy)}
2(μ-O)] (
7, pdc = 2,6-pyridinedicarboxylato, bipy= 2,2′-bipyridine) [
78]; [Ru
II2(μ-L)(μ-Cl)(pic)
4]
3+ (
8, L = 6-di-(6′-[1″,8″-naphthyrid-2″-yl]-pyridin-2′-yl)pyrazine, pic = 4-picoline) [
71]; [Ru(pic)
3(μ-cppd)Ru(pic)
3]
+ (
9, ccpd = 3,6-bis-(6′-carboxypyrid-2′-yl)-pyridazine) [
72]; [Ru(pic)
2(μ-Cl)(μ-cpptz)Ru(pic)
2]
+ (
10, cpptz = 1,4-bis-(6′-carboxypyrid-2′-yl)-phthalazine) [
73].

Table 1.
Select catalytic parameters and experimental conditions for dinuclear ruthenium catalysts 1–10, which are active in water oxidation. Electrochemical and chemical water oxidation using CAN are abbreviated as ‘electrochem WO’ and ‘chem WO’, respectively. The TOF values that are not listed in this table are not available in literature.
Table 1.
Select catalytic parameters and experimental conditions for dinuclear ruthenium catalysts 1–10, which are active in water oxidation. Electrochemical and chemical water oxidation using CAN are abbreviated as ‘electrochem WO’ and ‘chem WO’, respectively. The TOF values that are not listed in this table are not available in literature.
| Binuclear Ru Complex | TON | TOF (s−1) | Experimental Conditions | Reference |
|---|
| 1 | 13.2 | 0.0042 | chem WO | [49,61,62] |
| 2 | 21 | - | electrochem WO | [74] |
| 3 | 512 | 0.014 | chem WO | [69] |
| 4 | 75 | - | chem WO | [75] |
| 5 | 106 | - | chem WO | [75] |
| 6 | 22.6 | 0.068 | chem WO | [70] |
| 7 | inactive | inactive | electrochem WO | [78] |
| 8 | 538 | 0.046 | chem WO | [50,71,79] |
| 9 | 4700 | 0.28 | chem WO | [72,73] |
| 10 | 10,400 | 1.2 | chem WO | [73] |
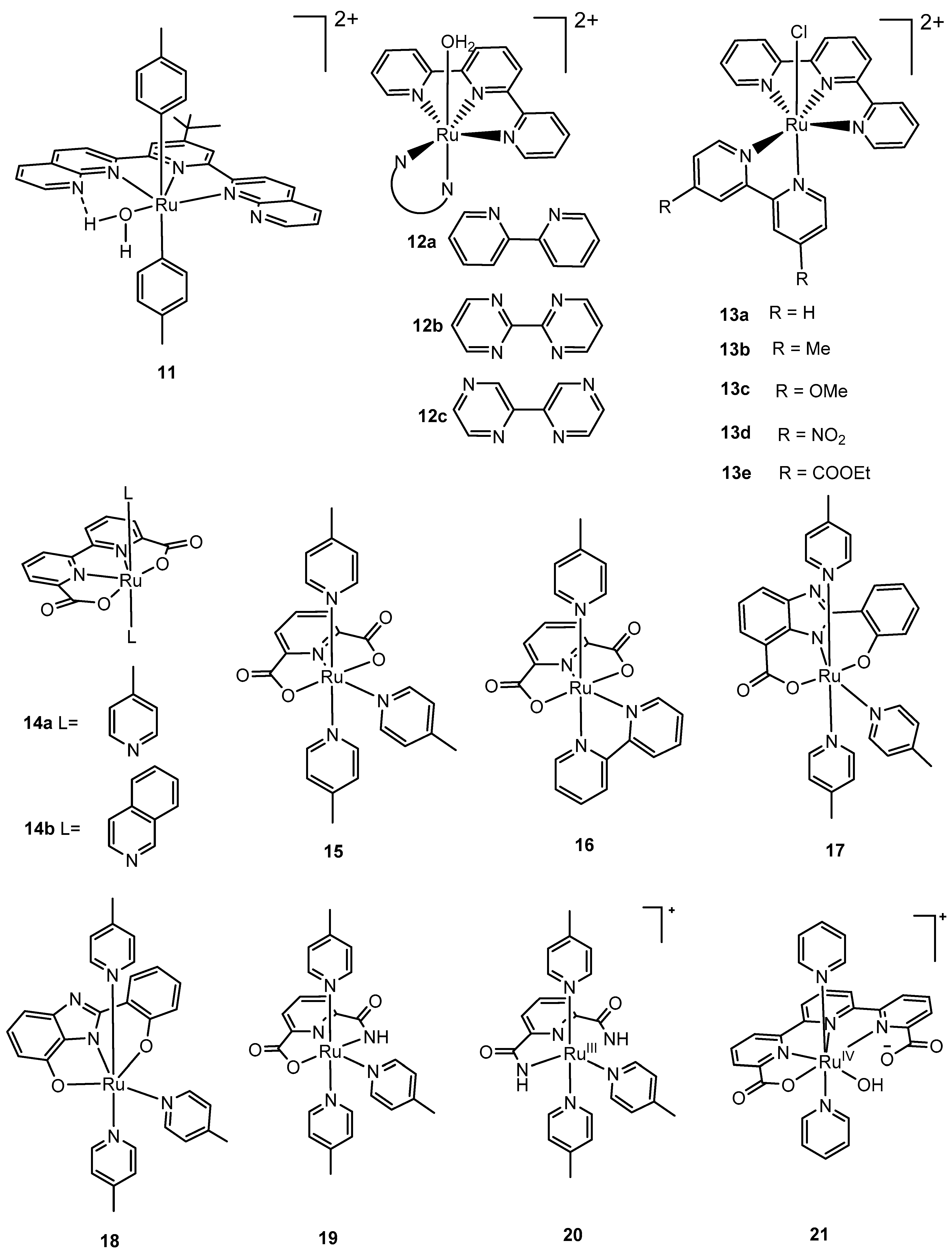
2.2. Mononuclear Ruthenium Catalysts
The success of the blue dimer [
49] in catalyzing water oxidation and the lower catalytic activity of mononuclear ruthenium complexes in early studies [
61] led to the assumption that multinuclear catalysts were required for the successful conversion of water to dioxygen. However, the design of a series of mononuclear ruthenium complexes that were catalytically active for water oxidation challenged this hypothesis [
71]. The ease of the chemical syntheses led to a shift towards the design of effective mononuclear ruthenium catalysts for water oxidation. The complex [(ntp)(pic)
2Ru(H
2O)]
2+ (
11) (where, ntp = 2,6-di(1,8-naphthyridin-2-yl)-4-tert-butylpyridine) (
Figure 4), reported by Thummel and coworkers, displayed catalytic activity for water oxidation (
Table 2) and paved the way for the development of mononuclear ruthenium complexes for water oxidation. Subsequently, a study by Meyer and coworkers on the mononuclear ruthenium complexes [Ru(terpy)(bpm)(OH
2)]
2+ (
12b) and [Ru(terpy)(bpz)(OH
2)]
2+ (
12c) (where, bpm = 2,2′-bipyrimidine; bpz = 2,2′-bipyrazine) demonstrated conclusively that a single Ru site is sufficient for catalytic water oxidation [
82,
83]. A large number of monomeric ruthenium complexes for water oxidation have been reported since the initial findings by Thummel, Meyer and coworkers. The mononuclear ruthenium catalysts that have been reported in the literature can broadly be divided into two classes based on the equatorial and axial ligands that were employed in the respective syntheses.
