

Emerging Treatment Options for Neuroendocrine Neoplasms of Unknown Primary Origin: Current Evidence and Future Perspectives

 ,
,  , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
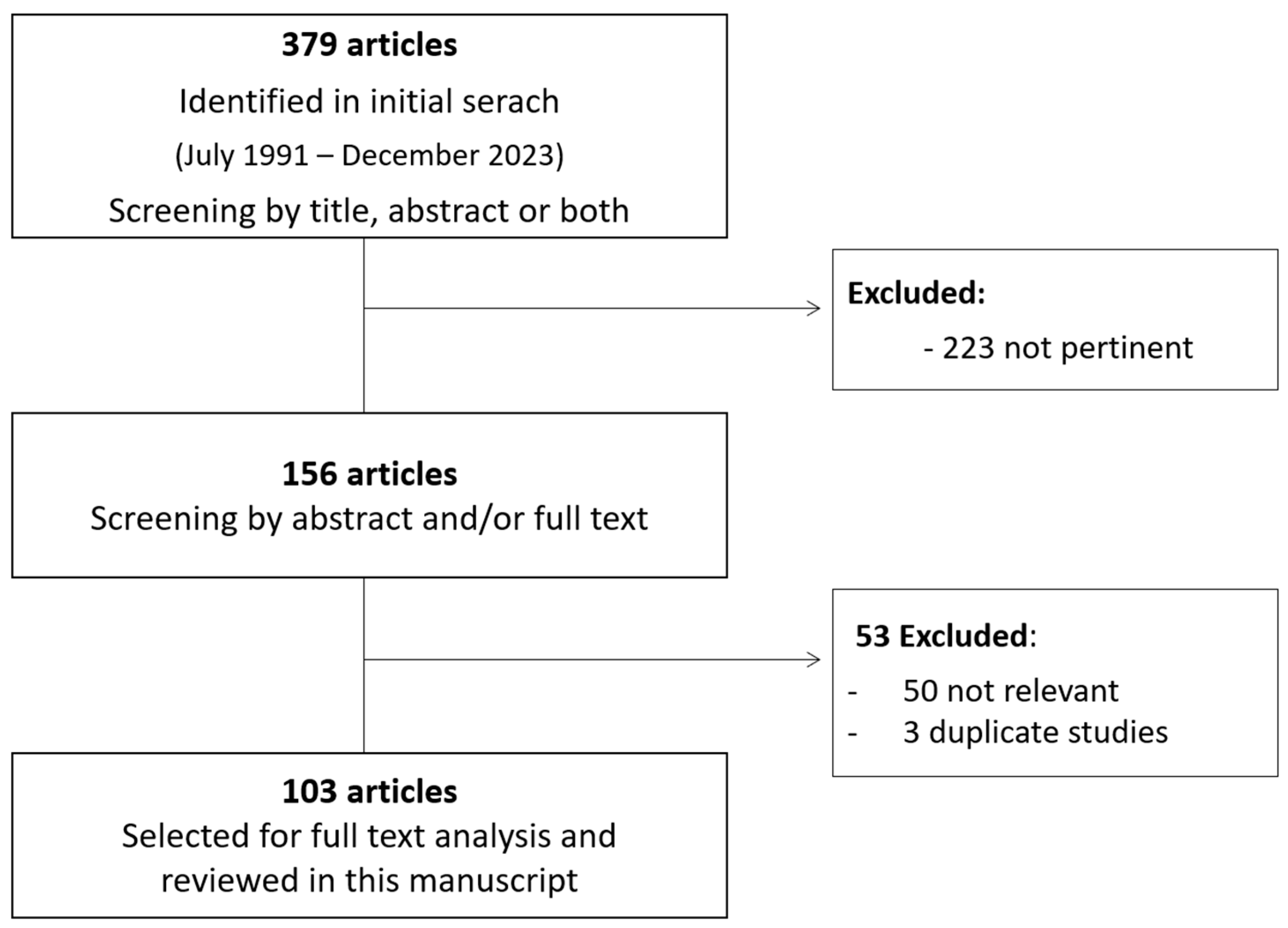
2. Materials and Methods
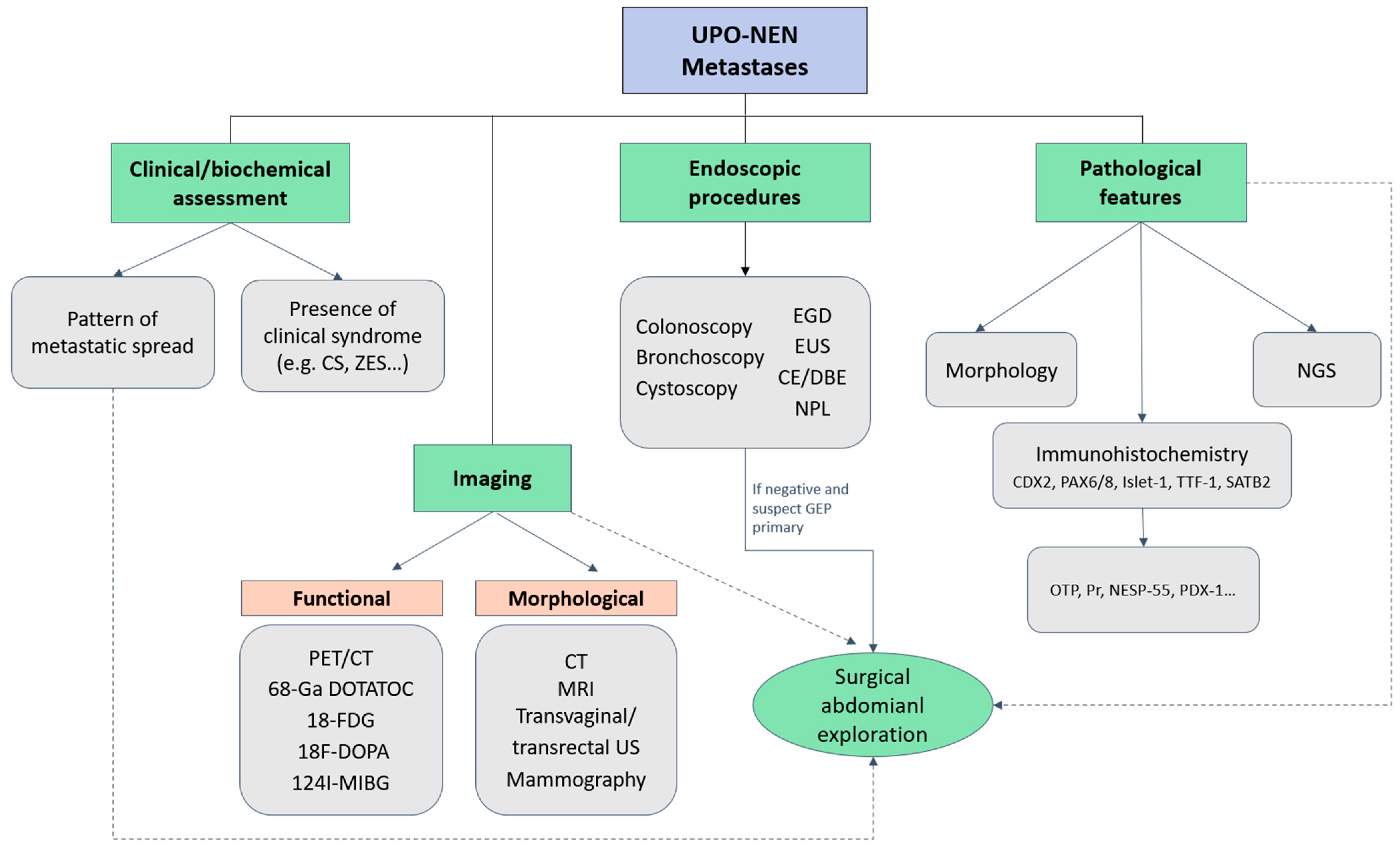
3. Diagnostic Approach to UPO-NEN
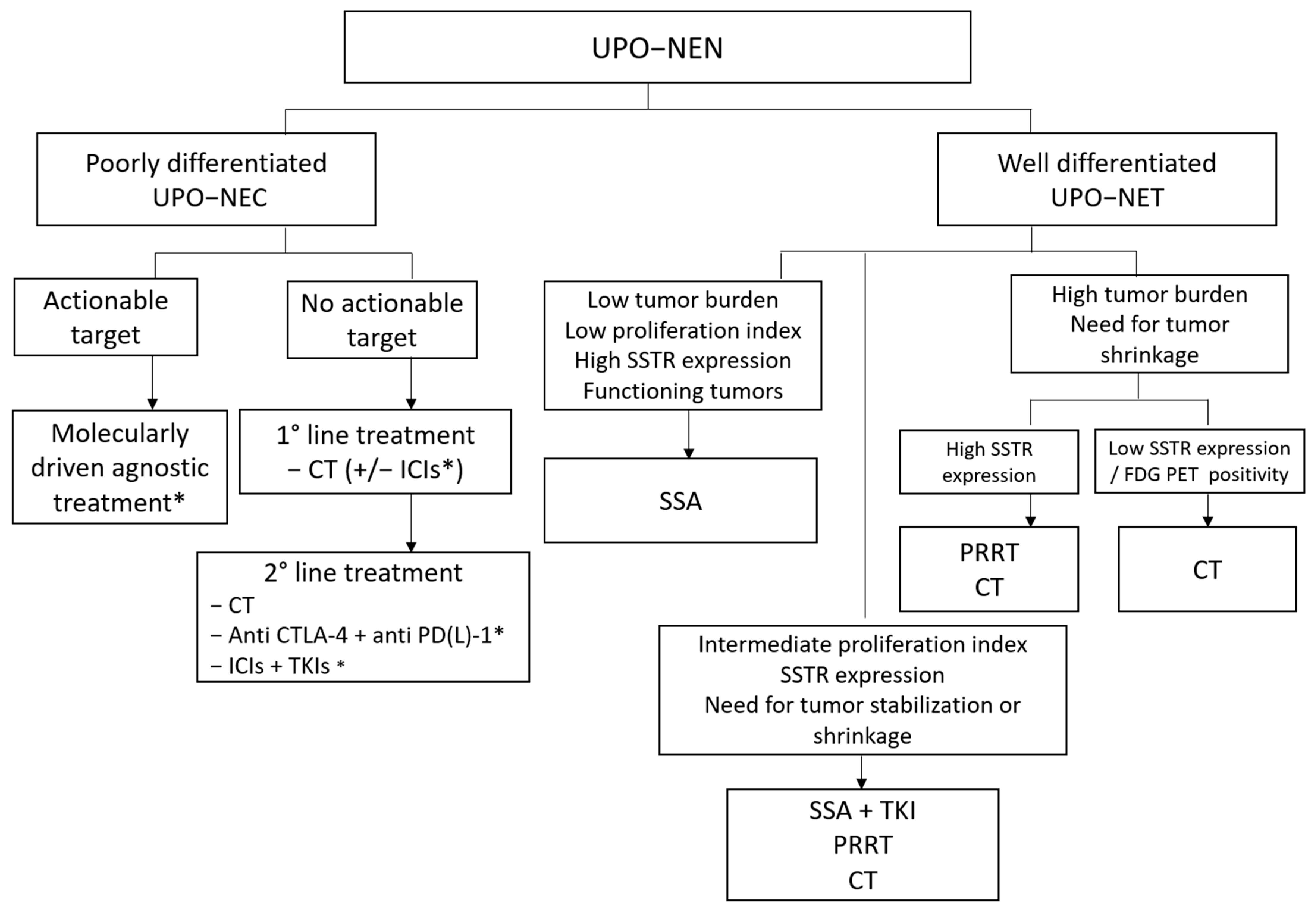
4. Therapeutic Approach to UPO-NEN
4.1. Treatment of Poorly Differentiated NEC of Unknown Origin
4.1.1. First-Line Setting
4.1.2. Second-Line Setting
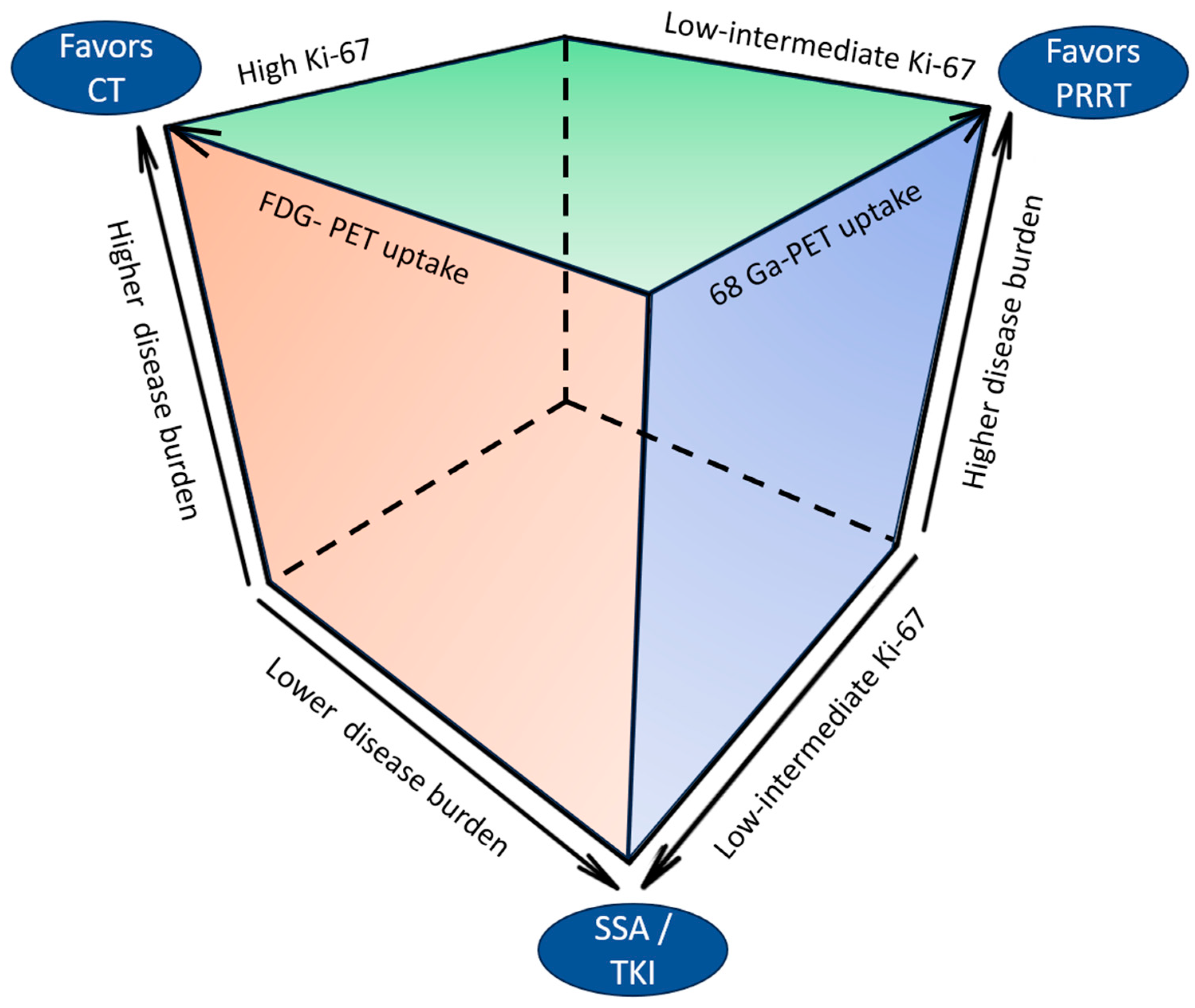
4.2. Treatment of Well-Differentiated NETs of Unknown Origin
4.3. Special Situations: Liver-Limited Disease
5. Unraveling Molecular Characterization of UPO-NENs: Time for an Agnostic Approach?
6. Discussion
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Taal, B.; Visser, O. Epidemiology of neuroendocrine tumours. Neuroendocrinology 2004, 80 (Suppl. S1), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Polish, A.; Vergo, M.T.; Agulnik, M. Management of neuroendocrine tumors of unknown origin. J. Natl. Compr. Cancer Netw. 2011, 9, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Perren, A.; Couvelard, A.; Scoazec, J.-Y.; Costa, F.; Borbath, I.; Fave, G.D.; Gorbounova, V.; Gross, D.; Grossman, A.; Jensen, R.T.; et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Pathology-diagnosis and prognostic stratification. Neuroendocrinology 2017, 105, 196–200. [Google Scholar] [CrossRef]
- Fernandez, C.J.; Agarwal, M.; Pottakkat, B.; Haroon, N.N.; George, A.S.; Pappachan, J.M. Gastroenteropancreatic neuroendocrine neoplasms: A clinical snapshot. World J. Gastrointest. Surg. 2021, 13, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Berner, A.M.; Pipinikas, C.; Ryan, A.; Dibra, H.; Moghul, I.; Webster, A.; Luong, T.V.; Thirlwell, C. Diagnostic Approaches to Neuroendocrine Neoplasms of Unknown Primary Site. Neuroendocrinology 2020, 110, 563–573. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Capdevila, J.; Crespo-Herrero, G.; Díaz-Pérez, J.A.; del Prado, M.P.M.; Orduña, V.A.; Sevilla-García, I.; Villabona-Artero, C.; Beguiristain-Gómez, A.; Llanos-Muñoz, M.; et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): Results from the National Cancer Registry of Spain (RGETNE). Ann. Oncol. 2010, 21, 1794–1803. [Google Scholar] [CrossRef]
- Bhosale, P.; Shah, A.; Wei, W.; Varadhachary, G.; Johnson, V.; Shah, V.; Kundra, V. Carcinoid tumours: Predicting the location of the primary neoplasm based on the sites of metastases. Eur. Radiol. 2013, 23, 400–407. [Google Scholar] [CrossRef]
- Kurl, S.; Rytkönen, H.; Farin, P.; Ala-Opas, M.; Soimakallio, S. A primary carcinoid tumor of the kidney: A case report and review of the literature. Abdom. Imaging 1996, 21, 464–467. [Google Scholar] [CrossRef]
- Stroosma, O.B.; Delaere, K.P. Carcinoid tumours of the testis. BJU Int. 2008, 101, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Radfar, L.; Fatahzadeh, M. Neuroendocrine carcinoma of the oral cavity: A case report and review of the literature. Gen. Dent. 2008, 56, 714–718. [Google Scholar] [PubMed]
- Widmeier, E.; Fuellgraf, H.; Waller, C.F. Complete remission of Cdx-2 positive primary testicular carcinoid tumor: 10-years follow-up and literature review. BMC Urol. 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Okasho, K.; Ogawa, O.; Akamatsu, S. Narrative review of challenges in the management of advanced neuroendocrine prostate cancer. Transl. Androl. Urol. 2021, 10, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Jagiella-Lodise, O.; Jagiella, V.; Weitman, E. An abdominal wall neuroendocrine tumor of unknown primary origin: A case report and review of the literature. Cancer Rep. 2022, 5, e1610. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; van Zante, A.; Granados, M.L. Combined Neuroendocrine and Squamous Cell Carcinoma of the Sinonasal Tract: A Morphologic and Immunohistochemical Analysis and Review of Literature. Head Neck Pathol. 2022, 16, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Dai, S.; Xu, J.; Liu, L.; Yu, J.; Sun, T. Primary Neuroendocrine Tumor of the Breast: Current Understanding and Future Perspectives. Front. Oncol. 2022, 12, 848485. [Google Scholar] [CrossRef] [PubMed]
- Miquelestorena-Standley, E.; Dujardin, F.; Arbion, F.; Touzé, A.; Machet, L.; Velut, S.; Guyétant, S. Recurrent primary cutaneous mucinous carcinoma with neuroendocrine differentiation: Case report and review of the literature. J. Cutan. Pathol. 2014, 41, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Batra, V.V.; Majumdar, K.; Javed, A.; Agarwal, A.K.; Sakhuja, P. Signet ring cell neuroendocrine tumor liver with mesenteric metastasis: Description of a rare phenomenon, with literature review. J. Cancer Res. Ther. 2015, 11, 658. [Google Scholar] [CrossRef]
- La Salvia, A.; Persano, I.; Trevisi, E.; Parlagreco, E.; Muratori, L.; Scagliotti, G.V.; Brizzi, M.P. Ocular metastases from neuroendocrine tumors: A literature review. Semin. Oncol. 2020, 47, 144–147. [Google Scholar] [CrossRef]
- Pavel, M.; O’Toole, D.; Costa, F.; Capdevila, J.; Gross, D.; Kianmanesh, R.; Krenning, E.; Knigge, U.; Salazar, R.; Pape, U.-F.; et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology 2016, 103, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; de Herder, W.W. ENETS Consensus Guidelines for the Standard of Care in Neuroendocrine Tumors. Neuroendocrinology 2017, 105, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Turner, G.; King, B.; Jones, L.; Culliford, D.; McCance, D.; Ardill, J.; Johnston, B.T.; Poston, G.; Rees, M.; et al. Midgut neu-roendocrine tumours with liver metastases: Results of the UKINETS study. Endocr. Relat. Cancer 2009, 16, 885–894. [Google Scholar] [CrossRef]
