2.1. Catalytic Deoxygenation of UCO Over Ni/Al2O3 Promoted with Cu, Fe, and Pt
The composition of the UCO employed in this work is shown in
Table A1 within
Appendix A. The feed is mostly triolein (~95%) with a small amount (~5%) of oleic acid. This feed was upgraded in a fixed bed reactor using a WHSV of 1 h
−1 and a reaction temperature of 375 °C (see
Section 3.3) in order to investigate and compare the relative effect of Cu, Fe and Pt promotion on the performance of Ni/Al
2O
3 in the conversion of UCO to diesel-like hydrocarbons. The results of the gas chromatography-mass spectrometry (GC-MS) analysis of the liquid products collected at representative times on stream are summarized in
Figure 1, and are presented in more detail in
Appendix A (
Table A2,
Table A3 and
Table A4), while the gaseous products are shown in
Figure 2. In addition, a blank (sans catalyst) run was performed using an identical set of conditions in order to assess the extent of thermal (as opposed to catalytic) contributions to UCO conversion and diesel yield. The GC-MS analysis of the liquid products obtained in this blank run (see
Table A5 in
Appendix A) revealed the vast majority (>79%) of the products to be fatty acids and monolein stemming from the thermal conversion of triolein. In addition, the amount of hydrocarbons obtained was <21%, and olefins represented the vast majority of hydrocarbon products irrespective of TOS, which is unsurprising in the absence of a hydrogenation catalyst. Thus, it can be concluded that under the experimental conditions employed, thermal contributions to the conversion of UCO to diesel-like hydrocarbons are relatively minor.
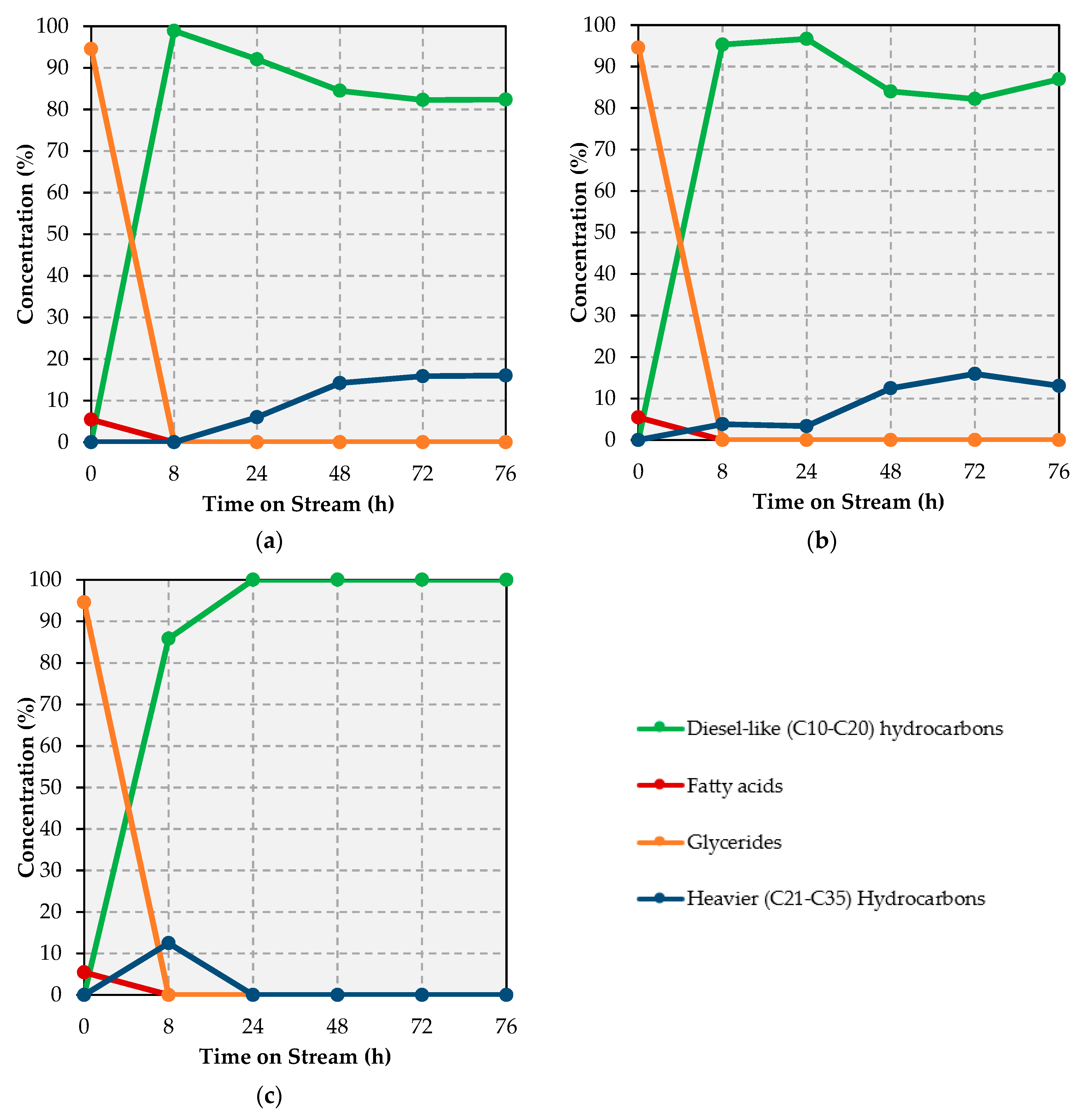
Remarkably, complete deoxygenation occurs over all catalysts tested (see
Figure 1 and
Table A2,
Table A3 and
Table A4), the concentration of diesel-like (C10–C20) hydrocarbons in the reaction products being >82% irrespective of both catalyst and TOS, heavier (C21–C35) hydrocarbons comprising the remainder of the product mixtures. Whereas heavier hydrocarbons stem from the deoxygenation of long-chain ester intermediates [
21], diesel-like hydrocarbons are the result of the deoxygenation of the triglycerides and the fatty acids constituting the UCO feed (as well as of the cracking of heavier hydrocarbons) [
11,
18].
It is also worth noting that the Ni-Pt catalyst afforded liquid product mixtures comprised solely of diesel-like hydrocarbons after 24 h on stream. In contrast, the Ni-Cu and Ni-Fe catalysts showed lower diesel amounts along with a higher yield of heavier (C21–C35) hydrocarbons (particularly at ≥48 h on stream) in their liquid products, which suggests that Ni-Pt disfavors the production of long-chain ester intermediates and/or favors cracking reactions. A closer look at the individual components of the liquid products in the diesel range, namely, C10, C11, C14, C15, and C16 (C12 and C13 could not be determined due to the interference of the reaction solvent) provides valuable insights vis-à-vis the changes in selectivity that take place as the reaction progresses over each catalyst (see