Berlinguette and coworkers investigated a series of structurally related mononuclear ruthenium catalysts that were formulated as [Ru(terpy)(L)(OH
2)]
2+ (where, L = 2,2′-bipyridine (bipy) (
12a), 4,4′-dimethoxy-2,2′-bipyridine (bipy-OMe), and 4,4′-dicarboxy-2,2′-bipyridine (bipy-COOH)) [
84]. The goal of this study was to determine the effect of the electronic density at the active site on the catalytic performance while holding the balance of the structure at parity. The effects of the systematic modification of the substituent groups on the bipy ligand of the complex indicated that while the presence of electron-withdrawing groups (EWG), such as –Cl and –COOH, suppressed the rate of the reaction, k
obs, and enhanced the catalytic TON, the installation of electron-donating groups (EDG), such as –OMe, accelerated the catalytic rate while decreasing the stability of the catalyst [
85]. The observation of a reverse relationship between the rate of the reaction and the TON was similar to prior observations by Thummel and coworkers [
86]. However, a study by Berlinguette and coworkers suggested that chemical water oxidation driven by Ce
IV as an oxidant led to reaction pathways that diverged from the prevailing “acid-base” mechanism for single-site catalysts. The catalysts displayed complicated pathways that involved the incorporation of O atoms from different sources [
87,
88].
Thummel and coworkers (2008) assessed a series of chloro-coordinated mononuclear ruthenium complexes using terpy, bipy, and related derivatives as ligands that demonstrated high catalytic activity for water oxidation [
86]. These studies demonstrated that the presence of an EDG led to an increase in the rate of the reaction with a decrease in the TON, whereas an EWG yielded a higher TON with a decrease in the rate [
86]. The parent complex [Ru
II(terpy)(bipy)(Cl)]
+ (
13a) in this study was shown to be catalytically active for water oxidation with a TON of 390 using CAN as an oxidant, and it was suggested that the mechanism for complex
13a involved a seven-coordinate intermediate retaining the Ru–Cl bond [
86]. However, in contrast to these observations, Sakai and coworkers demonstrated that complex
13a was inactive in the presence of NaCl in solution. This was thought to be due to a dominant shift of the substitution equilibrium, [Ru
II(terpy)(bipy)Cl]
+ + Solv ⇌ [Ru
II(terpy)(bipy)(Solv)]
2+ + Cl
−, towards the reactant. Moreover, since the oxygen evolution as a function of time suggested that the chloro species was inactive, the real catalyst responsible for dioxygen evolution was inferred to be the aqua species, [Ru
II(terpy)(bipy)(Solv)]
2+, in solution. This suggested that the conversion of the [Ru(terpy)(bipy)Cl]
+ complex (
13a) to the [Ru(terpy)(bipy)(H
2O)]
2+ species (
12a) may have been involved in the mechanism [
75].
Although several studies have proposed a tentative mechanism for water oxidation involving seven-coordinate ruthenium intermediates [
82,
86,
89], it was not possible to isolate and characterize these proposed complexes. This left an open question as to the interaction of water molecules with ruthenium in mononuclear catalysts. The use of negatively charged ligands was thought to be an appropriate means to capture high-valent ruthenium intermediates as they can stabilize higher oxidation states. Given the enhanced catalytic performance of dinuclear ruthenium complexes (
9) and (
10) with a dicarboxylato ligand where the introduction of negatively charged ligands dramatically lowered the oxidation potential of Ru
II to Ru
III [
72,
73], equatorial backbone ligands with terminating carboxylato groups were also introduced in the design of mononuclear ruthenium complexes. The synthesis of a mononuclear ruthenium complex, [Ru(bda)(pic)
2] (
14a) (where, bda
2− = 2,2′-bipyridine-6,6′-dicarboxylate), was shown to stabilize a possible seven-coordinate Ru
IV dimeric intermediate with a proposed [HOHOH]
− bridging ligand [
90]. This supported the hypothesis that the O–O bond formation could arise from the coupling of two Ru
IV=O units, termed as the “interaction between two metal oxo units” or the intermolecular bimolecular (I2M) pathway for water oxidation.