- Citterio, D.; Pusceddu, S.; Facciorusso, A.; Coppa, J.; Milione, M.; Buzzoni, R.; Bongini, M.; Debraud, F.; Mazzaferro, V. Primary tumour resection may improve survival in functional well-differentiated neuroendocrine tumours metastatic to the liver. Eur. J. Surg. Oncol. (EJSO) 2016, 43, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Capurso, G.; Rinzivillo, M.; Bettini, R.; Boninsegna, L.; Fave, G.D.; Falconi, M. Systematic review of resection of primary midgut carcinoid tumour in patients with unresectable liver metastases. Br. J. Surg. 2012, 99, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Keutgen, X.M.; Nilubol, N.; Glanville, J.; Sadowski, S.M.; Liewehr, D.J.; Venzon, D.J.; Steinberg, S.M.; Kebebew, E. Resection of primary tumor site is associated with prolonged survival in metastatic nonfunctioning pancreatic neuroendocrine tumors. Surgery 2016, 159, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Corti, F.; Pusceddu, S.; Milione, M.; Coppa, J.; Masoni, B.; Oldani, S.; Sabella, G.; Cafaro, P.; Repici, A. Multidisciplinary Approach to the Diagnosis of Occult Primary Neuroendocrine Neoplasm: A Clinical Challenge. J. Clin. Med. 2023, 12, 5537. [Google Scholar] [CrossRef]
- Bellizzi, A.M. Immunohistochemistry in the diagnosis and classification of neuroendocrine neoplasms: What can brown do for you? Hum. Pathol. 2020, 96, 8–33. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Zedenius, J.; Höög, A. Metastatic Neuroendocrine Neoplasms of Unknown Primary: Clues from Pathology Workup. Cancers 2022, 14, 2210. [Google Scholar] [CrossRef] [PubMed]
- Savelli, G.; Lucignani, G.; Seregni, E.; Marchian, A.; Serafini, G.; Aliberti, G.; Villano, C.; Maccauro, M.; Bombardieri, E. Feasibility of somatostatin receptor scintigraphy in the detection of occult primary gastro-entero-pancreatic (GEP) neuroendocrine tumours. Nucl. Med. Commun. 2004, 25, 445–449. [Google Scholar] [CrossRef]
- Santhanam, P.; Chandramahanti, S.; Kroiss, A.; Yu, R.; Ruszniewski, P.; Kumar, R.; Taïeb, D. Nuclear imaging of neuroendocrine tumors with unknown primary: Why, when and how? Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Kunikowska, J.; Baudin, E.; Bodei, L.; Bouvier, C.; Capdevila, J.; Cremonesi, M.; de Herder, W.W.; Dromain, C.; Falconi, M.; et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur. J. Cancer 2021, 146, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Balogova, S.; Talbot, J.-N.; Nataf, V.; Michaud, L.; Huchet, V.; Kerrou, K.; Montravers, F. 18F-Fluorodihydroxyphenylalanine vs other radiopharmaceuticals for imaging neuroendocrine tumours according to their type. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 943–966. [Google Scholar] [CrossRef] [PubMed]
- Menda, Y.; O’Dorisio, T.M.; Howe, J.R.; Schultz, M.; Dillon, J.S.; Dick, D.; Watkins, G.L.; Ginader, T.; Bushnell, D.L.; Sunderland, J.J.; et al. Localization of Unknown Primary Site with 68Ga-DOTATOC PET/CT in Patients with Metastatic Neuroendocrine Tumor. J. Nucl. Med. 2017, 58, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.M.; Gu, X.; Ginader, T.; Breheny, P.; Sunderland, J.J. 68Ga-DOTATOC Imaging of Neuroendocrine Tumors: A Systematic Review and Metaanalysis. J. Nucl. Med. 2017, 58, 1452–1458. [Google Scholar] [CrossRef]
- Sanli, Y.; Garg, I.; Kandathil, A.; Zanetti, M.J.B.; Kuyumcu, S.; Subramaniam, R.M. Neuroendocrine Tumor Diagnosis and Man-agement: 68Ga-DOTATATE PET/CT. AJR Am. J. Roentgenol. 2018, 211, 267–277. [Google Scholar] [CrossRef]
- De Dosso, S.; Treglia, G.; Pascale, M.; Tamburello, A.; Santhanam, P.; Kroiss, A.S.; Mestre, R.P.; Saletti, P.; Giovanella, L. Detection rate of unknown primary tumour by using somatostatin receptor PET/CT in patients with metastatic neuroendocrine tumours: A meta-analysis. Endocrine 2019, 64, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.; McNeely, P.; Menda, Y. Nuclear Imaging of Neuroendocrine Tumors. Surg. Oncol. Clin. N. Am. 2020, 29, 209–221. [Google Scholar] [CrossRef]
- Ma, H.; Kan, Y.; Yang, J.-G. Clinical value of 68Ga-DOTA-SSTR PET/CT in the diagnosis and detection of neuroendocrine tumors of unknown primary origin: A systematic review and meta-analysis. Acta Radiol. 2021, 62, 1217–1228. [Google Scholar] [CrossRef]
- Hubalewska-Dydejczyk, A.; Kulig, J.; Szybinski, P.; Mikolajczak, R.; Pach, D.; Sowa-Staszczak, A.; Fröss-Baron, K.; Huszno, B. Radio-guided surgery with the use of [99mTc-EDDA/HYNIC]octreotate in intra-operative detection of neuroendocrine tumours of the gastrointestinal tract. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1545–1555. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Chauhan, A.; Rau, J.; Diebold, A.E.; Opoku-Boateng, A.; Ramcharan, T.; Boudreaux, J.P.; Woltering, E.A. Neuroendocrine tumors (NETs) of unknown primary: Is early surgical exploration and aggressive debulking justifiable? Chin. Clin. Oncol. 2016, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Kvols, L.K.; O’Connell, M.J.; Rubin, J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 1991, 68, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mitry, E.; Baudin, E.; Ducreux, M.; Sabourin, J.-C.; Rufié, P.; Aparicio, T.; Lasser, P.; Elias, D.; Duvillard, P.; Schlumberger, M.; et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br. J. Cancer 1999, 81, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Fjällskog, M.L.; Granberg, D.P.; Welin, S.L.; Eriksson, C.; Oberg, K.E.; Janson, E.T.; Eriksson, B.K. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001, 92, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Brandi, G.; Paragona, M.; Campana, D. Good performance of platinum-based chemotherapy for high-grade gastroentero-pancreatic and unknown primary neuroendocrine neoplasms. J. Chemother. 