
Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. The DNA Damage Response (DDR)
3. IR-Induced DNA Damage Repair
4. Targeting Cell-Cycle Checkpoints and DNA Repair Pathways to Enhance Radiosensitivity in CRC
4.1. ATM and ATR
4.2. CHK1 and CHK2
4.3. WEE1
4.4. DNA-PKcs/NHEJ
4.5. HR
4.6. PARP1
5. Clinical Trials of DDR and DNA Repair Pathway Inhibitors in CRC Patients
6. Combination Therapies
6.1. Combination of Different DDR Inhibitors
6.2. Combination of DDR Inhibitors with Immunotherapy and Radiotherapy
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Arnold, D.; Taniguchi, H.; Pentheroudakis, G.; Yamazaki, K.; Xu, R.H.; Kim, T.W.; Ismail, F.; Tan, I.B.; Yeh, K.H.; et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 44–70. [Google Scholar] [CrossRef]
- Cercek, A.; Roxburgh, C.S.D.; Strombom, P.; Smith, J.J.; Temple, L.K.F.; Nash, G.M.; Guillem, J.G.; Paty, P.B.; Yaeger, R.; Stadler, Z.K.; et al. Adoption of Total Neoadjuvant Therapy for Locally Advanced Rectal Cancer. JAMA Oncol. 2018, 4, e180071. [Google Scholar] [CrossRef]
- Patel, P.A. Evolution of 5-fluorouracil-based chemoradiation in the management of rectal cancer. Anti-Cancer Drugs 2011, 22, 311–316. [Google Scholar] [CrossRef]
- Toulany, M.; Rodemann, H.P. Membrane receptor signaling and control of DNA repair after exposure to ionizing radiation. Nuklearmedizin. Nucl. Med. 2010, 49 (Suppl. S1), S26–S30. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.W.; Wang, J.Y.; Hung, W.C.; Peng, G.; Tsai, Y.L.; Chang, T.M.; Chai, C.Y.; Lin, C.H.; Pan, M.R. G9a governs colon cancer stem cell phenotype and chemoradioresistance through PP2A-RPA axis-mediated DNA damage response. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2017, 124, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef]
- Anuja, K.; Chowdhury, A.R.; Saha, A.; Roy, S.; Rath, A.K.; Kar, M.; Banerjee, B. Radiation-induced DNA damage response and resistance in colorectal cancer stem-like cells. Int. J. Radiat. Biol. 2019, 95, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef]
- García-Santisteban, I.; Llopis, A.; Krenning, L.; Vallejo-Rodríguez, J.; van den Broek, B.; Zubiaga, A.M.; Medema, R.H. Sustained CHK2 activity, but not ATM activity, is critical to maintain a G1 arrest after DNA damage in untransformed cells. BMC Biol. 2021, 19, 35. [Google Scholar] [CrossRef]
- Mian, E.; Wiesmüller, L. Phenotypic Analysis of ATM Protein Kinase in DNA Double-Strand Break Formation and Repair. Methods Mol. Biol. 2017, 1599, 317–334. [Google Scholar] [CrossRef]
- Ma, M.; Rodriguez, A.; Sugimoto, K. Activation of ATR-related protein kinase upon DNA damage recognition. Curr. Genet. 2020, 66, 327–333. [Google Scholar] [CrossRef]
- Ditano, J.P.; Sakurikar, N.; Eastman, A. Activation of CDC25A phosphatase is limited by CDK2/cyclin A-mediated feedback inhibition. Cell Cycle 2021, 20, 1308–1319. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Rubin, S.M.; Sage, J.; Skotheim, J.M. Integrating Old and New Paradigms of G1/S Control. Mol. Cell 2020, 80, 183–192. [Google Scholar] [CrossRef]
- Peng, G.; Cao, R.B.; Li, Y.H.; Zou, Z.W.; Huang, J.; Ding, Q. Alterations of cell cycle control proteins SHP-1/2, p16, CDK4 and cyclin D1 in radioresistant nasopharyngeal carcinoma cells. Mol. Med. Rep. 2014, 10, 1709–1716. [Google Scholar] [CrossRef][Green Version]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Hernández-Monge, J.; Rousset-Roman, A.B.; Medina-Medina, I.; Olivares-Illana, V. Dual function of MDM2 and MDMX toward the tumor suppressors p53 and RB. Genes Cancer 2016, 7, 278–287. [Google Scholar] [CrossRef][Green Version]