Subsequently, two mononuclear ruthenium complexes, [Ru
II(pdc)(pic)
3] (
15) and [Ru
II(pdc)(bipy)(pic)] (
16) (where, H
2pdc = 2,6-pyridinedicarboxylic acid), were investigated for their catalytic activity in chemical water oxidation [
91]. Complex
15 displayed a TON of 553 and a TOF of 0.23 s
−1, which was better than complex
16, which had a TON of 17 and a TOF of 7.2 × 10
−3 s
−1 at pH 1 for chemical water oxidation in the presence of Ce
IV ions [
91]. Although both
15 and
16 employed tridentate equatorial backbone ligands containing negatively charged biscarboxylato groups, they were not as catalytically active as [Ru(bda)(pic)
2] (
14a) [
53], which displayed a TON of 2000 and a TOF of 41 s
−1 under similar reaction conditions. Upon closer examination, the tetradentate equatorial backbone with two axial picoline ligands in complex
14a formed a highly distorted octahedral configuration with an “open coordination site” (O–Ru–O angle of 123°) that greatly facilitated the access of an aqua ligand [
50,
90]. The isolation of a Ru
IV dimeric intermediate with a [HOHOH]
− bridging ligand from water oxidation catalyzed by complex
14a suggested that radical coupling of Ru=O units led to O–O bond formation [
90]. As a result of this finding, isoquinolines were employed as axial ligands to facilitate the non-covalent attraction between them and lower the barrier of interaction for the Ru=O units. This strategy succeeded as the complex [Ru(bda)(isq)
2] (
14b) (where, isq = isoquinoline) and led to a TON of 8369 and a TOF of 303 s
−1 [
53].
The studies involving mononuclear ruthenium catalysts described thus far required a powerful sacrificial oxidant, CAN, for chemical water oxidation. In principle, it should be possible to use a light-absorbing photosensitizer to conduct sustainable light-driven water oxidation. As described by Åkermark and coworkers [
92], a major obstacle that is frequently encountered in light-driven water oxidation is the mismatch between the relatively high redox potential at which a catalyst assumes its active state and the lower potential attainable with a photosensitizer. One way to decrease the redox potential of the active catalyst is to involve PCET, which is a fundamental process that is employed in nature by the OEC of PSII [
25,
27]. It involves the simultaneous transfer of an electron and a proton, which has a profound effect on the energetics of the water oxidation reaction. As mentioned in the Introduction section, PCET allows for redox leveling at the catalytic site, which is a prerequisite for carrying out the four-electron water oxidation reaction. Additionally, another means of altering the redox potential of the active catalyst is to coordinate electron-donating and redox-active ligands to the metal centers, which would influence the balance between efficiency and stability of the water oxidation catalysts [
92]. Thus, Åkermark and coworkers demonstrated that the introduction of imidazole and phenol motifs, in combination with carboxylate groups, facilitated PCET and the formation of high-valent metal–oxo catalytic intermediates at low potentials. This strategy was implemented by the development of two mononuclear ruthenium complexes, [Ru
III(L)(pic)
3] (where, L = 2-(2-hydroxyphenyl)-1H-benzimidazole-7-carboxylate (
17) and L = 2-(2-hydroxyphenyl)-1H-benzimidazol-7-ol (
18)), which contained negative equatorial backbone ligands comprised of imidazole and phenol motifs with a carboxylate group [
92]. By using the imidazole motif, it was possible to introduce a combined redox and proton-transfer mediator, a highly active and essential element, into the mononuclear ruthenium catalysts. Complex
17, with a single carboxylate and phenol moiety, displayed a TON of up to 4000 and a TOF of 7.4 s
−1 with [Ru(bipy)
3]
3+ as an oxidant for chemical water oxidation, and a postulated [Ru
V=O]
n+ intermediate of
18 was characterized by high-resolution mass spectrometry [
92]. Moreover, to evaluate the possibility of performing light-driven water oxidation under homogeneous, neutral conditions at pH 7.2, the authors employed a three-component system consisting of complex
17 or
18, a photosensitizer ([Ru(bipy)
3]
2+ or [Ru(bipy)
2(deeb)]
2+ (where, deeb = 4,4′-di(ethoxycarbonyl)-2,2′-bipyridine)), and a sacrificial electron acceptor (Na
2S
2O
8). Successful evolution of dioxygen was detected upon visible-light illumination of this system. The [Ru(bipy)
3]
2+ photosensitizer displayed a low TON of approx. 20, whereas, replacing [Ru(bipy)
3]
2+ (
E [Ru
III/Ru
II] = 1.26 V vs. NHE) with the more strongly oxidizing photosensitizer [Ru(bipy)
2(deeb)]
2+ (
E [Ru
III/Ru
II] = 1.4 V vs. NHE) yielded a significantly higher TON of ~200 [
92]. Similarly, [Ru(bda)(pic)
2] (
14a), [Ru
II(pdc)(pic)
3] (
15) and [Ru
II(pdc)(bipy)(pic)] (
16) also demonstrated moderate catalytic performance for photochemical water oxidation using [Ru(bipy)
3]
2+ or [Ru(bipy)
2(dcb)]
2+ (dcb = 4,4′-dicarboxyethyl-2,2′-bipyridine) as a photosensitizer and [Co(NH
3)
5Cl]Cl
2, or Na
2S
2O
8 as a sacrificial electron acceptor [
91,
93].
The design of a mononuclear ruthenium complex with a carboxylate-amide motif, [Ru
II(HL)(pic)
3] (
19) (where, L = 6-carbamoylpicolinic acid), was also shown to catalyze water oxidation (TON of 280 and TOF of 1.16 s
−1) at a neutral pH of 7.2 using [Ru(bipy)
3]
3+ as a mild chemical oxidant [
94]. This complex was similar to [Ru
II(pdc)(pic)
3] (
15), with the difference that one of the carboxylate ligands was replaced by an amide group. The crystal structure of complex
19 revealed a Ru
III ion due to the strong electron-donating ability of the 6-carbamoylpicolinic acid ligand. The presence of the carboxylate-amide ligand in
19 lowered the redox potential of the complex to an extent where catalytic water oxidation could take place under neutral conditions with the mild [Ru(bipy)
3]
3+ oxidant [
94]. In comparison with the mononuclear ruthenium complex,
19, catalysts based on neutral nitrogen containing heterocyclic ligands were generally not compatible with the mild oxidant, [Ru(bipy)
3]
3+. This study once again highlighted the importance of incorporating anionic backbone ligands to decrease the redox potential of ruthenium catalysts.