2018, 30, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Lobins, R.; Floyd, J. Small cell carcinoma of unknown primary. Semin. Oncol. 2007, 34, 39–42. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Li, J.; Zhang, X.; Zhou, J.; Wang, X.; Peng, Z.; Shen, L.; Lu, M. Etoposide and cisplatin versus irinotecan and cisplatin as the first-line therapy for patients with advanced, poorly differentiated gastroenteropancreatic neuroendocrine carcinoma: A randomized phase 2 study. Cancer 2020, 126 (Suppl. S9), 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann. Oncol. 2012, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Eads, J.R.; Catalano, P.J.; Fisher, G.A.; Rubin, D.; Iagaru, A.; Klimstra, D.S.; Konda, B.; Kwong, M.S.; Chan, J.A.; De Jesus-Acosta, A.; et al. Randomized phase II study of platinum and etoposide (EP) versus temozolomide and capecitabine (CAPTEM) in patients (pts) with advanced G3 non-small cell gastroenteropancreatic neuroendocrine neoplasms (GEPNENs): ECOG-ACRIN EA2142. J. Clin. Oncol. 2022, 40 (Suppl. S16), 4020. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Erland, J.B.; Kalman, L.A.; Schreeder, M.T.; Greco, F.A. Carcinoma of unknown primary site: Treatment with 1-hour paclitaxel, carboplatin, and extended-schedule etoposide. J. Clin. Oncol. 1997, 15, 2385–2393. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Spigel, D.R.; Litchy, S.; Greco, F.A. Phase II Trial of Paclitaxel, Carboplatin, and Etoposide in Advanced Poorly Differentiated Neuroendocrine Carcinoma: A Minnie Pearl Cancer Research Network Study. J. Clin. Oncol. 2006, 24, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Afchain, P.; Walter, T.; Tougeron, D.; Hautefeuille, V.; Monterymard, C.; Lorgis, V.; Thuillier, F.; Baudin, E.; Scoazec, J.Y.; et al. FOLFIRINEC: A randomized phase II trial of mFOLFIRINOX vs platinum-etoposide for metastatic neuroendocrine carcinoma of gastroenteropancreatic or unknown origin. Dig. Liver Dis. 2021, 53, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Riesco Martinez, M.C.; Capdevila Castillon, J.; Alonso, V.; Jimenez-Fonseca, P.; Teule, A.; Grande, E.; Sevilla, I.; Benavent, M.; Alonso-Gordoa, T.; Custodio, A.; et al. Final overall survival results from the NICE-NEC trial (GETNE-T1913): A phase II study of nivolumab and platinum-doublet chemotherapy (CT) in untreated advanced G3 neuroendocrine neoplasms (NENs) of gastro-enteropancreatic (GEP) or unknown (UK) origin. Ann. Oncol. 2022, 33 (Suppl. S7), S769. [Google Scholar] [CrossRef]
- Hentic, O.; Hammel, P.; Couvelard, A.; Rebours, V.; Zappa, M.; Palazzo, M.; Maire, F.; Goujon, G.; Gillet, A.; Lévy, P.; et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide–platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr. Relat. Cancer 2012, 19, 751–757. [Google Scholar] [CrossRef]
- Welin, S.; Sorbye, H.; Sebjornsen, S.; Knappskog, S.; Busch, C.; Öberg, K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 2011, 117, 4617–4622. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Malka, D.; Planchard, D.; Scoazec, J.Y.; Caramella, C.; Guigay, J.; Boige, V.; Leboulleux, S.; Burtin, P.; Berdelou, A.; et al. Post-first-line FOLFOX chemotherapy for grade 3 neuroendocrine carcinoma. Endocr. Relat. Cancer 2015, 22, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Takeda, Y.; Okubo, N.; Suzuki, A.; Tokuhisa, M.; Hiroshima, Y.; Ichikawa, Y. Phase II study of temozolomide monotherapy in patients with extrapulmonary neuroendocrine carcinoma. Cancer Sci. 2021, 112, 1936–1942. [Google Scholar] [CrossRef]
- Walter, T.; Lievre, A.; Coriat, R.; Malka, D.; Elhajbi, F.; Di Fiore, F.; Hentic, O.; Smith, D.; Hautefeuille, V.; Roquin, G.; et al. Bevacizumab plus FOLFIRI after failure of platinum-etoposide first-line chemotherapy in patients with advanced neuroendocrine carcinoma (PRODIGE 41-BEVANEC): A randomised, multicentre, non-comparative, open-label, phase 2 trial. Lancet Oncol. 2023, 24, 297–306. [Google Scholar] [CrossRef]
- McNamara, M.G.; Swain, J.; Craig, Z.; Sharma, R.; Faluyi, O.; Wadsley, J.; Morgan, C.; Wall, L.R.; Chau, I.; Reed, N.; et al. NET-02: A randomised, non-comparative, phase II trial of nal-IRI/5-FU or docetaxel as second-line therapy in patients with progressive poorly differentiated extra-pulmonary neuroendocrine carcinoma. eClinicalMedicine 2023, 60, 102015. [Google Scholar] [CrossRef]
- Hadoux, J.; Walter, T.; Kanaan, C.; Hescot, S.; Hautefeuille, V.; Perrier, M.; Tauveron, I.; Laboureau, S.; Cao, C.D.; Petorin, C.; et al. Second-line treatment and prognostic factors in neuroendocrine carcinoma: The RBNEC study. Endocr. Relat. Cancer 2022, 29, 569–580. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in Patients with Chemotherapy-Refractory Metastatic Merkel Cell Carcinoma: A Multicentre, Single-Group, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-Line Avelumab Treatment in Patients with Stage IV Metastatic Merkel Cell Carcinoma: A Pre-planned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Chan, J.A.; Wang, S.J.; Aggarwal, R.R.; Calabrese, S.; DeMore, A.; Fong, L.; Grabowsky, J.; Hope, T.A.; Kolli, K.P.; et al. Pembrolizumab alone and pembrolizumab plus chemotherapy in previously treated, extrapulmonary poorly differentiated neuroendocrine carcinomas. Br. J. Cancer 2023, 129, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergia, N.; Dasari, A.; Deng, M.; Litwin, S.; Al-Toubah, T.; Alpaugh, R.K.; Dotan, E.; Hall, M.J.; Ross, N.M.; Runyen, M.M.; et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: Joint analysis of two prospective, non-randomised trials. Br. J. Cancer 2020, 122, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Fottner, C.; Apostolidis, L.; Ferrata, M.; Krug, S.; Michl, P.; Schad, A.; Roth, W.; Jaeger, D.; Galle, P.R.; Weber, M.M. A phase II, open label, multicenter trial of avelumab in patients with advanced, metastatic high-grade neuroendocrine carcinomas NEC G3 (WHO 2010) progressive after first-line chemotherapy (AVENEC). J. Clin. Oncol. 2019, 37 (Suppl. S15), 4103. [Google Scholar] [CrossRef]