Table A2,
Table A3 and
Table A4). While the vast majority of the feed comprised triolein and oleic acid, the most abundant product obtained over all the catalysts is heptadecane (C17), suggesting that the reaction proceeds mainly via deCO
x as opposed to HDO, which would afford octadecane (C18). Nevertheless, C18 is also produced in significant amounts, which indicates that HDO also occurs. Whereas the amount of C17 and C18 produced over the Ni-Cu and Ni-Fe catalysts remains relatively stable throughout, the corresponding values drop considerably beyond 28 h on stream over the Ni-Pt formulation. Tellingly, the amount of lighter (C10–C14) diesel-like hydrocarbons—which is the result of (and, thus, a proxy for) cracking activity—remains low and fairly stable, drops significantly, and increases considerably with TOS over the Ni-Cu, Ni-Fe and Ni-Pt catalysts, respectively.
Differences in the evolution of the incondensable gas products observed over each catalyst (see
Figure 2) provide insights that are both consistent and complementary to those drawn from the composition of the liquid product mixtures. Briefly, whereas H
2 represents the reaction atmosphere, CO and CO
2 are produced from glycerides and fatty acids via decarbonylation and decarboxylation, respectively. Butane, propane, and ethane are produced through the internal chain cracking of glycerides, fatty acids, and long-chain hydrocarbons, while propane can also stem from the triglyceride backbone and its cracking can afford additional ethane and methane. Lastly, methane is also produced from the methanation of CO
x as well as from the cracking of glycerides, fatty acids, and long-chain hydrocarbons via terminal carbon loss, the main chain shortening mechanism according to a previous report [
22]. With this in mind, the first thing worth noting is the small amount of CO
x detected. Indeed, the amount of CO
x is practically negligible in the gaseous products evolved over both the Fe- and Pt-promoted catalysts (see
Figure 2b,c), indicating that the entirety of these gases is converted to methane and/or remain adsorbed on the surface of these formulations. In contrast, a small amount of CO
2 is detected in the gaseous products evolved over the Cu-promoted catalyst (see
Figure 2a), particularly after a brief induction period observed in the first hours of the experiment. Parenthetically, this induction period has been observed in previous work and attributed to the accumulation of CO
2 on the catalyst surface as alumina-bound carbonates [
22]. While CO
2 eventually breaks through and is detected in the gaseous products, CO remains undetected, likely indicating its full conversion to methane, to CO
2, and coke via the Boudouard reaction, or its strong and irreversible adsorption on the surface of the Ni-Cu catalyst (see
Section 2.3).