- Li, S.J.; Liang, X.Y.; Li, H.J.; Li, W.; Zhou, L.; Chen, H.Q.; Ye, S.G.; Yu, D.H.; Cui, J.W. Low-dose irradiation promotes proliferation of the human breast cancer MDA-MB-231 cells through accumulation of mutant P53. Int. J. Oncol. 2017, 50, 290–296. [Google Scholar] [CrossRef][Green Version]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef]
- Cheng, Q.; Cross, B.; Li, B.; Chen, L.; Li, Z.; Chen, J. Regulation of MDM2 E3 ligase activity by phosphorylation after DNA damage. Mol. Cell. Biol. 2011, 31, 4951–4963. [Google Scholar] [CrossRef]
- Mansilla, S.F.; de la Vega, M.B.; Calzetta, N.L.; Siri, S.O.; Gottifredi, V. CDK-Independent and PCNA-Dependent Functions of p21 in DNA Replication. Genes 2020, 11, 593. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Q.; Guo, Z.; Wang, Y.; Wang, L.; Liu, X.; Lu, M.; Ju, L.; Xiao, Y.; Wang, X. Inhibition of MELK produces potential anti-tumour effects in bladder cancer by inducing G1/S cell cycle arrest via the ATM/CHK2/p53 pathway. J. Cell. Mol. Med. 2020, 24, 1804–1821. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef]
- Errico, A.; Costanzo, V. Mechanisms of replication fork protection: A safeguard for genome stability. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 222–235. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G(2) checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef]
- Sadeghi, H.; Golalipour, M.; Yamchi, A.; Farazmandfar, T.; Shahbazi, M. CDC25A pathway toward tumorigenesis: Molecular targets of CDC25A in cell-cycle regulation. J. Cell. Biochem. 2019, 120, 2919–2928. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef]
- Stukenberg, P.T.; Burke, D.J. Connecting the microtubule attachment status of each kinetochore to cell cycle arrest through the spindle assembly checkpoint. Chromosoma 2015, 124, 463–480. [Google Scholar] [CrossRef]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef]
- Allan, L.A.; Clarke, P.R. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 2007, 26, 301–310. [Google Scholar] [CrossRef]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Geuting, V.; Löbrich, M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2011, 101, 7–12. [Google Scholar] [CrossRef]
- Rouhani, M. Modeling the interplay between DNA-PK, Artemis, and ATM in non-homologous end-joining repair in G1 phase of the cell cycle. J. Biol. Phys. 2019, 45, 127–146. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef]
- Andrade, P.; Martín, M.J.; Juárez, R.; López de Saro, F.; Blanco, L. Limited terminal transferase in human DNA polymerase mu defines the required balance between accuracy and efficiency in NHEJ. Proc. Natl. Acad. Sci. USA 2009, 106, 16203–16208. [Google Scholar] [CrossRef]
- Capp, J.P.; Boudsocq, F.; Bertrand, P.; Laroche-Clary, A.; Pourquier, P.; Lopez, B.S.; Cazaux, C.; Hoffmann, J.S.; Canitrot, Y. The DNA polymerase lambda is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006, 34, 2998–3007. [Google Scholar] [CrossRef]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Piazza, A.; Heyer, W.D. Homologous Recombination and the Formation of Complex Genomic Rearrangements. Trends Cell Biol. 2019, 29, 135–149. [Google Scholar] [CrossRef]
- Li, J.; Holzschu, D.L.; Sugiyama, T. PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase δ, η, and ζ activities. Proc. Natl. Acad. Sci. USA 2013, 110, 7672–7677. [Google Scholar] [CrossRef]
- Maloisel, L.; Fabre, F.; Gangloff, S. DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol. Cell. Biol. 2008, 28, 1373–1382. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef]