- Yao, J.C.; Strosberg, J.; Fazio, N.; Pavel, M.E.; Bergsland, E.; Ruszniewski, P.; Halperin, D.M.; Li, D.; Tafuto, S.; Raj, N.; et al. Spartalizumab in metastatic, well/poorly-differentiated neuroendocrine neoplasms. Endocr.-Relat. Cancer 2021, 28, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.; Rodriguez-Freixinos, V.; Doherty, M.; Wasson, K.; Iscoe, N.; Raskin, W.; Hallet, J.; Myrehaug, S.; Law, C.; Thawer, A.; et al. Avelumab in unresectable/metastatic, progressive, grade 2-3 neuroendocrine neoplasms (NENs): Combined results from NET-001 and NET-002 trials. Eur. J. Cancer 2022, 169, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Raj, N.P.; Aggarwal, R.R.; Calabrese, S.; DeMore, A.; Dhawan, M.S.; Fattah, D.; Fong, L.; Grabowsky, J.; Hope, T.A.; et al. Phase II study of pembrolizumab-based therapy in previously treated extrapulmonary poorly differentiated neuroendocrine carcinomas: Results of Part B (pembrolizumab + chemotherapy). J. Clin. Oncol. 2021, 39 (Suppl. S15), 4148. [Google Scholar] [CrossRef]
- Patel, S.P.; Mayerson, E.; Chae, Y.K.; Strosberg, J.; Wang, J.; Konda, B.; Hayward, J.; McLeod, C.M.; Chen, H.X.; Sharon, E.; et al. A phase II basket trial of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART) SWOG S1609: High-grade neuroendocrine neoplasm cohort. Cancer 2021, 127, 3194–3201. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Markman, B.; Michael, M.; Underhill, C.; Carlino, M.S.; Jackett, L.; Lum, C.; Scott, C.; Nagrial, A.; et al. Immunotherapy of ipilimumab and nivolumab in patients with advanced neuroendocrine tumours: A subgroup analysis of the CA209–538 clinical trial for rare cancers. Clin. Cancer Res. 2020, 26, 4454–4459. [Google Scholar] [CrossRef]
- Capdevila, J.; Hernando, J.; Teule, A.; Lopez, C.; Garcia-Carbonero, R.; Benavent, M.; Custodio, A.; Garcia-Alvarez, A.; Cubillo, A.; Alonso, V.; et al. Durvalumab plus tremelimumab for the treatment of advanced neuroendocrine neoplasms of gastroentero-pancreatic and lung origin. Nat. Commun. 2023, 14, 2973. [Google Scholar] [CrossRef]
- Pavel, M.E.; Fischer von Weikersthal, L.; Klöppel, G.; Krause, K.; Apostolidis, L. Safety and efficacy of everolimus (EVE) as second–line treatment in neuroendocrine neoplasms G3 (NEN G3)—An AIO phase II study (EVINEC). Ann. Oncol. 2023, 34 (Suppl. S2), S702. [Google Scholar] [CrossRef]
- Weber, M.; Apostolidis, L.; Krug, S.; Rinke, A.; Gruen, B.; Michl, P.; Gress, T.; Wagner, D.; Roth, W.; Mettler, E.; et al. Activity and safety of avelumab alone or in combination with cabozantinib in patients with advanced high grade neuroendocrine neoplasias (NEN G3) progressing after chemotherapy. The phase II, open-label, multicenter AVENEC and CABOAVENEC trials. Ann. Oncol. 2023, 34 (Suppl. S2), S702. [Google Scholar] [CrossRef]
- Capdevila Castillon, J.; Molina-Cerrillo, J.; Benavent Viñuales, M.; Garcia-Carbonero, R.; Teule, A.; Custodio, A.; Jimenez-Fonseca, P.; Lopez, C.; Hierro, C.; Carmona-Bayonas, A.; et al. Cabozantinib plus Atezolizumab in Advanced and Progressive Neoplasms of the Endocrine System: A multi-cohort Basket Phase II Trial (CABATEN/GETNE-T1914). Ann. Oncol. 2023, 34 (Suppl. S2), S498. [Google Scholar] [CrossRef]
- Lu, M.; Wang, Y.; Zhang, P.; Shen, L. Surufatinib combined with sintilimab and IBI310 in the treatment of high-grade advanced-neuroendocrine neoplasm: A single arm, open-label, multicenter study. Ann. Oncol. 2022, 33 (Suppl. S7), S770. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Phan, A.T.; Ćwikła, J.B.; Sedláčková, E.; Thanh, X.T.; Wolin, E.M.; Ruszniewski, P.; CLARINET Investigators. Lanreotide autogel/depot in advanced enteropancreatic neuroendocrine tumours: Final results of the CLARINET openlabel ex-tension study. Endocrine 2021, 71, 502–551. [Google Scholar] [CrossRef] [PubMed]
- Faiss, S.; Pape, U.F.; Böhmig, M.; Dörffel, Y.; Mansmann, U.; Golder, W.; Riecken, E.O.; Wiedenmann, B.; International Lanreotide and Interferon Alfa Study Group. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors—The International Lanreo-tide and Interferon Alfa Study Group. J. Clin. Oncol. 2003, 21, 2689–2696. [Google Scholar]
- Ito, T.; Honma, Y.; Hijioka, S.; Kudo, A.; Fukutomi, A.; Nozaki, A.; Kimura, Y.; Motoi, F.; Isayama, H.; Komoto, I.; et al. Phase II study of lanreotide autogel in Japanese patients with unresectable or metastatic well-differentiated neuroendocrine tumors. Investig. New Drugs 2017, 35, 499–508. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Mueller-Brand, J.; Paganelli, G.; Anthony, L.B.; Pauwels, S.; Kvols, L.K.; O’Dorisio, T.M.; Valkema, R.; Bodei, L.; Chinol, M.; et al. Overview of results of peptide receptor radionuclide therapy with 3 radiolabeled somatostatin analogs. J. Nucl. Med. 2005, 46, 62S–66S. [Google Scholar]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Saravana-Bawan, B.; Bajwa, A.; Paterson, J.; McEwan, A.J.B.; McMullen, T.P.W. Efficacy of 177Lu Peptide Receptor Radionuclide Therapy for the Treatment of Neuroendocrine Tumors: A Meta-analysis. Clin. Nucl. Med. 2019, 44, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Hertelendi, M.; Belguenani, O.; Cherfi, A.; Folitar, I.; Kollar, G.; Polack, B.D. Efficacy and Safety of [177Lu]Lu-DOTA-TATE in Adults with Inoperable or Metastatic Somatostatin Receptor-Positive Pheochromocytomas/Paragangliomas, Bronchial and Unknown Origin Neuroendocrine Tumors, and Medullary Thyroid Carcinoma: A Systematic Literature Review. Biomedicines 2023, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Forrer, F.; Waldherr, C.; Maecke, H.R.; Mueller-Brand, J. Targeted radionuclide therapy with 90Y-DOTATOC in patients with neuroendocrine tumors. J. Anticancer Res. 2006, 26, 703–707. [Google Scholar]