The above results led to the design of the complex [Ru
III(H
2pdca)(pic)
3]
+ (
20) (where, H
4pdca = 2,6-pyridine-dicarboxamide), which was also shown to catalyze water oxidation at a low redox potential using [Ru(bipy)
3]
3+ at pH 7.2 with a TON of 400 and a TOF of 1.6 s
−1 [
95]. The improvement of the catalytic activity in terms of TOF was attributed to the presence of a flexible equatorial backbone ligand. This was followed by the synthesis of a seven-coordinate mononuclear ruthenium complex, [Ru
IV(OH)(tda-κ-N
3O)(py)
2]
+ (
21) (where, tda
2− = 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate) that was found to be an active and robust catalyst with a maximum TOF (TOF
max) of 50,000 s
−1 at pH 10 using a foot-of-wave analysis (FOWA) [
54]. Based on density functional theory (DFT) calculations, it was proposed that the carboxylate moiety in the dianionic ligand, tda
2−, stabilized seven-coordinate intermediates in the high-valent oxidation state of the catalyst. Moreover, the dangling carboxylate group was a putative hydrogen-bonding site that could function as a proton acceptor and hence favor WNA. This could lower the free energy of the activation and lead to O–O bond formation [
54]. To our best knowledge, the catalytic activity of complex
21 is the highest that has been reported in literature, albeit it uses FOWA.
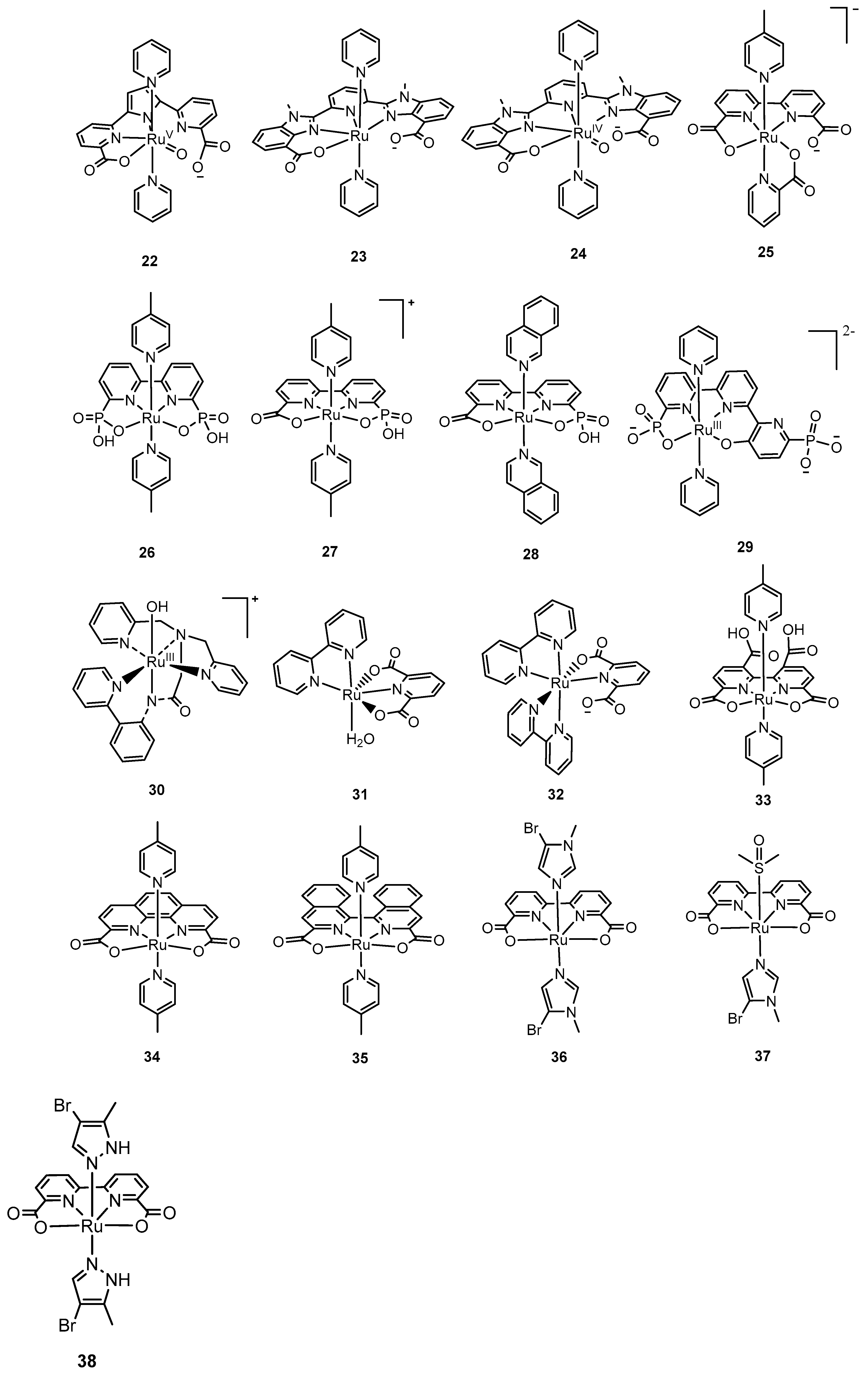
The trianionic mononuclear ruthenium complex, [Ru
V(O)(t5a-κ-N
2O)(py)
2] (
22) (where, t5a
3− = 2,5-bis(6-carboxylatopyridin-2-yl)pyrrol-1-ide and py = pyridine), was demonstrated to be an efficient catalyst with a TOF
max of 9400 s
−1 at pH 7 via the FOWA [
96]. In this case, the highly anionic nature of the backbone could reduce the redox potential of the Ru
IV/Ru
V couple, and the flexibility of the carboxylate moiety could facilitate intramolecular proton transfer to facilitate O–O bond formation through WNA. However, unlike complex
21, which required the formation of a seven-coordinate Ru
V=O intermediate during water oxidation, complex
22 was not thought to require a seven-coordinate intermediate for O–O formation. This was due to the geometrical distortion of 12° and increased anionic nature of
22 in comparison with
21 (
Table 3) [
51,
96]. The ligands tda
2− and t5a
3− were shown to be flexible, adaptive, multidentate, and equatorial and were thus termed as “FAME ligands” [
51]. We would like to refer the readers to a recent review article that is focused on ruthenium-based molecular catalysts with the ability to achieve seven-coordinate intermediates and unprecedented activity [
51].
