
BRAF Inhibitors in Non-Small Cell Lung Cancer
 ,
,  , ,
, ,  and
and 
Simple Summary
Abstract
1. Introduction
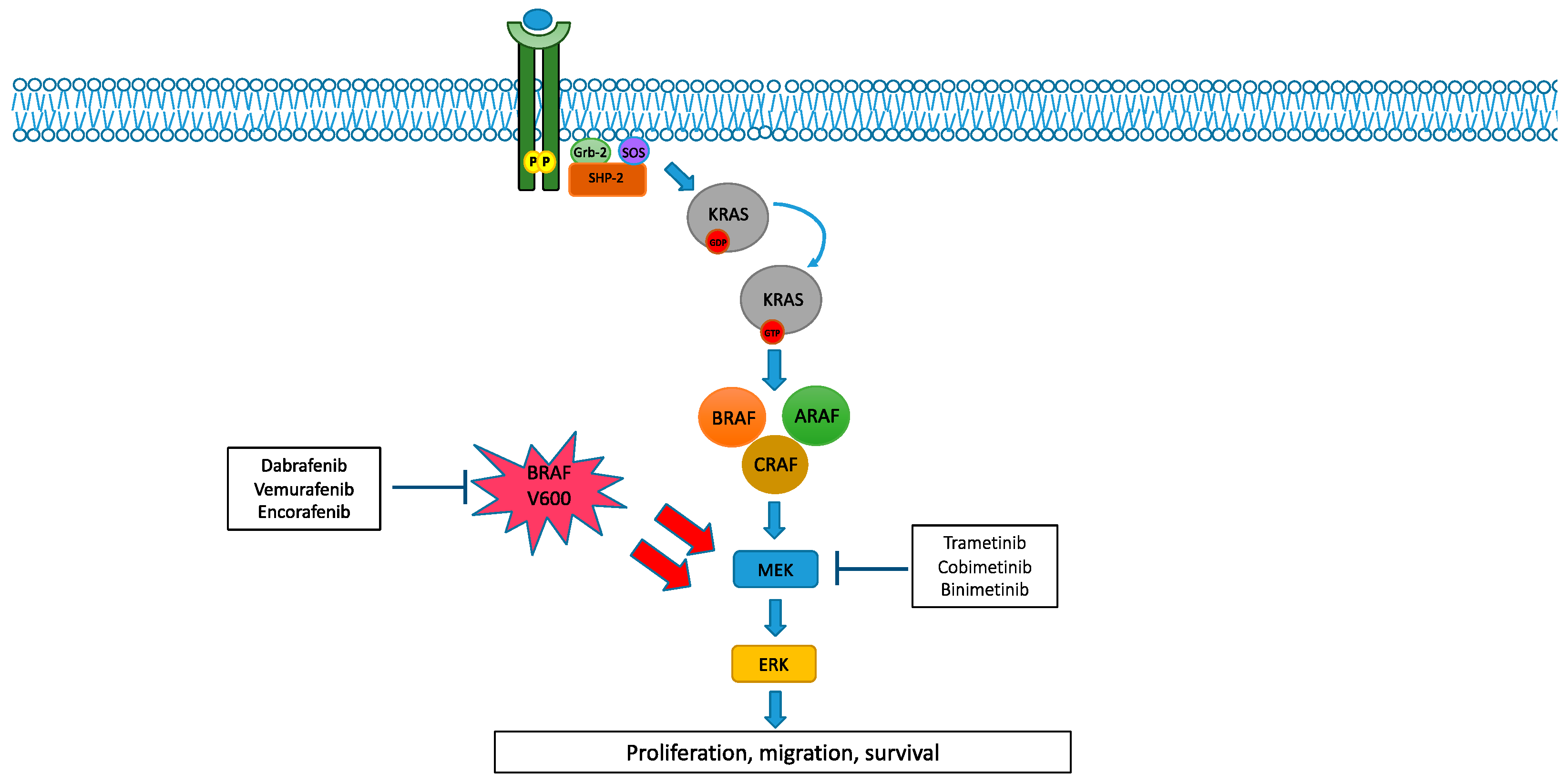
2. MAPK Pathway and Its Alterations
2.1. BRAF and MEK Alterations in NSCLC
2.2. BRAF and MEK Inhibitors in NSCLC
3. Discussion

{kind=link}
| Trial | Phase | Setting | Stage | Pts | Treatment | Primary Endpoints |
|---|---|---|---|---|---|---|
| NCT04526782 (ENCO-BRAF Trial) [59] | Phase 2 | Naïve or subsequent lines (two different arms) | Extensive stage | 119 | Encorafenib + Binimetinib | ORR |
| NCT03915951 [60] | Phase 2 | Any line of treatment | Extensive stage | 97 | Encorafenib + binimetinib | ORR |
| NCT04585815 (Landscape 1011 trial) [62] | Phase 1/2 | Any line of treatment | Extensive stage | 375 | Sasalnimab + encorafenib + binimetinib | Durable objective response rate |
| NCT04591431 (ROME trial) [63] | Phase 2 | Subsequent lines (no more than two treatments are allowed) | Extensive stage | 384 | Vemurafenib + cobimetinib vs. SOC * | ORR |
| NCT03178552 (BFAST Trial) [64] | Phase 2/3 | First line | Extensive stage | 1000 | Vemurafenib + Cobimetinib+ Atezolizumab | Time in response (TIR) |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; van Schil, P.E.; Hellmann, M.D.; et al. ESMO Guidelines Committee. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, J.R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Papint, C.; Denouel-Galy, A.; Laugier, D.; Caloty, G.; Eychene, A. Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J. Biol. Chem. 1998, 273, 24939–24947. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, J.R. MEK1/2 dual-specificity protein kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2012, 417, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, S.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef]
- Danker, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutation. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Alvarez, J.G.B.; Otterson, G.A. Agents to treat BRAF-mutant lung cancer. Drugs Context 2019, 8, 212566. [Google Scholar] [CrossRef]
- Negrao, M.V.; Raymond, V.M.; Lanman, R.B.; Robichaux, J.P.; He, J.; Nilsson, M.B.; Ng, P.K.S.; Amador, B.E.; Roarty, E.B.; Nagy, R.J.; et al. Molecular landscape of BRAF-Mutant NSCLC reveals an association between clonality and driver mutations and identifies targetable non-V600 driver mutations. J. Thorac. Oncol. 2020, 15, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Janne, P.J. Clinical, pathological and biological features associated with BRAF mutations in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef] [PubMed]
- Brustugun, O.T.; Khattak, A.M.; Tromborg, A.K.; Beigi, M.; Beiske, K.; Lund-Iversen, M.; Helland, A. BRAF-mutations in non-small cell lung cancer. Lung Cancer 2014, 84, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Felicioni, L.; Malatesta, S.; Sciarrotta, M.G.; Guetti, L.; Chella, A.; Viola, P.; Pullara, C.; Mucilli, F.; Buttitta, F. Clinical features and outcome of patients with Non–Small-Cell Lung Cancer harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 3574–3579. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, L.Q.; Huang, J.F.; Liu, K.; Chuai, Z.R.; Yang, Z.; Wang, Y.X.; Shi, D.C.; Liu, Q.; Huang, Q.; et al. BRAF mutations in patients with Non-Small Cell Lung Cancer: A systematic review and meta-Analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef]