- Seregni, E.; Maccauro, M.; Chiesa, C.; Mariani, L.; Pascali, C.; Mazzaferro, V.; De Braud, F.; Buzzoni, R.; Milione, M.; Lorenzoni, A.; et al. Treatment with tandem [90Y]DOTATATE and [177Lu]DOTA-TATE of neuroendocrine tumours refractory to conventional therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Frilling, A.; Weber, F.; Saner, F.; Bockisch, A.; Hofmann, M.; Mueller-Brand, J.; Broelsch, C.E. Treatment with (90)Y- and (177)Lu-DOTATOC in patients with metastatic neuroendocrine tumors. Surgery 2006, 140, 968–977; Discussion 976–977. [Google Scholar] [CrossRef] [PubMed]
- Thang, S.P.; Lung, M.S.; Kong, G.; Hofman, M.S.; Callahan, J.; Michael, M.; Hicks, R.J. Peptide receptor radionuclidetherapy (PRRT) in European Neuroendocrine Tumour Society (ENETS)grade 3 (G3) neuroendocrine neoplasia (NEN)–a single-institution retro-spective analysis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 262–277. [Google Scholar] [CrossRef]
- Carlsen, E.A.; Fazio, N.; Granberg, D.; Grozinsky-Glasberg, S.; Ahmadzadehfar, H.; Grana, C.M.; Zandee, W.T.; Cwikla, J.; Walter, M.A.; Oturai, P.S.; et al. Peptide receptor radionuclidetherapy in gastroenteropancreatic NEN G3: A multicenter cohortstudy. Endocr. Relat. Cancer 2019, 26, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, S.; Severi, S.; Ianniello, A.; Sansovini, M.; Ambrosetti, A.; Bongiovanni, A.; Scarpi, E.; Di Mauro, F.; Rossi, A.; Matteucci, F.; et al. Investigation of receptor radionuclide therapy with (177)Lu-DOTATATE in patients with GEP-NEN and a high Ki-67 proliferation index. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 923–930. [Google Scholar] [CrossRef]
- Zhang, J.; Kulkarni, H.R.; Singh, A.; Niepsch, K.; Müller, D.; Baum, R.P. Peptide receptor radionuclide therapy in grade 3 neuro-endocrine neoplasms: Safety and survival analysis in 69 patients. J. Nucl. Med. 2019, 60, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Phan, A.T.; Chang, D.Z.; Wolff, R.A.; Hess, K.; Gupta, S.; Jacobs, C.; Mares, J.E.; Landgraf, A.N.; Rashid, A.; et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase II trial. J. Clin. Oncol. 2008, 26, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Catena, L.; Pusceddu, S.; Spada, F.; Iannacone, C.; Sarno, I.; Di Menna, G.; Dottorini, L.; Marte, A.M. Everolimus in Combination with Octreotide Long-Acting Repeatable in a First-Line Setting for Patients with Neuroendocrine Tumors: A 5-Year Update. Neuroendocrinology 2018, 106, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Carnaghi, C.; Buzzoni, R.; Pommier, R.F.; Raderer, M.; Tomasek, J.; Lahner, H.; Valle, J.W.; Voi, M.; Bubuteishvili-Pacaud, L.; et al. Everolimus in Neuroendocrine Tumors of the Gastrointestinal Tract and Unknown Primary. Neuroendocrinology 2018, 106, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, L.; Zhou, Z.; Li, J.; Bai, C.; Chi, Y.; Li, Z.; Xu, N.; Li, E.; Liu, T.; et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Geyer, S.; Ou, F.S.; Knopp, M.; Behr, S.; Zemla, T.; Acoba, J.; Shergill, A.; Wolin, E.M.; Halfdanarson, T.R.; et al. Alliance A021602: Phase III, double-blinded study of cabozantinib versus placebo for advanced neuroendocrine tumors (NET) after progression on prior therapy (CABINET). Ann. Oncol. 2023, 34 (Suppl. S2), S1292. [Google Scholar] [CrossRef]
- Chan, J.A.; Stuart, K.; Earle, C.C.; Clark, J.W.; Bhargava, P.; Miksad, R.; Blaszkowsky, L.; Enzinger, P.C.; Meyerhardt, J.A.; Zheng, H.; et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2012, 30, 2963–2968. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Farooqui, Z.; Murray, L.A.; Weiss, H.L.; Myint, Z.W.; Raajasekar, A.K.A.; Evers, B.M.; Arnold, S.; Anthony, L. Capecitabine and Temozolomide in Neuroendocrine Tumor of Unknown Primary. J. Oncol. 2018, 2018, 3519247. [Google Scholar] [CrossRef] [PubMed]
- Spada, F.; Maisonneuve, P.; Fumagalli, C.; Marconcini, R.; Gelsomino, F.; Antonuzzo, L.; Campana, D.; Puliafito, I.; Rossi, G.; Faviana, P.; et al. Temozolomide alone or in combination with capecitabine in patients with advanced neuroendocrine neoplasms: An Italian multicenter real-world analysis. Endocrine 2020, 72, 268–278. [Google Scholar] [CrossRef]
- Cives, M.; Ghayouri, M.; Morse, B.; Brelsford, M.; Black, M.; Rizzo, A.; Meeker, A.; Strosberg, J. Analysis of potential response pre-dictors to capecitabine/temozolomide in metastatic pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 759–767. [Google Scholar] [CrossRef]
- Kulke, M.H.; Hornick, J.L.; Frauenhoffer, C.; Hooshmand, S.; Ryan, D.P.; Enzinger, P.C.; Meyerhardt, J.A.; Clark, J.W.; Stuart, K.; Fuchs, C.S.; et al. O 6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin. Cancer Res. 2009, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.; Lecomte, T.; Hadoux, J.; Niccoli, P.; Saban-Roche, L.; Gaye, E.; Guimbaud, R.; Baconnier, M.; Hautefeuille, V.; Cao, C.D.; et al. Alkylating agent-based vs oxaliplatin-based chemotherapy in neuroendocrine tumours according to the O6-methylguanine-DNA methyltransferase (MGMT) status: A randomized phase II study (MGMT-NET) on behalf of the French Group of Endocrine Tumors (GTE) and ENDOCAN-RENATEN network. Ann. Oncol. 2023, 34 (Suppl. S2), S1292–S1293. [Google Scholar]