Based on the above considerations, Llobet and coworkers developed the complex, [Ru
II(mcbp)(py)
2] (
23) (where, mcbp
2− = 2,6-bis(1-methyl-4-(carboxylate)-benzimidazol-2-yl)pyridine), which also contained flexible anionic carboxylate ligands [
97]. The active species, [Ru
IV(O)(mcbp)(py)
2] (
24), was generated by the controlled potential electrolysis (CPE) of complex
23 and displayed improved activity for water oxidation with TOF
max of 40,000 s
−1 at pH 9 [
97]. Additionally, the complex [Ru(bda)(pic)(pyC)] (
25) (where, pyC = 2-pyridinecarboxylate), with a similar backbone as the bda family of ligands (e.g., complex
14) but with carboxylate groups on the axial pyridine rather than equatorial backbone ligands, has also been reported in the literature. Complex
25 contained a dangling carboxylate ligand similar to complex
21, which was suggested to facilitate a WNA pathway [
98]. However, the catalytic performance of complex
25 was low with a TOF
max of 0.63–0.74 s
−1 measured via FOWA at pH 7, which was ascribed to its geometric features [
98]. Unlike complex
14 and the parent complex
21, [Ru
II(tda-κ-N
3O)(py)
2], which were shown to have a distorted octahedral geometry with a large O–Ru–O angle of 123° (or an O–Ru–N angle of 125°), complex
25 displayed a near perfect octahedral geometry with an O–Ru–O angle of 93.72° [
51,
90,
98].
Subsequently, Concepcion and coworkers incorporated phosphate ligands in a bipyridine backbone to generate complexes such as [Ru
II(bpaH
2)(pic)
2]
+ (
26) (where, bpaH
4 = 2,2′-bipyridine-6,6′-diphosphonic acid), [Ru
III(bpHc)(pic)
2]
+ (
27), and [Ru
II(bpHc)(isq)
2] (
28) (where, bpH
2cH = 2,2′-bipyridine-6-phosphonic acid- 6′-carboxylic acid) [
99,
100]. Complex
28, with a carboxylate-phosphonate moiety, exhibited the highest activity among these complexes, with a TOF of 107 s
−1 under acidic conditions and using CAN as an oxidant. However, the incorporation of a diphosphonate ligand in complex
26 drastically decreased the activity to 0.65 s
−1 [
100]. The complex, [Ru
III(tPaO-κ-N
2O
PO
C)(py)
2]
2− (
29) (where, tPaO
5− = 3-(hydroxo-[2,2′:6′,2″-terpyridine]-6,6″-diyl)bis(phosphonate)), was derived from a seven-coordinate H
4tPa-based ruthenium complex, [Ru
IV(H
2tPa-κ-N
3O
2)(py)
2]
2+ (where, H
4tPa = 2,2′:6′,2′′-terpyridine-6,6′′-diphosphonic acid), under neutral and basic conditions, where an exogenous OH
− ion from the solvent was coordinated to the complex [Ru
IV(H
2tPa-κ-N
3O
2)(py)
2]
2+. This led to the formation of the six-coordinate complex [Ru
IV(OH)(tPa-κ-N
2O)(py)
2]
− or [Ru
IV(O)(HtPa-κ-N
2O)(py)
2]
−. In this case, it was proposed that the Ru
V=O intermediate undergoes intramolecular oxygen atom insertion into the CH bond of a non-coordinated pyridyl ring to generate the catalytically active complex
29 with a TOF
max of 16,000 s
−1, measured via FOWA at pH 7.2 [
101]. Most recently, there was an interesting complex, [(L
N5−)Ru
III–OH]
+ (
30), with a redox-active electron-rich polypyridyl ligand that was reported for electrochemical catalytic water oxidation at neutral pH [
102]. Complex
30 was generated from [(L
N5−)Ru
III–Cl] by an oxidative-induced ligand exchange at neutral pH, and this species was electrochemically oxidized to form the active intermediate [(L
N5−)
+•Ru
IV=O]
2+, with a surprisingly low overpotential of 183 mV for O–O formation through a WNA pathway [
102]. In this case, ligand oxidation was proposed to lower overpotential (1.0 V vs. NHE), which was supported by DFT calculations [
102].