- Lin, Q.; zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef]
- Mendoza, D.P.; Dagogo-Jack, I.; Chen, T.; Padole, A.; Shepherd, J.O.A.; Shaw, A.T.; Digumarthy, S.R. Imaging characteristics of BRAF-mutant non-small cell lung cancer by functional class. Lung Cancer 2019, 129, 80–84. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant Lung Cancer. Clin. Cancer Res. 2018, 25, 158–165. [Google Scholar] [CrossRef]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF new world. Crit. Rev. Oncol. Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic characterization of concurrent alterations in Non-Small Cell Lung Cancer (NSCLC) harboring actionable mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhao, C.; Li, J.; Su, C.; Chen, X.; Ren, S.; Li, X.; Zhou, C. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019, 8, 1858–2866. [Google Scholar] [CrossRef] [PubMed]
- Martorell, P.M.; Huerta, M.; Quilis, A.C.; Abellan, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Beltran, D.D.; Keranen, S.R.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA mutations and ALK rearrangement in a comprehensive cohort of 326 consecutive spanish non squamous NSCLC patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C.H. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thor. Oncol. 2016, 12, 567–572. [Google Scholar] [CrossRef]
- Dudnik, E.; Peled, N.; Nechushtan, H.H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF mutant lung cancer: Programmed Death Ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitor. J. Thor. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef]
- Rihawi, K.; Giannarelli, D.; Galetta, D.; Delmonte, A.; Giavarra, M.; Turci, D.; garassino, M.; Tiseo, M. BRAF Mutant NSCLC and immune checkpoint inhibitors: Results from a real-world experience. J. Thor. Oncol. 2019, 14, e57–e59. [Google Scholar] [CrossRef]
- Li, S.D.; martial, A.; Schrock, A.B.; Liu, J.J. Extraordinary clinical benefit to sequential treatment with targeted therapy and immunotherapy of a BRAF V600E and PD-L1 positive metastatic lung adenocarcinoma. Exp. Hematol. Oncol. 2017, 6, 29. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Reddy, V.P.; Gay, L.M.; Elvin, J.A.; Vergilio, J.A.; Suh, J.; Ramkissoon, S.; Daniel, S.; Severson, E.A.; Ali, S.M.; Schrock, A.B.; et al. BRAF fusions in clinically advanced non-small cell lung cancer: An emerging target for anti-BRAF therapies. J. Clin. Oncol. 2017, 35, 9072. [Google Scholar] [CrossRef]
- Marks, J.L.; Gong, Y.; Chitale, D.; Golas, B.; McLellan, M.D.; Kasai, Y.; Ding, L.; Mardis, E.R.; Wilson, R.K.; Soliti, D.; et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008, 68, 5524–5528. [Google Scholar] [CrossRef] [PubMed]