- Meyer, T.; Qian, W.; Caplin, M.E.; Armstrong, G.; Lao-Sirieix, S.-H.; Hardy, R.; Valle, J.W.; Talbot, D.C.; Cunningham, D.; Reed, N.; et al. Capecitabine and streptozocin±cisplatin in advanced gastroenteropancreatic neuroendocrine tumours. Eur. J. Cancer 2014, 50, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Catena, L.; Procopio, G.; De Dosso, S.; Bichisao, E.; Ferrari, L.; Martinetti, A.; Platania, M.; Verzoni, E.; Formisano, B.; et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother. Pharmacol. 2007, 59, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Spada, F.; Antonuzzo, L.; Marconcini, R.; Radice, D.; Antonuzzo, A.; Ricci, S.; Di Costanzo, F.; Fontana, A.; Gelsomino, F.; Luppi, G.; et al. Oxaliplatin-Based Chemotherapy in Advanced Neuroendocrine Tumors: Clinical Outcomes and Preliminary Correlation with Biological Factors. Neuroendocrinology 2016, 103, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, S.; Fonck, M.; Bécouarn, Y.; Brunet, R. Dacarbazine, fluorouracil, and leucovorin in patients with advanced neuroen-docrine tumors: A phase II trial. Am. J. Clin. Oncol. 1998, 21, 237–240. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.P.; Shapira-Frommer, R.; Bergsland, E.; Shah, M.; Fakih, M.; Takahashi, S.; et al. Efficacy and safety of pembrolizumab in previously treated advanced neuroendocrine tumors: Results from the phase 2 KEYNOTE-158 study. Clin. Cancer Res. 2020, 26, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Othus, M.; Chae, Y.K.; Giles, F.J.; Hansel, D.E.; Singh, P.P.; Fontaine, A.; Shah, M.H.; Kasi, A.; Baghdadi, T.A.; et al. A phase II basket trial of dual anti–CTLA-4 and anti–PD-1 blockade in rare tumors (DART SWOG 1609) in patients with non-pancreatic neuroendocrine tumors. Clin. Cancer Res. 2020, 26, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, P.; Zhang, Y.; Li, Z.; Gong, J.; Li, J.; Li, J.; Li, Y.; Zhang, X.; Lu, Z.; et al. Efficacy, safety, and biomarkers of toripalimab in patients with recurrent or metastatic neuroendocrine neoplasms: A multiple-center phase Ib trial. Clin. Cancer Res. 2020, 26, 2337–2345. [Google Scholar] [CrossRef]
- Cavalcoli, F.; Rausa, E.; Conte, D.; Nicolini, A.F.; Massironi, S. Is there still a role for the hepatic locoregional treatment of meta-static neuroendocrine tumors in the era of systemic targeted therapies? World J. Gastroenterol. 2017, 23, 2640–2650. [Google Scholar] [CrossRef]
- Kim, H.S.; Shaib, W.L.; Zhang, C.; Nagaraju, G.P.; Wu, C.; Alese, O.B.; Chen, Z.; Brutcher, E.; Renfroe, M.; El-Rayes, B.F. Phase 1b study of pasireotide, everolimus, and selective internal radioembolization therapy for unresectable neuroendocrine tumors with hepatic metastases. Cancer 2018, 124, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Quinn, R.; Glenn, D.M.; Janssen, J.; Tong, D.; Liaw, W.; Morris, D.L. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer 2008, 113, 921–929. [Google Scholar] [CrossRef]
- Blonski, W.C.; Reddy, K.R.; Shaked, A.; Siegelman, E.; Metz, D.C. Liver transplantation for metastatic neuroendocrine tumor: A case report and review of the literature. World J. Gastroenterol. 2005, 11, 7676–7683. [Google Scholar] [CrossRef] [PubMed]
- Moradi, A.; Lamsehchi, N.; Khaki, S.; Nasiri-Toosi, M.; Jafarian, A. Liver Transplant for Patients with Neuroendocrine Tumor: A Report of 2 Exceptional Cases and Literature Review. Exp. Clin. Transplant. 2023, 21, 578–585. [Google Scholar]
- Puccini, A.; Poorman, K.; Salem, M.E.; Soldato, D.; Seeber, A.; Goldberg, R.M.; Shields, A.F.; Xiu, J.; Battaglin, F.; Berger, M.D.; et al. Comprehensive Genomic Profiling of Gastroenteropancreatic Neuroendocrine Neoplasms (GEP-NENs). Clin. Cancer Res. 2020, 26, 5943–5951. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Anton-Pascual, B.; Modrego, A.; Del Carmen Riesco-Martinez, M.; Lens-Pardo, A.; Carretero-Puche, C.; Ru-bio-Cuesta, B.; Soldevilla, B. Advances in the Treatment of Gastroenteropancreatic Neuroendocrine Carcinomas: Are we Moving Forward? Endocr. Rev. 2023, 44, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Basturk, O.; Sue, J.J.B.; Klimstra, D.S. A Practical Approach to the Classification of WHO Grade 3 (G3) Well-differentiated Neuroendocrine Tumor (WD-NET) and Poorly Differentiated Neuroendocrine Carcinoma (PD-NEC) of the Pancreas. Am. J. Surg. Pathol. 2016, 40, 1192–1202. [Google Scholar] [CrossRef]
- Derks, J.L.; Leblay, N.; Lantuejoul, S.; Dingemans, A.C.; Speel, E.M.; Fernandez-Cuesta, L. New Insights into the Molecular Characteristics of Pulmonary Carcinoids and Large Cell Neuroendocrine Carcinomas, and the Impact on Their Clinical Management. J. Thorac. Oncol. 2018, 13, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Corti, F.; Milione, M.; Centonze, G.; Prinzi, N.; Torchio, M.; de Braud, F. Are Cyclin-Dependent Kinase 4/6 Inhibitors Without Future in Neuroendocrine Tumors? Oncologist 2020, 25, e1257–e1258. [Google Scholar] [CrossRef]
- Hendifar, A.E.; Ramirez, R.A.; Anthony, L.B.; Liu, E. Current Practices and Novel Techniques in the Diagnosis and Management of Neuroendocrine Tumors of Unknown Primary. Pancreas 2019, 48, 1111–1118. [Google Scholar] [CrossRef]