The amount of ethane, propane, and butane in the gaseous products is also telling. Over both the Cu- and Fe-promoted catalysts, the amount of these gases detected at the beginning of the reaction is practically negligible, gradually increasing over the first 24 h on stream (see
Figure 2a,b). Beyond this point, the amount of these gases evolved over the Cu-promoted catalyst remains relatively stable for the remainder of the experiment, whereas it becomes negligible once again towards the end of the run over the Fe-promoted formulation. In contrast, the amount of C2–C4 gaseous products evolved over the Pt-promoted catalyst is both higher and constant throughout the entirety of the run (see
Figure 2c). Nevertheless, the amount of ethane, propane, and butane is always smaller than that of methane irrespective of both catalyst and TOS; the amount of methane detected being particularly informative. Whereas the quantity of methane evolved over the Ni-Cu catalyst is both small (< 0.6%) and stable (see
Figure 2a), the latter only applies to the end of the experiment involving the Ni-Fe formulation (see
Figure 2b). At the beginning of the run performed over the Fe-promoted catalysts, the amount of methane in the gaseous products varies from 71% at t = 0 h to 64% at t = 3 h (results not shown), before dropping precipitously to 4% at t = 4 h and then more gradually to reach stability around 0.4% at t = 54 h. In stark contrast, the amount of methane in the gaseous products evolved over the Ni-Pt catalyst varies widely and can be as high as ~75% at t = 24, 48, and 72 h (see
Figure 2c).
All of these trends indicate that while the Ni-Cu and Ni-Fe catalysts retain their deoxygenation activity within the time period investigated, cracking activity either remains constant or declines with TOS, consistent with results reported in other studies [
11,
17,
23]. Although the Ni-Pt catalyst also retains its deoxygenation activity throughout the entire experiment, cracking reactions remain prevalent during the entirety of the run. In short, a comparison of the results obtained with 20% Ni–5% Cu/Al
2O
3, 20% Ni–5% Fe/Al
2O
3, and 20% Ni-0.5% Pt/Al
2O
3 catalysts suggests that Cu- and Fe-promoted catalysts are preferable to Ni-Pt formulations. Indeed, the latter is rendered disadvantageous by its higher price and cracking activity, which would reduce the cost and carbon efficiency of a process designed to convert UCO to diesel-like hydrocarbons.
2.2. Characterization of Fresh and Spent Catalysts
The textural properties of the catalysts used in this study are compiled in
Table 1. The surface area, pore volume, and pore size of all catalysts fall in very narrow ranges, which is consistent with their total metal loadings and particle sizes (
vide infra) and the fact that all catalysts were prepared using the same alumina support. These results indicate that the effects of differences in these properties on catalyst performance should be minimal.
Figure 3 includes the X-ray diffractograms of the catalysts employed in this study. Since diffractograms were acquired using the fresh catalysts in their oxidized form—catalysts were subjected to XRD after the calcination in air constituting the final step of their preparation (see
Section 3.1)—the fact that all Ni detected is present as NiO is unsurprising. Indeed, the three diffractograms display several peaks (at 37.2°, 43.3°, 62.9°, 75.4°, and 79.4°) assigned to NiO [
24]. The fact that diffraction peaks attributed to Fe
3O
4 and to Ni-Fe alloy phases [
17,
19,
25,
26] are absent from the diffractogram corresponding to 20% Ni–5% Fe/Al
2O
3 (
Figure 3a) is unsurprising since these phases would only be expected in reduced (as opposed to oxidized) catalysts. Peaks associated with Fe
2O
3 are also absent from this diffractogram, indicating that Fe is highly dispersed. As previously reported [
18], the fact that peaks at 35.5° and 38.7° corresponding to a CuO phase [
27] are not observed in the diffractogram of 20% Ni–5% Cu/Al
2O
3 (
Figure 3b) can be similarly attributed to the high dispersion of the Cu phase [
24,
28]. Likewise, no distinct Pt-related features (peaks or peak shifts) can be observed in the diffractogram corresponding to 20% Ni-0.5% Pt/Al
2O
3 (
Figure 3c).