- Arcila, M.E.; Drilon, A.; Sylvester, B.E.; Lovly, C.M.; Borsu, L.; Reva, B.; Kris, M.; Solit, D.B.; Ladanyi, M. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin. Cancer Res. 2015, 21, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Gautschi, O.; Pauli, C.; Strobel, K.; Hirschmann, A.; Printzen, G.; Aebi, S.; Diebold, J. A Patient with BRAF V600E Lung Adenocarcinoma Responding to Vemurafenib. J. Thorac. Oncol. 2012, 7, e23–e24. [Google Scholar] [CrossRef]
- Peters, S.; Michielin, O.; Zimmermann, S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J. Clin. Oncol. 2013, 31, e341–e344. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Cabarrou, B.; Bluthgen, M.V.; Besse, B.; Smit, E.F.; Wolf, J.; Peters, S.; Früh, M.; Koeberle, D.; et al. Targeted therapy for patients with BRAF-mutant lung cancer results from the European EURAF cohort. J. Thorac. Oncol. 2015, 10, 1451–1457. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Subbiah, V.; Gervais, R.; Riely, G.J.; Hollebecque, A.; Blay, J.-Y.; Felip, E.; Schuler, M.; Gonçalves, A.; Italiano, A.; Keedy, V.; et al. Efficacy of Vemurafenib in Patients with Non–Small-Cell Lung Cancer with BRAF V600 Mutation: An Open-Label, Single-Arm Cohort of the Histology-Independent VE-BASKET Study. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.J.; Barlesi, F.; Baik, C.S.; et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients with BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef]
- Robinson, S.D.; O’Shaughnessy, J.A.; Lance, C.C.; Konduri, K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer 2014, 85, 326–330. [Google Scholar] [CrossRef]
- Fernandes, M.G.; Costa, J.; Reis, J.; Jacob, M.; Moura, C.; Machado, J.; Hespanhol, V. OA08.07 BRAF-V600E Advanced lung adenocarcinoma with leptomeningeal (LM) disease treated with vemurafenib. J. Thorac. Oncol. 2017, 12, S274–S275. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role played by signalling pathways in overcoming BRAF inhibitor resistance in melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E.; et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. 2012, 30, 316–321. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The Raf inhibitor paradox: Unexpected consequences of targeted drugs. Cancer Cell 2010, 17, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Approved Drugs—FDA Grants Regular Approval to Dabrafenib and Trametinib Combination for Metastatic NSCLC with BRAF V600E Mutation 2017. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm564331.htm (accessed on 10 November 2017).
- Trametinib in Combination with Dabrafenib Is Indicated for the Treatment of Adult Patients with Advanced Non-Small Cell Lung Cancer with a BRAF V600 Mutation 2017. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002643/WC500222159.pdf (accessed on 23 November 2017).
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04526782 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03915951 (accessed on 4 October 2022).