There is a family of mononuclear ruthenium complexes, [Ru
II(pdc-κ
3-N
1O
2)(bipy)(H
2O)] (
31) and [Ru
II(pdc-κ
2-N
1O
1)(bipy)
2] (
32), containing the tridentate dianionic meridional pyridyl-2,6-dicarboxylato (pdc
2−) ligand that have been studied for their electrochemical activity towards water oxidation [
78,
103]. Complex
31 has been shown to electrochemically catalyze water oxidation with a low overpotential of 240 mV under acidic conditions (pH of 1), due to the presence of two carboxylate groups on the pdc
2− ligand. The complex
32 was shown to generate a Ru
IV intermediate, [Ru
IV(O)(pdc-κ
2-N
1O
1)(bipy)
2], upon the addition of Ce
IV ions in solution and a WNA mechanism was proposed for O–O bond formation [
103]. Complex
32 was studied electrochemically with a TOF of 3400 s
−1, and the high-valent Ru
IV=O involved in the catalytic cycle had a seven-coordinate intermediate with a dangling carboxylate group, which could facilitate O–O bond formation by intramolecular proton transfer and thus decrease the activation energy [
103]. In 2021, Ahlquist et al. reported a mononuclear catalyst, [Ru(bnda)(pic)
2] (
33) (where, H
2bnda = 2,2′-bi(nicotinic acid)-6,6′-dicarboxylic acid), to investigate the effect of steric hindrance and hydrophilicity of the bda backbone [
104]. The comparison of the parent backbone of complex
14 and [Ru(pda)(pic)
2] (
34) (where, pda
2− = 1,10-phenanthroline-2,9-dicarboxylate, pic = 4-picoline) and [Ru(biqa)(pic)
2] (
35) (where, biqa
2− = (1,1′-biisoquinoline)-3,3′-dicarboxylate) indicated a switching of the mechanism of O–O bond formation between the WNA and I2M pathway [
104]. Based on experimental studies, catalyst
33 undergoes I2M, whereas complexes
34 and
35 follow the WNA pathway, although DFT calculations of complexes
33–
35 have indicated that I2M is a more favorable pathway. This difference may be due to failure to consider solvation effects and the collision of Ru
V=O species in the DFT calculations [
104].
The modification of the axial ligands to enhance the catalytic performance of mononuclear ruthenium complexes was explored by Sun and coworkers. They designed the complexes, [Ru(bda)(Im)
2] (
36) (where, Im = imidazole) and [Ru(bda)(Im)(DMSO)] (
37) (where, DMSO = dimethylsulfoxide) [
105,
106], which contained both imidazole and DMSO as axial ligands. Complex
36, with two axial imidazole ligands, yielded a TON of 1150 and a TOF of 4.5 s
−1 for chemical water oxidation [
105]. In contrast, the complex [Ru(bda)(Im)(DMSO)] (
37), which contained an imidazole and DMSO axial ligand, exhibited better stability and improved catalytic activity with a TON of 4050 and a TOF of up to 176.5 s
−1 [
105]. Detailed mechanistic investigations of the catalytic water oxidation reaction using kinetics, electrochemistry, high-resolution mass spectrometry, and density functional theory (DFT) calculations suggested the in situ formation of a Ru
II complex with an accessible seventh coordination site. The measured catalytic activity and kinetics revealed the influence of the axial ligands on the catalytic activity, where the increase of catalytic activity for complex
37 with an axial imidazole and DMSO ligands was attributed to the unhindered coupling between terminal oxygen atoms [
105]. The catalytic activity of mononuclear ruthenium complexes was shown to be further enhanced with a TON of 6200 and TOF of 506 s
−1 by employing two bromo substituted pyrazole-based axial ligands, [Ru
II(bda)(L)
2] (
38) (where bda
2− = 2,2′-bypyridine-6,6′-dicarboxylate and L = 4-Br-3-methyl pyrazole). The enhanced catalytic activity of
38 was ascribed to the high hydrophobicity of the complex, which tended to favor dimerization and, hence, facilitate the I2M reaction pathway [
48,
106]. Complexes
36–
38 presented the possibility of simultaneously observing the effects of the axial and equatorial ligand modifications. However, the modification of the equatorial backbone ligand, bda
2−, used in these catalysts has not been fully explored to date. This is most likely due to the challenges that are involved in the synthesis of substituted bda
2− backbone ligands [
50]. This is an avenue that could lead to further improvements of the catalytic performance as the introduction of substituents on the bda
2− backbone has been shown to influence the mechanistic pathways of mononuclear catalysts [
50].
Table 2.
Selected catalytic parameters and experimental conditions for Ru catalysts 11–38 in water oxidation. Electrochemical and chemical water oxidation using [Ce(NO3)6][(NH4)2] (CAN) are abbreviated as ‘electrochem WO’ and ‘chem WO’, respectively. The TON or TOF values that are not listed in this table are unavailable in literature *.
Table 2.
Selected catalytic parameters and experimental conditions for Ru catalysts 11–38 in water oxidation. Electrochemical and chemical water oxidation using [Ce(NO3)6][(NH4)2] (CAN) are abbreviated as ‘electrochem WO’ and ‘chem WO’, respectively. The TON or TOF values that are not listed in this table are unavailable in literature *.
| Ru Complex | TON | TOF (s−1) | Experimental Conditions | Reference |
|---|
| 11 | 260 | 0.014 | chem WO | [50,71,86] |
| 12a | 320 | 0.0296 | chem WO | [85] |
| 13a | 390 | - | chem WO | [86] |
| 13b | 190 | - | chem WO | [86] |
| 13c | 110 | - | chem WO | [86] |
| 13d | 260 | - | chem WO | [86] |
| 13e | 570 | - | chem WO | [86] |
| 14a | 2000 | 41 | chem WO | [53] |
| 14b | 8360 | 303 | chem WO | [53] |
| 15 | 553 | 0.23 | chem WO | [91] |
| 16 | 17 | 7.2 × 10−3 | chem WO | [91] |
| 17 | 4000 | 7.4 | chem WO (w/[Ru(bipy)3]3+) | [92] |
| 18 | 180 | 0.3 | chem WO (w/[Ru(bipy)3]3+) | [92] |
| 19 | 280 | 1.16 | chem WO (w/[Ru(bipy)3]3+) | [94] |
| 20 | 400 | 1.6 | chem WO (w/[Ru(bipy)3]3+) | [95] |
| 23 | n.a | - | inactive | [97] |
| 26 | 5.0 | 0.65 | chem WO | [100] |
| 27 | 3.8 | 58 | chem WO | [100] |
| 28 | 3.8 | 107 | chem WO | [100] |
| 30 | 21 | - | electrochem WO | [102] |
| 31 | 1.2 | 0.2 | chem WO | [78] |
| 32 | n.a | 3400 | electrochemWO | [103] |
| 33 | 480 | 10 | chem WO | [104] |
| 34 | 310 | 0.102 | chem WO | [107] |
| 35 | 87 | 0.63 | chem WO | [108] |
| 36 | 1150 | 4.5 | chem WO | [105] |
| 37 | 4050 | 176.5 | chem WO | [105] |
| 38 | 6200 | 506 | chem WO | [106] |
Figure 4.