- Saller, J.J.; Haider, M.; Al-Diffalha, S.; Coppola, D. Benefit of Gene Expression Profiling in Gastrointestinal Neuroendocrine Tumors of Unknown Primary Origin. Anticancer Res. 2022, 42, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Gershenhorn, B.; Tran, P.; Lee, T.K.; Erlander, M.G.; Gowen, K.; Schrock, A.B.; Morosini, D.; Ross, J.S.; Miller, V.A.; et al. BRAFV600E Mutations in High-Grade Colorectal Neuroendocrine Tumors May Predict Responsiveness to BRAF-MEK Combination Therapy. Cancer Discov. 2016, 6, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Burkart, J.; Owen, D.; Shah, M.H.; Abdel-Misih, S.R.Z.; Roychowdhury, S.; Wesolowski, R.; Haraldsdottir, S.; Reeser, J.W.; Samorodnitsky, E.; Smith, A.; et al. Targeting BRAF Mutations in High-Grade Neuroendocrine Carcinoma of the Colon. J. Natl. Compr. Cancer Netw. 2018, 16, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Ricco, G.; Seminerio, R.; Andrini, E.; Malvi, D.; Gruppioni, E.; Altimari, A.; Zagnoni, S.; Campana, D.; Lamberti, G. BRAF V600E-mutated large cell neuroendocrine carcinoma responding to targeted therapy: A case report and review of the literature. Anti-Cancer Drugs 2023, 34, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Tamragouri, K.B.; Chung, J.; Lin, X.; Miller, V.; Ali, S.M.; Giles, F.J. Large-Cell Neuroendocrine Carcinoma of the Lung: A Focused Analysis of BRAF Alterations and Case Report of a BRAF Non-V600–Mutated Tumor Responding to Targeted Therapy. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Balbach, M.L.; Eisenberg, R.; Iams, W.T. De Novo KRAS G12C-Mutant SCLC: A Case Report. JTO Clin. Res. Rep. 2022, 3, 100306. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Omori, C.; Morikawa, H.; Shinohara, K.; Shimamura, S.; Ohkoshi, H.; Uchida, Y.; Inoue, T.; Kondo, T.; Ikemura, S.; et al. The First Case Report of Effective Treatment with Sotorasib for Metastatic Atypical Lung Carcinoid Harboring KRAS G12C Mutation and Aggressive Disseminated Lung Metastasis: A Case Report. JTO Clin. Res. Rep. 2023, 5, 100620. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zheng, M.; Jin, Y.; Shen, X.; Shan, L.; Shen, L.; Sun, Y.; Chen, H.; Li, Y. ALK-Rearrangement Neuroendocrine Carcinoma of the Lung: A Comprehensive Study of a Rare Case Series and Review of Literature. OncoTargets Ther. 2018, 11, 4991–4998. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Fujita, A.; Saikai, T.; Takabatake, H.; Sotoshiro, M.; Sekine, K.; Kawana, A. Large Cell Neuroendocrine Carcinoma Harboring an Anaplastic Lymphoma Kinase (ALK) Rearrangement with Response to Alectinib. Intern. Med. 2018, 57, 713–716. [Google Scholar] [CrossRef]
- Shimizu, N.; Akashi, Y.; Fujii, T.; Shiono, H.; Yane, K.; Kitahara, T.; Ohta, Y.; Kakudo, K.; Wakasa, T. Use of ALK Immunohistochemistry for Optimal Therapeutic Strategy of Pulmonary Large-Cell Neuroendocrine Carcinoma and Identification of a Novel KIF5B–ALK Fusion Oncokinase. Anticancer Res. 2019, 39, 413–420. [Google Scholar] [CrossRef]
- Nakajima, M.; Uchiyama, N.; Shigemasa, R.; Matsumura, T.; Matsuoka, R.; Nomura, A. Atypical Carcinoid Tumor with Anaplastic Lymphoma Kinase (ALK) Rearrangement Successfully Treated by an ALK Inhibitor. Intern. Med. 2016, 55, 3151–3153. [Google Scholar] [CrossRef] [PubMed]
- Sigal, D.S.; Bhangoo, M.S.; Hermel, J.A.; Pavlick, D.C.; Frampton, G.; Miller, V.A.; Ross, J.S.; Ali, S.M. Comprehensive Genomic Profiling Identifies Novel NTRK Fusions in Neuroendocrine Tumors. Oncotarget 2018, 9, 35809–35812. [Google Scholar] [CrossRef] [PubMed]
- Bazhenova, L.; Liu, S.; Lin, J.; Lu, S.; Drilon, A.; Chawla, S.; Fakih, M.; Krzakowski, M.; Paz-Ares, L.; Blakely, C.; et al. Efficacy and safety of entrectinib in patients with locally advanced/metastatic NTRK fusion-positive (NTRK-FP) solid tumours. Ann. Oncol. 2021, 32 (Suppl. S5), S598–S599. [Google Scholar] [CrossRef]
- Demetri, G.D.; De Braud, F.; Drilon, A.; Siena, S.; Patel, M.R.; Cho, B.C.; Liu, S.; Ahn, M.-J.; Chiu, C.-H.; Lin, J.J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Patients With NTRK Fusion-Positive Solid Tumors. Clin. Cancer Res. 2022, 28, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Borazanci, E.; Shaw, A.T.; Katayama, R.; Shimizu, Y.; Zhu, V.W.; Sun, T.Y.; Wakelee, H.A.; Madison, R.; Schrock, A.B.; et al. Phase I First-In-Human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients With Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 4785–4794. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Chiosea, S.I.; Challinor, S.M.; Nikiforova, M.N.; Bauman, J.E. Primary RET-mutated lung neuroendocrine carcinoma in MEN2B: Response to RET-targeted therapy. Endocr. Relat. Cancer 2015, 22, L19–L22. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Zaemes, J.; Ozdemirli, M.; Kim, C. Response to selpercatinib in a patient with RET fusion-positive pulmonary large-cell neuroendocrine carcinoma: A case report. Front. Oncol. 2023, 13, 1134151. [Google Scholar] [CrossRef] [PubMed]
- Prisciandaro, M.; Antista, M.; Raimondi, A.; Corti, F.; Morano, F.; Centonze, G.; Sabella, G.; Mangogna, A.; Randon, G.; Pagani, F.; et al. Biomarker Landscape in Neuroendocrine Tumors with High-Grade Features: Current Knowledge and Future Perspective. Front. Oncol. 2022, 12, 780716. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Hong, D.S.; Hann, C.L.; Farago, A.F.; Beltran, H.; Waqar, S.N.; Hendifar, A.E.; Anthony, L.B.; Taylor, M.H.; Bryce, A.H.; et al. A phase I/II study of rovalpituzumab tesirine in delta-like 3—Expressing advanced solid tumors. npj Precis. Oncol. 2021, 5, 74. [Google Scholar] [CrossRef]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab tesirine as a maintenance therapy after first-line platinum-based chemotherapy in patients with exten-sive-stage-SCLC: Results from the phase 3 MERU study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1570–1581. [Google Scholar]