- Giovannini, S.; Weller, M.C.; Repmann, S.; Moch, H.; Jiricny, J. Synthetic lethality between BRCA1 deficiency and poly(ADP-ribose) polymerase inhibition is modulated by processing of endogenous oxidative DNA damage. Nucleic Acids Res. 2019, 47, 9132–9143. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 2014, 21, 251–259. [Google Scholar] [CrossRef]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef]
- Wang, X.; Chu, H.; Lv, M.; Zhang, Z.; Qiu, S.; Liu, H.; Shen, X.; Wang, W.; Cai, G. Structure of the intact ATM/Tel1 kinase. Nat. Commun. 2016, 7, 11655. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Lu, Y.; Gao, J.; Lu, Y. Downregulated Ku70 and ATM associated to poor prognosis in colorectal cancer among Chinese patients. OncoTargets Ther. 2014, 7, 1955–1961. [Google Scholar] [CrossRef]
- Tang, S.; Li, Z.; Yang, L.; Shen, L.; Wang, Y. A potential new role of ATM inhibitor in radiotherapy: Suppressing ionizing Radiation-Activated EGFR. Int. J. Radiat. Biol. 2020, 96, 461–468. [Google Scholar] [CrossRef]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef]
- Lin, C.; Yu, Y.; Zhao, H.G.; Yang, A.; Yan, H.; Cui, Y. Combination of quercetin with radiotherapy enhances tumor radiosensitivity in vitro and in vivo. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2012, 104, 395–400. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Petermann, E.; Yates, E.; Brown, J.; Lau, A.; Stankovic, T. Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet 2015, 385 (Suppl. S1), S58. [Google Scholar] [CrossRef]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- González Besteiro, M.A.; Gottifredi, V. The fork and the kinase: A DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 2015, 763, 168–180. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Matthews, T.P.; Jones, A.M.; Collins, I. Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies. Expert Opin. Drug Discov. 2013, 8, 621–640. [Google Scholar] [CrossRef]
- Chen, Z.; Xiao, Z.; Gu, W.Z.; Xue, J.; Bui, M.H.; Kovar, P.; Li, G.; Wang, G.; Tao, Z.F.; Tong, Y.; et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int. J. Cancer 2006, 119, 2784–2794. [Google Scholar] [CrossRef]
- Kleiman, L.B.; Krebs, A.M.; Kim, S.Y.; Hong, T.S.; Haigis, K.M. Comparative analysis of radiosensitizers for K-RAS mutant rectal cancers. PLoS ONE 2013, 8, e82982. [Google Scholar] [CrossRef]
- Tao, Y.; Leteur, C.; Yang, C.; Zhang, P.; Castedo, M.; Pierré, A.; Golsteyn, R.M.; Bourhis, J.; Kroemer, G.; Deutsch, E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle 2009, 8, 1196–1205. [Google Scholar] [CrossRef]
- Manic, G.; Signore, M.; Sistigu, A.; Russo, G.; Corradi, F.; Siteni, S.; Musella, M.; Vitale, S.; De Angelis, M.L.; Pallocca, M.; et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut 2018, 67, 903–917. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Choudhuri, R.; Fabre, K.; Sowers, A.L.; Citrin, D.; Zabludoff, S.D.; Cook, J.A. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2076–2084. [Google Scholar] [CrossRef]
- Elbæk, C.R.; Petrosius, V.; Sørensen, C.S. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat. Res. 2020, 819–820, 111694. [Google Scholar] [CrossRef]
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef]
- Bridges, K.A.; Hirai, H.; Buser, C.A.; Brooks, C.; Liu, H.; Buchholz, T.A.; Molkentine, J.M.; Mason, K.A.; Meyn, R.E. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin. Cancer Res. 2011, 17, 5638–5648. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Chan, G.K.; Gamper, A.M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 2022, 12, 828684. [Google Scholar] [CrossRef]