The temperature-programmed reduction (TPR) profiles shown in
Figure 4 clearly illustrate that the three catalysts employed in this study display very different reduction behavior. As discussed in a previous report [
18], the TPR profile for 20% Ni–5% Cu/Al
2O
3 shows four distinct reduction events: (1) a sharp peak at 180 °C attributed to the reduction of copper oxide [
24,
29]; (2) a broader but well-defined peak with a maximum at 360 °C assigned to the reduction of a NiO-CuO phase [
30]; (3) a shoulder with a local maximum at 460 °C signaling the reduction of NiO [
31]; and (4) a weak and broad signal around 690 °C indicating the reduction of nickel aluminate (NiAl
2O
4) [
32]. The TPR profile for 20% Ni–5% Fe/Al
2O
3 also shows four (but less distinct) reduction events, namely: (1) a small signal with a maximum at 235 °C corresponding to large (10–50 nm) NiO ensembles (
vide infra); (2,3) a very large and broad peak ranging from 260 to 675 °C with a maximum at 350 °C, commingling the reduction of nickel and iron oxides (leading to the formation of a Ni-Fe alloy) [
17,
19]; and (4) a high-temperature tail of the latter peak, assigned to NiAl
2O
4 reduction. Lastly, as discussed in a recent report [
13] the TPR profile for 20% Ni-0.5% Pt/Al
2O
3 also displays several reduction events, including (1) a small and broad peak between 300 and 350 °C attributed both to the reduction of surface Pt and of large NiO particles in close proximity to Pt [
13,
33]; (2) an intense and well-defined signal with a maximum at 460 °C assigned to the Pt-assisted reduction of smaller NiO particles [
13]; and (3) a broad peak above 500 °C with a high temperature (>700 °C) shoulder attributed to the reduction of NiO and NiAl
2O
4, respectively.
Given that Ni-based formulations used in the deoxygenation of FOG to fuel-like hydrocarbons are known to be particularly susceptible to coking [
1], the spent catalysts were subjected to thermogravimetric analysis (TGA) in air, the resulting profiles being shown in
Figure 5.
The TGA profiles indicate that the total mass loss displayed by the spent catalysts follows the trend Ni-Fe (3.0%) < Ni-Cu (7.6%) < Ni-Pt (10.8%). In addition, the temperature at which mass loss takes place is also noteworthy, since mass loss events <400 °C can be attributed to strongly adsorbed reactants, intermediates and products (or soft coke) and mass loss events >400 °C can be assigned to more recalcitrant carbonaceous deposits (graphitic or hard coke). Tellingly, albeit the majority of the weight loss displayed by all spent catalyst takes place below 400 °C, the Ni-Pt formulation also shows distinct and significant weight loss above this temperature. The increased coking observed on the Ni-Pt catalyst is consistent with its considerably higher cracking activity, which is evinced by the copious amounts of methane produced by this formulation (see
Section 2.1). In turn, the fact that both the Ni-Cu and the Ni-Fe catalysts display lower amounts of carbonaceous deposits is in agreement with the known ability of both Cu and Fe to curb the hydrogenolysis activity of Ni via geometric effects (see
Section 1).
Table 2 shows the surface concentration (in at.%) of the elements detected via x-ray photoelectron spectroscopy (XPS) in the catalysts after (i) 76 h of TOS, followed by washing with dodecane and drying (spent); (ii) their subsequent calcination for 5 h at 450 °C under air (calcined); and (iii) their successive reduction for 3 h at 400 °C under H
2 (re-reduced). All XPS spectra can be found in
Appendix B (
Figure A1,
Figure A2,
Figure A3,
Figure A4,
Figure A5,
Figure A6,
Figure A7 and
Figure A8). Carbon in the samples can be divided into inorganic (carbide) and organic (coke) carbon, the inorganic carbon being mostly associated with the SiC used as a diluent in the upgrading experiments (see
Section 3.3) and being indicative of the relative amount of sample components—catalyst or diluent—analyzed during XPS measurements. The amount of inorganic carbon can considerably impact the interpretation of the data in
Table 2. This is also the case for the amount of organic carbon (calculated by subtracting the inorganic from the total carbon) associated with coke deposits and whose Ni-Pt > Ni-Cu > Ni-Fe trend in the spent catalysts is in agreement with the results of TGA (
vide supra). The fact that both spent Ni-Fe and Ni-Pt catalysts display a very similar surface concentration of Si and inorganic carbon indicates that a similar fraction of SiC diluent and catalyst is being analyzed, which in turn confirms that organic carbon (coke) deposits are much more abundant on the Pt- than on the Fe-promoted formulation. Similarly, while both the Si and inorganic carbon concentration of the spent Ni-Cu sample is lower, suggesting that a lower amount of C is contributed by SiC, the fact that the concentration of C
Tot (and, thus, of C
Org) in this formulation is higher than on Ni-Fe indicates that coke deposits on the surface of Ni-Cu are intermediate to those on Ni-Pt and Ni-Fe, which is also consistent with TGA data.