- Blackhall, F.; Jao, K.; Greillier, L.; Cho, B.C.; Penkov, K.; Reguart, N.; Majem, M.; Nackaerts, K.; Syrigos, K.; Hansen, K.; et al. Efficacy and safety of rovalpituzumab tesirine compared with topotecan as second-line therapy in DLL3-high SCLC: Results from the phase 3 TAHOE study. J. Thorac. Oncol. 2021, 16, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Sahnane, N.; Furlan, D.; Monti, M.; Romualdi, C.; Vanoli, A.; Vicari, E.; Solcia, E.; Capella, C.; Sessa, F.; La Rosa, S. Microsatellite Unstable Gastrointestinal Neuroendocrine Carcinomas: A New Clinicopathologic Entity. Endocr.-Relat. Cancer 2014, 22, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; Acosta, A.D.J.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: Updated analysis from the phase II KEYNOTE-158 study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Geurts, B.; Battaglia, T.; Henegouwen, J.v.B.; Zeverijn, L.; Hoes, L.; van der Wijngaart, H.; de Wit, G.; Roepman, P.; Jansen, A.; de Leng, W.; et al. Durvalumab in advanced, pre-treated microsatellite instability-high solid tumors: Results of a tumor-agnostic DRUP trial cohort. Ann. Oncol. 2022, 33 (Suppl. S7), S594. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D.; et al. Prevalence of High Tumor Mutational Burden and Association with Survival in Patients With Less Common Solid Tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Phase | Treatments | Setting | Primary Endpoint | Status | Estimated Completion Date |
|---|---|---|---|---|---|---|
| NCT04325425 (FOLFIRINEC) | II | mFOLFIRINOX vs. Platinum–Etoposide | 1st line GEP or UPO NEC | PFS | Recruiting | September 2024 |
| NCT03980925 (NICE-NEC) | II | Nivolumab/Carboplatin–Etoposide | 1st line G3 GEP or UPO NEN | 12 months-OS | Active, not recruiting | December 2023 |
| NCT02820857 (BEVANEC) | II | FOLFIRI/Bevacizumab vs. FOLFIRI | Pretreated GEP or UPO NEC | Proportion of pts alive after 6 months | Active, not recruiting | September 2024 |
| NCT03736720 | II | Liposomal Irinotecan/Fluorouracil/Leucovorin | Pretreated GEP or UPO NEC | ORR | Active, not recruiting | June 2026 |
| NCT04412629 | II | Cabozantinib | Pretreated High Grade NENs including UPO | ORR | Recruiting | November 2024 |
| NCT04525638 | II | Nivolumab plus 177Lu-Dotatate | Pretreated SSTR positive NET/NEC G3 including UPO | ORR | Recruiting | September 2024 |
| NCT02628067 (Keynote 158) | II | Pembrolizumab | TMB-high /MSI-H solid tumors including NEN | ORR | Recruiting | October 2026 |
| NCT05882058 (DAREON™-5) | II | BI 764532 | NECs | ORR, TEAEs | Recruiting | July 2025 |
| NCT02925234 (DRUP) | II | Targeted therapies Basket trial | Solid tumors including NEN | % pts treated based on molecular profile; ORR; G ≥ 3/serious TRAEs | Recruiting | December 2027 |
| NCT04589845 (TAPISTRY) | II | Targeted therapies or immunotherapies Basket trial | Solid tumors including NEN | ORR | Recruiting | September 2032 |
| NCT02568267 (STARTRK-2) | II | Entrectinib | NTRK 1/2/3, ROS1, or ALK rearranged solid tumors including NEN | ORR | Active, not recruiting | April 2025 |
| NCT03157128 (LIBRETTO-001) | I/II | Selpercatinib | RET Fusion-Positive solid tumors including NEN | MTD, RP2D, ORR | Recruiting | February 2026 |
| NCT03037385 (ARROW) | I/II | Pralsetinib | RET altered solid tumors including NEN | MTD, safety, ORR | Active, not recruiting | December 2023 |
| NCT04427787 (LOLA) | II | Lanreotide/ Cabozantinib | Pretreated/not pretreated GEP, thoracic or UPO-NET | Safety, ORR | Recruiting | November 2023 |
| NCT04544098 | Early I | intraarterial/ intravenous 177Lu-DOTATATE | GEP, Bronchial or UPO NET | nr of pts who completed 2 IA injections; ORR | Recruiting | September 2024 |
| NCT05249114 | I | Cabozantinib plus 177Lu-Dotatate | SSTR2 positive NET including UPO NET | MTD | Recruiting | December 2027 |
| NCT05554003 (MeTe) | II | Metronomic Temozolomide | NETs including UPO NETs in unfit patients | PFS | Recruiting | December 2024 |
| Molecular Target | Targeted Therapies | Level of Evidence | References |
|---|---|---|---|
| BRAF V600E | BRAF-MEK inhibitors | Phase I/II trials Case reports | [122,123,124,125] |
| KRAS | KRAS G12C inhibitors | Case-reports | [126,127] |
| ALK | ALK inhibitors | Case-series | [128,129,130,131] |
| NTRK | NTRK inhibitors, NTRK/ROS1 inhibitors | Phase II trials | [132,133,134,135] |
| RET | RET kinase inhibitors | Phase I/II trials | [136,137,138] |
| DLL3 | DLL3-targeted antibody-drug conjugate, DLL3-targeting T-cell engager | Phase I/II trials Phase III trials | [140,141,142] |
| H-MSI | ICIs | Phase II trials | [144,145] |
| TMB | ICIs | Phase II trials | [147] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corti, F.; Rossi, R.E.; Cafaro, P.; Passarella, G.; Turla, A.; Pusceddu, S.; Coppa, J.; Oldani, S.; Guidi, A.; Longarini, R.; et al. Emerging Treatment Options for Neuroendocrine Neoplasms of Unknown Primary Origin: Current Evidence and Future Perspectives. Cancers 2024, 16, 2025. https://doi.org/10.3390/cancers16112025
Corti F, Rossi RE, Cafaro P, Passarella G, Turla A, Pusceddu S, Coppa J, Oldani S, Guidi A, Longarini R, et al. Emerging Treatment Options for Neuroendocrine Neoplasms of Unknown Primary Origin: Current Evidence and Future Perspectives. Cancers. 2024; 16(11):2025. https://doi.org/10.3390/cancers16112025
Chicago/Turabian StyleCorti, Francesca, Roberta Elisa Rossi, Pietro Cafaro, Gaia Passarella, Antonella Turla, Sara Pusceddu, Jorgelina Coppa, Simone Oldani, Alessandro Guidi, Raffaella Longarini, and et al. 2024. "Emerging Treatment Options for Neuroendocrine Neoplasms of Unknown Primary Origin: Current Evidence and Future Perspectives" Cancers 16, no. 11: 2025. https://doi.org/10.3390/cancers16112025
APA StyleCorti, F., Rossi, R. E., Cafaro, P., Passarella, G., Turla, A., Pusceddu, S., Coppa, J., Oldani, S., Guidi, A., Longarini, R., & Cortinovis, D. L. (2024). Emerging Treatment Options for Neuroendocrine Neoplasms of Unknown Primary Origin: Current Evidence and Future Perspectives. Cancers, 16(11), 2025. https://doi.org/10.3390/cancers16112025