- Vakili-Samiani, S.; Khanghah, O.J.; Gholipour, E.; Najafi, F.; Zeinalzadeh, E.; Samadi, P.; Sarvarian, P.; Pourvahdani, S.; Kelaye, S.K.; Hamblin, M.R.; et al. Cell cycle involvement in cancer therapy; WEE1 kinase, a potential target as therapeutic strategy. Mutat. Res. 2022, 824, 111776. [Google Scholar] [CrossRef]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, Q.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, P.; Tao, K. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol. Med. Rep. 2018, 17, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Mårtensson, S.; Moshinsky, D.; Rice, A.; Tang, C.; Howlett, A.; McMahon, G.; Hammarsten, O. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene 2004, 23, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Investig. 2020, 130, 258–271. [Google Scholar] [CrossRef]
- Sun, X.; Yang, C.; Liu, H.; Wang, Q.; Wu, S.X.; Li, X.; Xie, T.; Brinkman, K.L.; Teh, B.S.; Butler, E.B.; et al. Identification and characterization of a small inhibitory peptide that can target DNA-PKcs autophosphorylation and increase tumor radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 1212–1219. [Google Scholar] [CrossRef]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef]
- Smithson, M.; Irwin, R.K.; Williams, G.; McLeod, M.C.; Choi, E.K.; Ganguly, A.; Pepple, A.; Cho, C.S.; Willey, C.D.; Leopold, J.; et al. Inhibition of DNA-PK may improve response to neoadjuvant chemoradiotherapy in rectal cancer. Neoplasia 2022, 25, 53–61. [Google Scholar] [CrossRef]
- Stachelek, G.C.; Peterson-Roth, E.; Liu, Y.; Fernandez, R.J., 3rd; Pike, L.R.; Qian, J.M.; Abriola, L.; Hoyer, D.; Hungerford, W.; Merkel, J.; et al. YU238259 Is a Novel Inhibitor of Homology-Dependent DNA Repair That Exhibits Synthetic Lethality and Radiosensitization in Repair-Deficient Tumors. Mol. Cancer Res. MCR 2015, 13, 1389–1397. [Google Scholar] [CrossRef]
- Oh, M.; McBride, A.; Yun, S.; Bhattacharjee, S.; Slack, M.; Martin, J.R.; Jeter, J.; Abraham, I. BRCA1 and BRCA2 Gene Mutations and Colorectal Cancer Risk: Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2018, 110, 1178–1189. [Google Scholar] [CrossRef]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef]
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Conte, B.; Martel, S.; Maurer, C.; Tagliamento, M.; Viglietti, G.; Del Mastro, L.; de Azambuja, E.; et al. Single-agent PARP inhibitors for the treatment of patients with BRCA-mutated HER2-negative metastatic breast cancer: A systematic review and meta-analysis. ESMO Open 2018, 3, e000361. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.C.; Conti, D.V.; Siegmund, K.D.; Corral, R.; Yuan, J.M.; Koh, W.P.; Yu, M.C. DNA repair single-nucleotide polymorphisms in colorectal cancer and their role as modifiers of the effect of cigarette smoking and alcohol in the Singapore Chinese Health Study. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2007, 16, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Vormoor, B.; Schlosser, Y.T.; Blair, H.; Sharma, A.; Wilkinson, S.; Newell, D.R.; Curtin, N. Sensitizing Ewing sarcoma to chemo- and radiotherapy by inhibition of the DNA-repair enzymes DNA protein kinase (DNA-PK) and poly-ADP-ribose polymerase (PARP) 1/2. Oncotarget 2017, 8, 113418–113430. [Google Scholar] [CrossRef]
- Hirai, T.; Saito, S.; Fujimori, H.; Matsushita, K.; Nishio, T.; Okayasu, R.; Masutani, M. Radiosensitization by PARP inhibition to proton beam irradiation in cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 234–240. [Google Scholar] [CrossRef]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef]