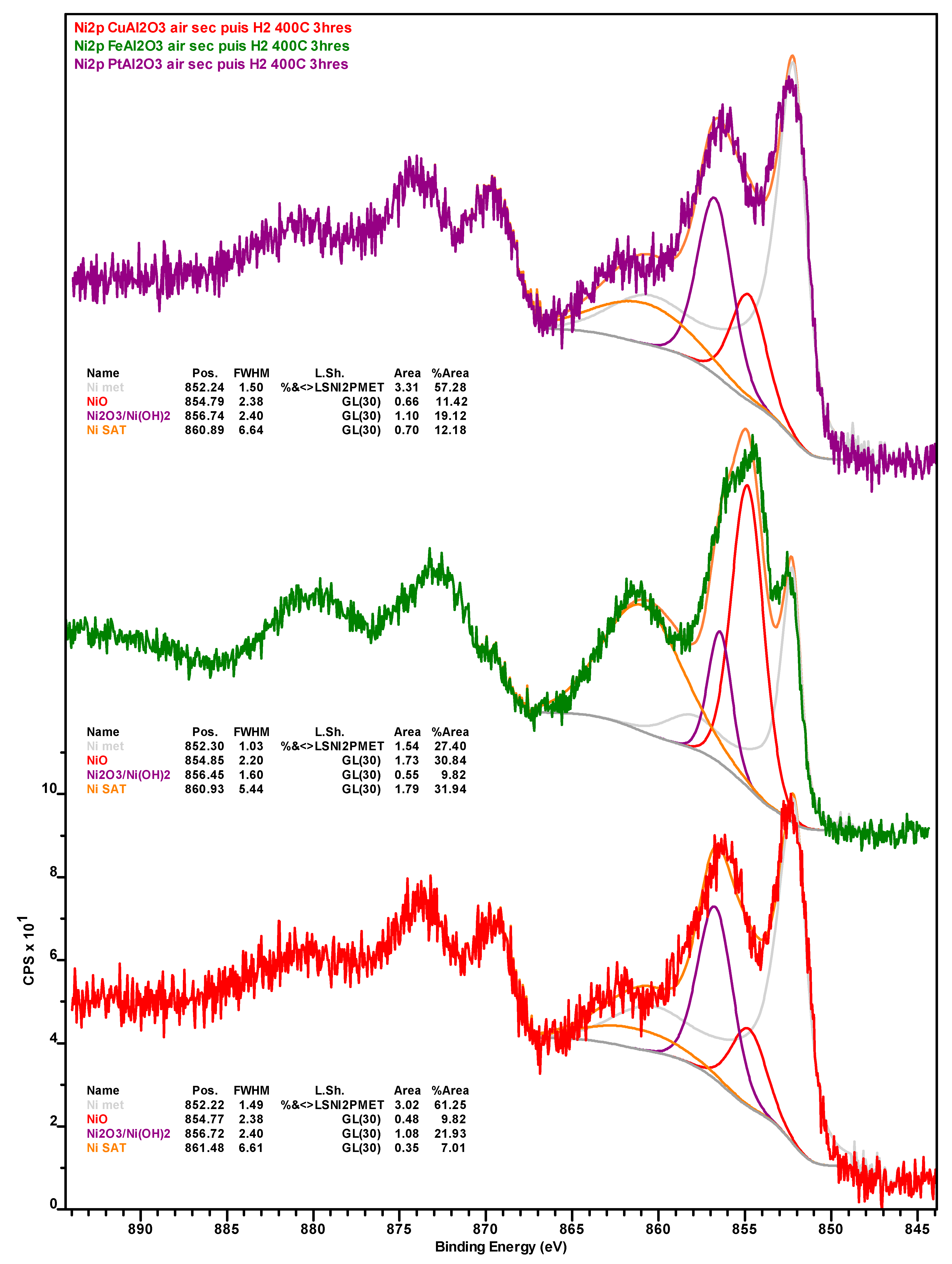
Notably, the amount of Ni in general and Ni
0 in particular, is considerably higher on the surface of the spent Ni-Fe catalyst than on the other two formulations, although the observed trend (Ni-Fe >> Ni-Cu > Ni-Pt) likely stems—at least in part—from the relative amount of coke deposits on the surface of these catalysts. Considering the promoter metals, it is worth noting that a much higher amount of Fe in the Ni-Fe catalyst (relative to Cu in the Ni-Cu formulation) is detected at the surface. While this cannot be attributed to the amount of SiC artificially depressing the amount of Cu detected (since the Si and inorganic carbon concentration is higher in the Ni-Fe catalyst) or to the slightly lower atomic weight (A
r) of Fe—the A
r difference is too small—the higher amount of coke deposits on the surface of the Ni-Cu catalyst could partially explain the relatively low Cu concentration, as with the case of Ni. Lastly, while the entirety of Cu is present as Cu
+, which is indicative of Cu
2O as copper does not form a carbide phase, a small amount (ca. 13%) of Fe is in the metallic state, the remainder being present in oxidic form (see
Figure A7 and
Figure A8 in
Appendix B).