Selected mononuclear ruthenium complexes
11–38 for water oxidation: [(ntp)(pic)
2Ru(H
2O)]
2+ (
11, ntp = 2,6-di (1,8-naphthyridin-2-yl)-4-tert-butylpyridine, pic = 4-picoline) [
71,
86]; Ru(terpy)(bipy)(OH
2)]
2+ (
12a, bipy = 2,2′-bipyridine) [
85], [Ru(terpy)(bpm)(OH
2)]
2+ (
12b, terpy = 2,2′:6′,2″-terpyridine, bpm = 2,2′-bipyrimidine) [
82]; [Ru(terpy)(bpz)(OH
2)]
2+ (
12c, terpy = 2,2′:6′,2″-terpyridine, bpz = 2,2′-bipyrazine) [
82]; [Ru(terpy)(bipy)(Cl)]
+ (
13a, terpy = 2,2′:6′,2″-terpyridine, bipy = 2,2′-bipyridine); [Ru(terpy)(dmbipy)(Cl)]
+ (
13b, dmbipy = 4,4′-dimethyl-2,2′-bipyridine); [Ru(terpy)(dmxbipy)(Cl)]
+ (
13c, dmxbipy = 4,4′-dimethoxy-2,2′-bipyridine); [Ru(terpy)(dnbipy)(Cl)]
+ (
13d, dnbipy = 4,4′-dinitro-2,2′-bipyridine); [Ru(terpy)(dedcbipy)(Cl)]
+ (
13e, dedcbipy = diethyl-2,2′-bipyridine-4,4′-dicarboxylate) [
86]; [Ru(bda)(pic)
2] (
14a, bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = 4-picoline); [Ru(bda)(isq)
2] (
14b, isq = isoquinoline) [
53]; [Ru(pdc)(pic)
3] (
15, pdc = 2,6-pyridinedicarboxylate, pic = 4-picoline) [
91]; [Ru(pdc)(bipy)(pic)] (
16, pdc = 2,6-pyridinedicarboxylate, bipy = 2,2′-bipyridine, pic = 4-picoline) [
91], [Ru
III(L)(pic)
3] (
17, L = 2-(2-hydroxyphenyl)-1H-benzimidazole-7-carboxylate;
18, L = 2-(2-hydroxyphenyl)-1H-benzimidazol-7-ol) [
92]; [Ru(HL)(pic)
3] (
19, L = 6-carbamoylpicolinic acid) [
94]; [Ru
III(H
2pdca)(pic)
3]
+ (
20, H
4pdca = 2,6-pyridine-dicarboxamide) [
95]; [Ru
IV(OH)(tda-κ-N
3O(py)
2]
+ (
21, tda = 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate, py = pyridine) [
54]; {Ru
V(O)(t5a-κ-N
2O)(py)
2} (
22, t5a = 2,5-bis(6-carboxylatopyridin-2-yl)pyrrol-1-ide, py = pyridine) [
96]; [Ru(mcbp)(py)
2] (
23, mcbp = 2,6-bis(1-methyl-4-(carboxylate)-benzimidazol-2-yl)pyridine, py = pyridine); [Ru
IV(O)(mcbp)(py)
2] (
24) [
97]; [Ru(bda)(pic)(pyC)] (
25, bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = 4-picoline, pyC = 2-pyridinecarboxylate) [
98]; Ru(bpaH
2)(pic)
2] (
26, bpaH
2 = 2,2′-bipyridine-6,6′-diphosphonate, pic = 4-picoline); [Ru
III(bpHc)(pic)
2]
+ (
27, bpH
2cH = 2,2′-bipyridine-6-phosphonic acid- 6′-carboxylic acid); [Ru
II(bpHc)(isq)
2] (
28, isq = isoquinoline) [
100]; [Ru
III(tPaO-κ-N
2O
PO
C)(py)
2]
2− (
29, tPaO = 3-(hydroxo-[2,2′:6′,2″-terpyridine]-6,6”-diyl)bis(phosphonate) [
101]; [(L
N5−)Ru
III-OH]
+ (
30, L = 2-(bis-pyridin-2-ylmethyl-amino)-
N-(2-pyridin-2-yl-phenyl)-acetamide) [
102]; [Ru(pdc-κ
3-N
1O
2)(bipy)(H
2O)] (
31, pdc = pyridyl-2,6-dicarboxylato, bipy = 2,2′-bipyridine) [
78]; and [Ru(pdc-κ
2-N
1O
1)(bipy)
2] (
32, pdc = pyridyl-2,6-dicarboxylato, bipy = 2,2′-bipyridine) [
103]; [Ru(bnda)(pic)
2] (
33, bnda = 2,2′-bi(nicotinic acid)-6,6′-dicarboxylate) [
104], [Ru(pda)(pic)
2] (
34, pda = 1,10-phenanthroline-2,9-dicarboxylate, pic = 4-picoline) [
107]; [Ru(biqa)(pic)
2] (
35, biqa = (1,1′-biisoquinoline)-3,3′-dicarboxylate) [
108]; [Ru(bda)(Im)
2] (
36, bda = 2,2′-bipyridine-6,6′-dicarboxylate, Im = imidazole); [Ru(bda)(Im)(DMSO)] (
37, DMSO = dimethylsulfoxide) [
105]; [Ru(bda)(L)
2] (
38, bda = 2,2′-bypyridine-6,6′-dicarboxylate, L = 4-Br-3-methyl pyrazole) [
106].
Figure 4.