- Shelton, J.W.; Waxweiler, T.V.; Landry, J.; Gao, H.; Xu, Y.; Wang, L.; El-Rayes, B.; Shu, H.K. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Sakata, K.; Someya, M.; Matsumoto, Y.; Matsumoto, H.; Takahashi, A.; Hareyama, M. The combination of olaparib and camptothecin for effective radiosensitization. Radiat. Oncol. 2012, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Waqar, S.N.; Robinson, C.; Olszanski, A.J.; Spira, A.; Hackmaster, M.; Lucas, L.; Sponton, L.; Jin, H.; Hering, U.; Cronier, D.; et al. Phase I trial of ATM inhibitor M3541 in combination with palliative radiotherapy in patients with solid tumors. Investig. New Drugs 2022, 40, 596–605. [Google Scholar] [CrossRef]
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Bendell, J.C.; Bischoff, H.G.; Hwang, J.; Reinhardt, H.C.; Zander, T.; Wang, X.; Hynes, S.; Pitou, C.; Campbell, R.; Iversen, P.; et al. A phase 1 dose-escalation study of checkpoint kinase 1 (CHK1) inhibitor prexasertib in combination with p38 mitogen-activated protein kinase (p38 MAPK) inhibitor ralimetinib in patients with advanced or metastatic cancer. Investig. New Drugs 2020, 38, 1145–1155. [Google Scholar] [CrossRef]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target. Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Plummer, E.R.; Kristeleit, R.S.; Cojocaru, E.; Haris, N.M.; Carter, L.; Jones, R.H.; Blagden, S.P.; Evans, T.R.J.; Arkenau, H.-T.; Sarker, D.; et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3094. [Google Scholar] [CrossRef]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Esaki, T.; Hirai, F.; Arita, S.; Nosaki, K.; Makiyama, A.; Kometani, T.; Fujimoto, C.; Hamatake, M.; Takeoka, H.; et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 619–627. [Google Scholar] [CrossRef]
- Ho, A.L.; Bendell, J.C.; Cleary, J.M.; Schwartz, G.K.; Burris, H.A.; Oakes, P.; Agbo, F.; Barker, P.N.; Senderowicz, A.M.; Shapiro, G. Phase I, open-label, dose-escalation study of AZD7762 in combination with irinotecan (irino) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2011, 29, 3033. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef]
- Cohen, D.J.; Grabocka, E.; Bar-Sagi, D.; Godin, R.; Leichman, L.P. A phase Ib study combining irinotecan with AZD1775, a selective WEE 1 kinase inhibitor, in RAS/RAF mutated metastatic colorectal cancer patients who progressed on first line therapy. J. Clin. Oncol. 2017, 35, TPS3627. [Google Scholar] [CrossRef]
- Berlin, J.; Ramanathan, R.K.; Strickler, J.H.; Subramaniam, D.S.; Marshall, J.; Kang, Y.K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef]
- Chen, E.X.; Jonker, D.J.; Siu, L.L.; McKeever, K.; Keller, D.; Wells, J.; Hagerman, L.; Seymour, L. A Phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian Cancer Trials Group IND 187. Investig. New Drugs 2016, 34, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.D.; Garcia-Alfonso, P.; Nechaeva, M.; Cubillo Gracian, A.; Mangel, L.; Elez Fernandez, E.; Deming, D.A.; Ramanathan, R.K.; et al. A phase 2 randomised study of veliparib plus FOLFIRI±bevacizumab versus placebo plus FOLFIRI±bevacizumab in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [Google Scholar] [CrossRef] [PubMed]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.J.; et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncol. 2016, 21, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef]
- Smith, G.; Alholm, Z.; Coleman, R.L.; Monk, B.J. DNA Damage Repair Inhibitors-Combination Therapies. Cancer J. 2021, 27, 501–505. [Google Scholar] [CrossRef]