Upon calcination, the amount of C
Tot is significantly reduced mainly due to the combustion of coke deposits. Consistent with the more graphitic—and thus recalcitrant—nature of the coke on this formulation as indicated by TGA results (see
Figure 5), the amount of residual coke after calcination is highest for the Ni-Pt catalyst. Although the amount of Ni in the calcined catalysts is lowest for 20% Ni–5% Cu/Al
2O
3, the amount of Ni
0 follows the trend Ni-Cu > Ni-Pt > Ni-Fe, which is indicative of the resistance of surface Ni in each catalyst to oxidation. Regarding the metallic promoters within the calcined catalyst, which increase in concentration due to the removal of coke, Cu is present as a mixture of Cu
2O and CuO, the vast majority of Fe also being present in the oxidized form (only ~3% being present as Fe
0).
Changes observed upon the reduction of the calcined catalysts are also informative. The similar amounts of Si, Ni, and Ni
0 on the re-reduced Ni-Cu and Ni-Pt formulations and the changes in these values relative to those displayed by their calcined counterparts indicate that (i) the region of the samples analyzed are catalyst-rich and SiC-poor; and (ii) sintering takes place during reduction based on the loss of surface Ni relative to the calcined catalysts. Changes in the Ni/Al ratio—from 0.18 to 0.12 and from 0.65 to 0.27 for calcined to re-reduced Ni-Cu and Ni-Pt, respectively—also suggests that sintering takes place during reduction. Moreover, the amount of C
Tot and C
Inorg on the Ni-Pt catalyst is striking and suggests the formation of a considerable amount of metallic carbide(s). Unfortunately, the presence of the latter could not be conclusively confirmed since Pt could not be observed (due to the small amount of Pt and the overlapping of the Pt4f and the Al2p XPS regions) and a distinct nickel carbide signal is not resolved. It is also noteworthy that the surface concentration of Ni (and to a lesser degree that of Ni
0) on the re-reduced Ni-Fe catalyst is significantly higher than that of its Ni-Cu and Ni-Pt counterparts, particularly taking into account the considerably larger amount of SiC being analyzed alongside Ni-Fe. Finally, it is interesting to note that the surface concentration of Cu is lower on the re-reduced Ni-Cu catalyst than on its spent and calcined counterparts—particularly taking into account the lower amount of SiC analyzed alongside the re-reduced material and the higher amount of coke on the spent formulation—which may indicate the alloying of Cu with Ni. Regarding the oxidation state of the promoter metals after reduction, while the entirety of Cu is present in the metallic form, only ~9% of iron is present as Fe
0, the remainder being present as Fe
2+ or Fe
3+. Since it has been reported that Fe
2O
3 undergoes reduction to Fe
3O
4 in the ~300–400 °C range, while Fe
3O
4 is reduced between ~400 and 500 °C [
34], it is unsurprising that after reduction at 400 °C most of the Fe detected by means of XPS is oxidic.
In short, XPS results indicate that the trend related to the amount of organic (coke) deposits on the surface of spent catalysts (Ni-Pt >> Ni-Cu > Ni-Fe) explains the relative amounts of Ni and Ni0—as well as of promoter metals—on the surface of spent formulations (Ni-Fe >> Ni-Cu > Ni-Pt). Moreover, the trends related to the amount of Ni0 on the surface of calcined and re-reduced catalysts (Ni-Cu > Ni-Pt > Ni-Fe and Ni-Fe > Ni-Cu ≈ Ni-Pt, respectively) indicate that Ni displays distinct redox behavior within each formulation. Indeed, Ni on the surface of 20% Ni–5% Fe/Al2O3 is easier to oxidize and reduce than Ni on the surface of 20% Ni–5% Cu/Al2O3 or 20% Ni–0.5% Pt/Al2O3. Finally, XPS results evince that while the Ni-Cu and Ni-Pt catalysts experience metal particle sintering during re-reduction, Ni-Cu and Ni-Fe alloys form within the Cu- and Fe-promoted catalysts and metallic carbides may form within the Pt-promoted formulation.