Selected mononuclear ruthenium complexes
11–38 for water oxidation: [(ntp)(pic)
2Ru(H
2O)]
2+ (
11, ntp = 2,6-di (1,8-naphthyridin-2-yl)-4-tert-butylpyridine, pic = 4-picoline) [
71,
86]; Ru(terpy)(bipy)(OH
2)]
2+ (
12a, bipy = 2,2′-bipyridine) [
85], [Ru(terpy)(bpm)(OH
2)]
2+ (
12b, terpy = 2,2′:6′,2″-terpyridine, bpm = 2,2′-bipyrimidine) [
82]; [Ru(terpy)(bpz)(OH
2)]
2+ (
12c, terpy = 2,2′:6′,2″-terpyridine, bpz = 2,2′-bipyrazine) [
82]; [Ru(terpy)(bipy)(Cl)]
+ (
13a, terpy = 2,2′:6′,2″-terpyridine, bipy = 2,2′-bipyridine); [Ru(terpy)(dmbipy)(Cl)]
+ (
13b, dmbipy = 4,4′-dimethyl-2,2′-bipyridine); [Ru(terpy)(dmxbipy)(Cl)]
+ (
13c, dmxbipy = 4,4′-dimethoxy-2,2′-bipyridine); [Ru(terpy)(dnbipy)(Cl)]
+ (
13d, dnbipy = 4,4′-dinitro-2,2′-bipyridine); [Ru(terpy)(dedcbipy)(Cl)]
+ (
13e, dedcbipy = diethyl-2,2′-bipyridine-4,4′-dicarboxylate) [
86]; [Ru(bda)(pic)
2] (
14a, bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = 4-picoline); [Ru(bda)(isq)
2] (
14b, isq = isoquinoline) [
53]; [Ru(pdc)(pic)
3] (
15, pdc = 2,6-pyridinedicarboxylate, pic = 4-picoline) [
91]; [Ru(pdc)(bipy)(pic)] (
16, pdc = 2,6-pyridinedicarboxylate, bipy = 2,2′-bipyridine, pic = 4-picoline) [
91], [Ru
III(L)(pic)
3] (
17, L = 2-(2-hydroxyphenyl)-1H-benzimidazole-7-carboxylate;
18, L = 2-(2-hydroxyphenyl)-1H-benzimidazol-7-ol) [
92]; [Ru(HL)(pic)
3] (
19, L = 6-carbamoylpicolinic acid) [
94]; [Ru
III(H
2pdca)(pic)
3]
+ (
20, H
4pdca = 2,6-pyridine-dicarboxamide) [
95]; [Ru
IV(OH)(tda-κ-N
3O(py)
2]
+ (
21, tda = 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate, py = pyridine) [
54]; {Ru
V(O)(t5a-κ-N
2O)(py)
2} (
22, t5a = 2,5-bis(6-carboxylatopyridin-2-yl)pyrrol-1-ide, py = pyridine) [
96]; [Ru(mcbp)(py)
2] (
23, mcbp = 2,6-bis(1-methyl-4-(carboxylate)-benzimidazol-2-yl)pyridine, py = pyridine); [Ru
IV(O)(mcbp)(py)
2] (
24) [
97]; [Ru(bda)(pic)(pyC)] (
25, bda = 2,2′-bipyridine-6,6′-dicarboxylate, pic = 4-picoline, pyC = 2-pyridinecarboxylate) [
98]; Ru(bpaH
2)(pic)
2] (
26, bpaH
2 = 2,2′-bipyridine-6,6′-diphosphonate, pic = 4-picoline); [Ru
III(bpHc)(pic)
2]
+ (
27, bpH
2cH = 2,2′-bipyridine-6-phosphonic acid- 6′-carboxylic acid); [Ru
II(bpHc)(isq)
2] (
28, isq = isoquinoline) [
100]; [Ru
III(tPaO-κ-N
2O
PO
C)(py)
2]
2− (
29, tPaO = 3-(hydroxo-[2,2′:6′,2″-terpyridine]-6,6”-diyl)bis(phosphonate) [
101]; [(L
N5−)Ru
III-OH]
+ (
30, L = 2-(bis-pyridin-2-ylmethyl-amino)-
N-(2-pyridin-2-yl-phenyl)-acetamide) [
102]; [Ru(pdc-κ
3-N
1O
2)(bipy)(H
2O)] (
31, pdc = pyridyl-2,6-dicarboxylato, bipy = 2,2′-bipyridine) [
78]; and [Ru(pdc-κ
2-N
1O
1)(bipy)
2] (
32, pdc = pyridyl-2,6-dicarboxylato, bipy = 2,2′-bipyridine) [
103]; [Ru(bnda)(pic)
2] (
33, bnda = 2,2′-bi(nicotinic acid)-6,6′-dicarboxylate) [
104], [Ru(pda)(pic)
2] (
34, pda = 1,10-phenanthroline-2,9-dicarboxylate, pic = 4-picoline) [
107]; [Ru(biqa)(pic)
2] (
35, biqa = (1,1′-biisoquinoline)-3,3′-dicarboxylate) [
108]; [Ru(bda)(Im)
2] (
36, bda = 2,2′-bipyridine-6,6′-dicarboxylate, Im = imidazole); [Ru(bda)(Im)(DMSO)] (
37, DMSO = dimethylsulfoxide) [
105]; [Ru(bda)(L)
2] (
38, bda = 2,2′-bypyridine-6,6′-dicarboxylate, L = 4-Br-3-methyl pyrazole) [
106].


Table 3.
Comparison of complexes 21 and 22 that contain similar backbone ligands.
Table 3.
Comparison of complexes 21 and 22 that contain similar backbone ligands.
| Ru Complex | CCN Angle for Free Backbone Ligand | CCN Angle for Complex (ave.) * | Coordination Number | Activation Energy | Redox Potential (RuIV/RuIII, RuV=O/RuIV=O) |
|---|
| 21 (tda) | 120° | 113.9° | 7 | M11-L, 19.5 kcal/mol | 1.1 V, 1.43 V |
| 22 (t5a) | 126° | 111.5° | 6 | M06-L, 14.2 kcal/mol | 0.55 V, 1.41 V |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