- Fuchss, T.; Grädler, U.; Schiemann, K.; Kuhn, D.; Kubas, H.; Dahmen, H.; Zimmermann, A.; Zenke, F.; Blaukat, A. Abstract 3500: Highly potent and selective ATM kinase inhibitor M4076: A clinical candidate drug with strong anti-tumor activity in combination therapies. Cancer Res. 2019, 79, 3500. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Van Bijsterveldt, L.; Durley, S.C.; Maughan, T.S.; Humphrey, T.C. The Challenge of Combining Chemo- and Radiotherapy with Checkpoint Kinase Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 937–962. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- De’ Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’ Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Bio-Med. Atenei Parm. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Demaria, S.; Formenti, S.C. Radiation as an immunological adjuvant: Current evidence on dose and fractionation. Front. Oncol. 2012, 2, 153. [Google Scholar] [CrossRef] [PubMed]
- Herskind, C.; Wenz, F.; Giordano, F.A. Immunotherapy Combined with Large Fractions of Radiotherapy: Stereotactic Radiosurgery for Brain Metastases-Implications for Intraoperative Radiotherapy after Resection. Front. Oncol. 2017, 7, 147. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- Theelen, W.; Chen, D.; Verma, V.; Hobbs, B.P.; Peulen, H.M.U.; Aerts, J.; Bahce, I.; Niemeijer, A.L.N.; Chang, J.Y.; de Groot, P.M.; et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Respir. Med. 2021, 9, 467–475. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Huang, J.; Liang, D.; Hu, Y.; Mao, B.; Li, Q.; Sun, H.; Yang, Y.; Zhang, J.; Zhang, H.; et al. DNA Damage Repair Gene Mutations Are Indicative of a Favorable Prognosis in Colorectal Cancer Treated With Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 549777. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Zheng, J.; Mo, J.; Zhu, T.; Zhuo, W.; Yi, Y.; Hu, S.; Yin, J.; Zhang, W.; Zhou, H.; Liu, Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 2020, 19, 133. [Google Scholar] [CrossRef]
- Carter, L. Phase I modular study of AZD6738, a novel oral, potent and selective ataxia telangiectasia Rad3-related (ATR) inhibitor in combination (combo) with carboplatin, olaparib or durvalumab in patients (pts) with advanced cancers. Eur. J. Cancer 2016, 69, S2. [Google Scholar]
- Ahmad, S.S.; Crittenden, M.R.; Tran, P.T.; Kluetz, P.G.; Blumenthal, G.M.; Bulbeck, H.; Baird, R.D.; Williams, K.J.; Illidge, T.; Hahn, S.M.; et al. Clinical Development of Novel Drug-Radiotherapy Combinations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1455–1461. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]



{kind=link}
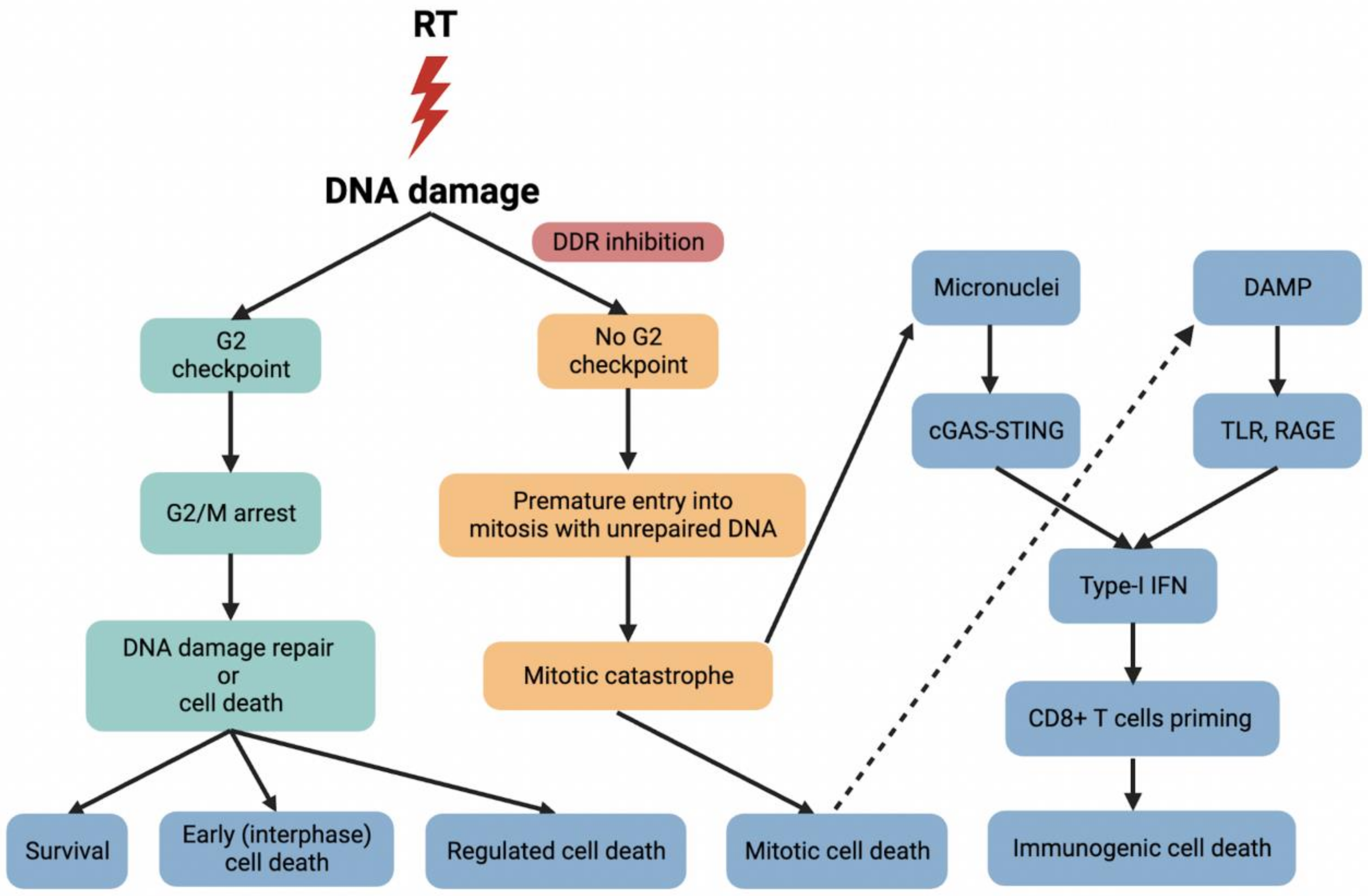{kind=link}
| Target | ClinicalTrials.gov Identifier, Authors and Reference | Phase | Status | Disease(s) | Treatment | Primary Outcome Measures | CRC Patients Enrolled | ORR (%) | MTD | Grade 3–4 AEs (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| ATM | NCT03225105 Waqar [114] | I | Completed | Solid tumors, including CRC | M3541 + RT | DLTs | 2 | NA | NA | 26.7 a |
| PARP | NCT01589419 Czito [115] | Ib | Locally advanced rectal cancer | Veliparib (ABT-888) + capecitabine + RT | MTD, RP2D | 32 | 9/32 (28.1) | NA | 12.5 | |
| DNA-PKcs | NCT03770689 | Ib | Locally advanced rectal cancer | Peposertib (M3814) + capecitabine + RT | DLTs | 19 | NA | NA | 36.8 | |
| NCT03724890 | I | Ongoing | Advanced solid tumors, including CRC | M3814 + avelumab ± RT | DLTs | NA | NA | NA | NA | |
| ATR | NCT02223923 | I | Solid tumors, including CRC | Ceralasertib (AZD6738) + RT | MTD | NA | NA | NA | NA |
| Target | ClinicalTrials.gov Identifier, Authors and Reference | Phase | Disease(s) | Treatment | CRC Patients Enrolled | ORR (%) | MTD | Grade 3–4 AEs (%) |
|---|---|---|---|---|---|---|---|---|
| ATR | NCT02157792 Middleton [116] | I | Advanced solid tumors, including CRC | Berzosertib (M6620, VX-970) + gemcitabne ± cisplatin | 22 | NA | NA | 79.3 a |
| NCT02157792 Shapiro [117] | I | Advanced solid tumors, including CRC | Berzosertib (M6620, VX-970) + cisplatin | 5 | NA | NA | 70.0 | |