The analysis of the fresh and spent catalysts via transmission electron microscopy-energy dispersive X-ray spectroscopy (TEM-EDS) also afforded significant insights. In the case of the Cu-promoted catalyst, TEM results indicate that the particle size distribution—which is narrow and centered around 4 nm particles in the fresh catalyst—is both broader and centered around larger particles in the spent formulation (see
Figure 6a), signaling particle sintering. The TEM-EDS results in
Figure 6b reveal that the metal particles in the fresh catalyst display a composition that is close to that of the bulk formulation (80% Ni-20% Cu considering only the metallic phase), albeit Ni-rich particles containing 85–95% Ni are also observed. Notably, the spent catalyst comprises particles slightly more enriched in Cu relative to those in the fresh formulation, all particles in the spent catalyst containing between 65 and 80% Ni. This indicates that particles not only grow in size but also become Cu-rich during the reaction, which is also in line with previously reported results [
11]. This conclusion is clearly illustrated by the TEM micrographs and the TEM-EDS elemental maps included in
Figure A9; the elemental maps also showing that Ni and Cu are present in close association on both the fresh and spent catalysts. The Cu map of the spent Ni-Cu catalyst shown in
Figure A9 also provides an example of the Cu-hollow space not observed in the fresh formulation, as Ni-Cu particles likely undergo Cu-hollowing through a mechanism based on the Kirkendall effect [
35]. These observations are consistent with the widely reported bulk and surface enrichment of Ni-Cu nanoparticles with Cu [
11,
36,
37].
In the case of the Fe-promoted catalyst, TEM results indicate that a similar particle size change takes place during reaction. Indeed, the particle size distribution—which is narrow in the fresh catalyst as the vast majority of particles range from 3 to 7 nm—is both broader and shifted to larger (8–30 nm) particles in the spent formulation (see
Figure 7a). However, the composition of the metal particles does not change much during the reaction in contrast with the Cu-promoted catalyst; the only change observed being the disappearance of Ni-rich (90–100% Ni) particles according to the TEM-EDS results in
Figure 7b. These observations are illustrated by the TEM micrographs and the TEM-EDS elemental maps in
Figure A10, which show the degree of association between Ni and Fe in both the fresh and the spent catalyst. The Fe maps included in
Figure A10 also evince Fe-hollow spaces on both the fresh and spent formulation, albeit the latter displays more of these spaces. Analogous to the case of the Ni-Cu catalyst, Fe-hollowing may occur through a mechanism based on the Kirkendall effect, which has also been reported for Ni-Fe bimetallic formulations [
38,
39].
In the case of the Pt-promoted catalyst, TEM results indicate a similar particle size change as those experienced during the reaction by the other formulations. Specifically, whereas the fresh catalyst shows a fairly narrow particle size distribution with the vast majority of particles falling within the 5–10 nm range, the spent formulations show a much broader distribution with particles as small as 3 nm and as large as 28 nm (see
Figure 8a). Nevertheless, changes in the composition of metal particles within the Pt-promoted catalyst are noteworthy (see
Figure 8b). Indeed, the fresh catalyst shows a significant amount of Pt-rich particles—relative to the bulk formulation (97.6 wt.% Ni-2.4 wt.% Pt or 99.3 at. % Ni-0.7 at.% Pt)—whereas the vast majority of metal particles in the spent catalyst show a composition very close to that of the bulk. The TEM micrographs and the TEM-EDS elemental maps in
Figure A11 support these conclusions, showing both the increase in metal particle size and the closer association of Ni and Pt in the spent catalysts than in the fresh state.
2.3. Structural and Activity Changes Observed during Catalysts Aging and Regeneration
As mentioned in the preceding section, the similarity between the textural properties (surface area, pore volume, and pore diameter) of all fresh catalysts suggests that the effect of these properties on catalyst performance should be minimal, at least at the onset. However, any variations in these and other properties with TOS may influence catalyst performance. Indeed, based on TGA results, the loss of surface area and porosity attributable to coking and fouling should follow the trend Ni-Fe < Ni-Cu < Ni-Pt, the latter catalyst showing a higher amount of more recalcitrant (graphitic) carbonaceous deposits. Notably, complete deoxygenation of the feed was maintained throughout the experiment for each of the catalysts, which suggests that losses in surface area and porosity due to coking, fouling, and sintering are not sufficient to noticeably impact the deoxygenation activity of these formulations in the time period investigated. Thus, differences in the cracking activity of the catalysts offer better insights vis-à-vis structure-activity relationships.
Looking at both the composition of the liquid and gaseous products, the cracking activity of the Cu-promoted catalyst—although always relatively low—is highest between 8 and 30 h on stream and progressively drops between 30 and 72 h on stream, at which point it becomes stable. Somewhat similarly, the cracking activity of the Fe-promoted formulation is considerably higher in the first 8 h of the experiment, becomes moderate between 24 and 52 h on stream, and is both negligible and stable beyond t = 72 h. Contrastingly, the cracking activity of the Pt-promoted formulation is higher at the end of the run than at its onset. Therefore, at least in the case of the Ni-Pt catalyst, it appears that neither the loss of surface area due to coking, fouling, and sintering, nor changes in the bulk or surface composition of the metal particles, reduce the cracking activity of this formulation in the time period investigated. The opposite is the case for the Cu- and Fe-promoted catalysts, which display a lower cracking activity towards the end of the experiment, this effect being more pronounced for the Ni-Fe formulation.