- Johnson, M.L.; Braiteh, F.; Grilley-Olson, J.E.; Chou, J.; Davda, J.; Forgie, A.; Li, R.; Jacobs, I.; Kazazi, F.; Hu-Lieskovan, S. Assessment of Subcutaneous vs Intravenous Administration of Anti-PD-1 Antibody PF-06801591 in Patients with Advanced Solid Tumors: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2019, 5, 999–1007. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04585815 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04591431 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03178552 (accessed on 4 October 2022).
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hong, K.; Streit, M. Molecular characterization of acquired resistance to the BRAF Inhibitor dabrafenib in a patient With BRAF Mutant Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, e41–e42. [Google Scholar] [CrossRef]
- Girotti, M.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.; Rizvi, N.; Wolchok, J.D.; Chan, T.A. Genomic profile, smoking, and response to anti-PD-1 therapy in non-small cell lung carcinoma. Mol. Cell. Oncol. 2016, 3, e1048929. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Somatic mutations and immunotherapy outcome with CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2230–2232. [Google Scholar] [CrossRef] [PubMed]
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and Safety of Anti-PD-1 Immunotherapy in Patients with Advanced NSCLC With BRAF, HER2, or MET Mutations or RET Translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]

| Study | Author | Setting | Pts | Treatment | Response Rate (%) | Disease Control Rate (%) | Progression-Free Survival (Months) | Overall Survival (Months) | Toxicity |
|---|---|---|---|---|---|---|---|---|---|
| Phase 2 (VE-BASKET study) | Subbiah, 2019 [41] | Advanced solid tumours, Cohort NSCLC BRAFV600 | 62 | Vemurafenib | 37.1 | - | 6.5 | 15.4 | Nausea, hyperkeratosis, decreased appetite, arthralgia, cutaneous SCC |
| Phase 2 (AcSé-BASKET study) | Mazieres, 2020 [42] | Advanced solid tumours, Cohort NSCLC: BRAFnonV600 BRAFV600 | 115 15 100 | Vemurafenib | 0 44.8 | - - | 1.8 5.2 | 5.2 10 | Asthenia, decreased appetite, acneiform dermatitis, nausea and diarrhoea |
| Phase 2 (BRF113928) | Planchard 2016; 2022 [43,44,45] | BRAFV600E advanced NSCLC: Cohort A pretreated Cohort B pretreated Cohort C naïve | 171 78 57 36 | Dabrafenib Dabrafenib+ Trametinib Dabrafenib+ Trametinib | 33 68.4 63.9 | 58 80.7 75 | 5.5 10.2 10.8 | 12.7 18.2 17.3 | Pyrexia, asthenia, hyperkeratosis, decreased appetite, nausea |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sforza, V.; Palumbo, G.; Cascetta, P.; Carillio, G.; Manzo, A.; Montanino, A.; Sandomenico, C.; Costanzo, R.; Esposito, G.; Laudato, F.; et al. BRAF Inhibitors in Non-Small Cell Lung Cancer. Cancers 2022, 14, 4863. https://doi.org/10.3390/cancers14194863
Sforza V, Palumbo G, Cascetta P, Carillio G, Manzo A, Montanino A, Sandomenico C, Costanzo R, Esposito G, Laudato F, et al. BRAF Inhibitors in Non-Small Cell Lung Cancer. Cancers. 2022; 14(19):4863. https://doi.org/10.3390/cancers14194863
Chicago/Turabian StyleSforza, Vincenzo, Giuliano Palumbo, Priscilla Cascetta, Guido Carillio, Anna Manzo, Agnese Montanino, Claudia Sandomenico, Raffaele Costanzo, Giovanna Esposito, Francesca Laudato, and et al. 2022. "BRAF Inhibitors in Non-Small Cell Lung Cancer" Cancers 14, no. 19: 4863. https://doi.org/10.3390/cancers14194863
APA StyleSforza, V., Palumbo, G., Cascetta, P., Carillio, G., Manzo, A., Montanino, A., Sandomenico, C., Costanzo, R., Esposito, G., Laudato, F., Damiano, S., Forte, C. A., Frosini, G., Farese, S., Piccirillo, M. C., Pascarella, G., Normanno, N., & Morabito, A. (2022). BRAF Inhibitors in Non-Small Cell Lung Cancer. Cancers, 14(19), 4863. https://doi.org/10.3390/cancers14194863