| NCT02157792 Yap [118] | I | Advanced solid tumors, including CRC | Berzosertib (M6620, VX-970) ± carboplatin | 11 | NA | 90 mg/m2 | 30.4 a | |
| CHK1 | NCT02860780 Bendell [119] | I | Advanced/metastatic cancer, including CRC | Prexasertib (LY2606368) + ralimetinib | 9 | NA | 105 mg/m2 | 33.3 a |
| NCT02124148 Moore [120] | Ib | Advanced/metastatic cancer, including CRC | Prexasertib (LY2606368) + cetuximab | 41 | 2/41 (4.9) | 80 mg/m2 | 53.7 | |
| NCT01115790 Hong [121] | I | Advanced cancer, including CRC | Prexasertib (LY2606368) | 9 | NA | 40 mg/m2; 105 mg/m2 | 88.9 a | |
| NCT02797964 Plummer [122] | I/II | Advanced solid tumors (including CRC), non-hodgkin’s lymphoma | SRA737 (CCT245737) | 32 | NA | 1000 mg/day | 44.9 a | |
| NCT01564251 Italiano [123] | I | Refractory solid tumors (including CRC), or lymphoma | GDC-575 (ARRY-575; RG7741) | 4 | NA | 60 mg/m2 | 49 a | |
| NCT00413686 Sausville [124] | I | US patients with advanced solid tumors, including CRC | AZD7762 ± gemcitabine | 11 | NA | 30 mg/m2 | 69.0 a | |
| NCT00937664 Seto [125] | I | Japanese patients with advanced solid tumors, including CRC | AZD7762 ± gemcitabine | 5 | NA | 21 mg/m2 | 60.0 a | |
| NCT00473616 Ho [126] | I | Advanced solid tumors, including CRC | AZD7762 + irinotecan | 29 | NA | 96 mg/m2 | 10.3 a | |
| WEE1 | NCT00648648 Leijen [127] | I | Advanced solid tumors, including CRC | Adavosertib (AZD1775, MK-1775) + gemcitabine + cisplatin or carboplatin | 15 | 1/15 (6.7) | 225 mg twice/day; 200 mg twice/day; 175 mg/day | 54.7 a |
| NCT01748825 Do [128] | I | Advanced solid tumors, including CRC | Adavosertib (AZD1775, MK-1775) | 2 | NA | 225 mg twice/day | 56.7 a | |
| NCT02906059 Cohen [129] | Ib | KRAS, NRAS or BRAF mutated metastatic CRC | Adavosertib (AZD1775, MK-1775) + irinotecan | 7 | NA | NA | NA | |
| PARP | NCT02033551 Berlin [130] | I | Advanced solid tumors, including CRC | Veliparib (ABT-888) + FOLFIRI | 10 | 2/10 (20.0) | NA | 38.0 a |
| NCT00535353 Chen [131] | I | Advanced or metastatic CRC | Olaparib (AZD-2281) + irinotecan | 25 | 0/25 (0.0) | NA | 76.0 b | |
| NCT02305758 Gorbunova [132] | II | Untreated metastatic CRC | Veliparib (ABT-888) + FOLFIRI ± bevacizumab | 65 | 37/65 (57) | NA | 59.0 b | |
| NCT00553189 Kummar [133] | I | Solid tumors (including CRC) and lymphomas | Veliparib (ABT-888) + topotecan | 5 | 0/5 (0.0) | 10 mg twice/day | 70.0 ab | |
| NCT00912743 Leichmann [134] | II | Chemorefractory metastatic CRC | Olaparib (AZD-2281) | 33 | 0/33 (0.0) | NA | 48.5 | |
| NCT01051596 Pishvaian [135] | II | Heavily pretreated metastatic CRC | Veliparib (ABT-888) + temozolomide | 75 | 2/75 (2.7) | NA | 18.7 | |
| NCT00516438 Samol [136] | I | Advanced solid tumors, including CRC | Olaparib (AZD-2281) + topotecan | 8 | 0/8 (0.0) | 100 mg twice/day | 47.4 a | |
| NCT03875313 | Ib/II | Solid tumors, including CRC | Talazoparib + CB-839 (Telaglenastat) | 4 | 0/4 (0.0) | NA | 18.2 a | |
| PARP, ATR | NCT02723864 Smith [137] | I | Refractory solid tumors, including CRC | veliparib (ABT-888) + berzosertib (M6620, VX-970) + cisplatin | 3 | NA | NA | 35.8 a |
| Target | ClinicalTrials.gov Identifier | Phase | Disease(s) | Treatment | Primary Outcome Measures |
|---|---|---|---|---|---|
| ATM | NCT02588105 | I | Advanced solid tumors, including CRC | AZD0156 ± olaparib/FOLFIRI | TRAEs |
| ATR | NCT03188965 | I | Advanced solid tumors, including CRC, and lymphomas | Elimusertib (BAY 1895344) | MTD, RP2D, DLTs, TEAEs |
| NCT04535401 | I | Advanced or metastatic CRC and gastric/gastroesophageal cancers | Elimusertib (BAY 1895344) + FOLFIRI | MTD | |