TPR measurements performed on the spent catalysts after calcination (see
Figure A12) provide valuable insights on the structural changes that occur during regeneration. The first thing worth noting is that all peaks (except for those assigned to NiAl
2O
4) are shifted to lower temperatures, which can be partially attributed to the formation of larger particles that are easier to reduce. However, the more substantial shifts (>100 °C) can only be fully explained by invoking a considerable increase in the association of Ni with the promoter metal. For the Cu-promoted catalyst, changes in the relative intensity of peaks attributed to the reduction of copper oxide, NiO-CuO, and NiO (with respect to that in the fresh formulation) point to a reduction in the amount of unalloyed Cu and the formation of Ni-Cu bimetallic particles with a Cu-rich surface. Indeed, the peaks with maxima at 245 and 375 °C likely correspond to the reduction of NiO-CuO at the surface and of NiO at the core of these particles, respectively. For the Fe-promoted formulation, the narrowness of the main peak relative to that in the fresh formulation indicates a closer association between the metals forming a Ni-Fe alloy, which is consistent with the disappearance of Ni-rich particles observed via TEM-EDS (see
Figure 7b). In addition, the intensity of this peak relative to that of peaks shown by other catalysts is in line with the higher amount of surface Ni in the Fe-promoted formulation measured via XPS (
vide infra). Lastly, for the Pt-promoted catalyst, the sizable shift (>200 °C) that the main peak displays between the fresh and the regenerated formulation, is indicative of a major increase in the association between Ni and Pt.
The results of the XPS measurements performed on calcined and re-reduced spent catalysts confirm these conclusions and offer additional insights. While most (if not all) of the coke is removed from Cu- and Fe-promoted spent catalysts, the regenerated Ni-Pt formulation displays both a significant amount of residual coke, as well as a possible metallic carbide phase. In addition, metal particle sintering takes place during the regeneration of spent Ni-Cu and Ni-Pt catalysts, likely explaining the lower amount of surface Ni and Ni0 these formulations display relative to their Fe-promoted counterpart.
Finally, the regenerated (i.e., calcined in air at 450 °C for 5 h and re-reduced under H
2 at 400 °C for 3 h) spent catalysts were subjected to in-situ diffuse reflectance infrared Fourier transform spectroscopy after CO adsorption (CO-DRIFTS) to gain additional information on their structure post-regeneration (see
Figure 9). Since the adsorption of CO on metals is highly temperature-dependent, adsorption was carried out at 25 °C in order to focus on the most active sites for CO adsorption and limit the confounding effects that the presence of multiple metals can have on M-CO spectra [
40]. Irrespective of its state (fresh or spent and regenerated), the Cu-promoted catalyst displays a very intense band at 2119–2100 cm
−1, which is attributed to CO adsorbed on Cu sites [
41,
42]. However, this band increases in intensity and shifts to higher wavenumbers on the regenerated catalyst, which signals an increase in the total quantity of Cu sites, as well as a change in their electronic properties. While the increase in intensity is in line with the enrichment of the surface with Cu, the shift in wavenumber may result from a greater extent of Ni-Cu alloying and from the rise of Cu-hollow spaces [
43]. Tellingly, while the fresh Ni-Fe catalyst shows a Ni-CO band at 2179 cm
−1, this band is absent from the corresponding spectrum post-regeneration. This suggests that the most coordinatively unsaturated Ni sites—which are the most active cracking sites [
44]—are irreversibly deactivated during reaction/regeneration. Similarly, while the fresh Ni-Pt catalyst shows a well-defined peak at ~2180 cm
−1 and a broad feature at ~2120 cm
−1 associated with CO on metallic Ni sites, as well as a large and well-defined peak at ~2077 cm
−1 assigned to CO on Pt sites [
13], none of these signals are observed post-regeneration. Since XPS results demonstrate the presence of Ni
0 on the regenerated Ni-Pt formulation, the dearth of signals can be explained by residual coke blocking the sites responsible for low-temperature CO adsorption.
These observations are reinforced by a recent report in which the performance of a regenerated Ni-Cu catalyst in fatty acid deoxygenation was found to be distinct from—and superior to—that of the fresh formulation [
11]. Thus, the need for additional work in which the regenerated catalysts are tested to study their performance (and the evolution thereof) in a second cycle post-regeneration is clearly indicated, particularly since these tests stand to shed light on the recyclability of these formulations and unveil additional structure-activity relationships.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