| NCT04704661 | I/Ib | Advanced solid tumors, including CRC that have a change (mutation) in the HER2 gene or protein | Ceralasertib (AZD6738) + trastuzumab deruxtecan (DS-8201a) | TRAEs, RP2D, PD profile | |
| NCT02595931 | I | Metastatic or unresectable solid tumors, including CRC | Berzosertib (M6620, VX-970) + irinotecan | MTD, RP2D | |
| NCT04266912 | I/II | DDR deficient metastatic or unresectable solid tumors, including CRC | Berzosertib (M6620, VX-970) + avelumab | AEs, SAEs, DLTs, MTD | |
| CHK1 | NCT02632448 | Ib/IIa | Solid tumors, including CRC | LY2880070 ± gemcitabine | MTD |
| WEE1 | NCT02465060 | II | Advanced refractory solid tumors (including CRC), lymphomas, or multiple myeloma | Adavosertib (AZD1775) + targeted therapy according to mutational status | ORR |
| NCT04158336 | I/II | Solid tumors, including CRC | ZN-c3 | MTD, RP2D, ORR | |
| NCT02617277 | I | Advanced solid tumors, including CRC | AZD1775 + durvalumab | DLTs | |
| PARP | NCT02484404 | I/II | Ovarian, triple negative breast, lung, prostate, CRC | Durvalumab (MEDI4736) + olaparib ± cediranib | OR, RP2D |
| NCT03851614 (DAPPER) | II | Mismatch repair proficient CRC, pancreatic adenocarcinoma, leiomyosarcoma | Durvalumab (MEDI4736) + olaparib + cediranib | Changes in genomic and immune biomarkers | |
| NCT04171700 (LODESTAR) | II | Solid tumors, including CRC | Rucaparib | ORR | |
| NCT03251612 | II | Metastatic CRC | Olaparib + therapy based on sensitivity analysis | PFS | |
| NCT03983993 (NIPAVect) | II | Advanced or metastatic CRC | Panitumumab + niraparib | CBR | |
| NCT03337087 | I/II | Metastatic pancreatic, CRC, gastroesophageal, or biliary cancer | Liposomal irinotecan + leucovorin calcium + fluorouracil + rucaparib | MTD, OR, BRR | |
| NCT04166435 | II | MGMT hypermethylated CRC | Temozolomide + olaparib | ORR | |
| NCT04456699 | III | Unresectable or metastatic CRC | Olaparib ± bevacizumab + 5-FU | PFS | |
| NCT04511039 | I | CRC or gastroesophageal cancer | Trifluridine/Tipiracil + talazoparib | AEs, MTD, RP2D | |
| NCT03842228 | Ib | Advanced solid tumors, including CRC | Olaparib + durvalumab + copanlisib (PI3K inhibitor) | MTD | |
| NCT04123366 | II | HRRm and HRD-positive advanced solid tumors, including CRC | Olaparib + pembrolizumab | ORR | |
| NCT03772561 | I | Advanced solid tumors, including CRC | Olaparib + durvalumab + AZD5363 (AKT inhibitor) | ORR | |
| PARP, ATR | NCT02264678 | I | Advanced solid tumors, including CRC | Olaparib + ceralasertib (AZD6738) + durvalumab + carboplatin | AEs, SAEs, ECG |
| NCT04497116 | I/IIa | Advanced solid tumors, including CRC | RP-3500 (ATR inhibitor) ± talazoparib±gemcitabine | MTD, DLTs | |
| PARP, ATR, WEE1 | NCT02576444 | II | Advanced solid tumors, including CRC | Olaparib + AZD6738 + AZD1775 + AZD5363 | ORR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, S.; Vlatkovic, T.; Li, M.; Zhan, T.; Veldwijk, M.R.; Herskind, C. Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers 2022, 14, 4874. https://doi.org/10.3390/cancers14194874
Deng S, Vlatkovic T, Li M, Zhan T, Veldwijk MR, Herskind C. Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers. 2022; 14(19):4874. https://doi.org/10.3390/cancers14194874
Chicago/Turabian StyleDeng, Siyao, Tijana Vlatkovic, Moying Li, Tianzuo Zhan, Marlon R. Veldwijk, and Carsten Herskind. 2022. "Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer" Cancers 14, no. 19: 4874. https://doi.org/10.3390/cancers14194874
APA StyleDeng, S., Vlatkovic, T., Li, M., Zhan, T., Veldwijk, M. R., & Herskind, C. (2022). Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers, 14(19), 4874. https://doi.org/10.3390/cancers14194874