
Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma



Abstract
Simple Summary
Abstract
1. Introduction
2. Tumour Immunity Cycle
3. Tumour Immune Escape
4. Mechanisms of Primary ICI Resistance
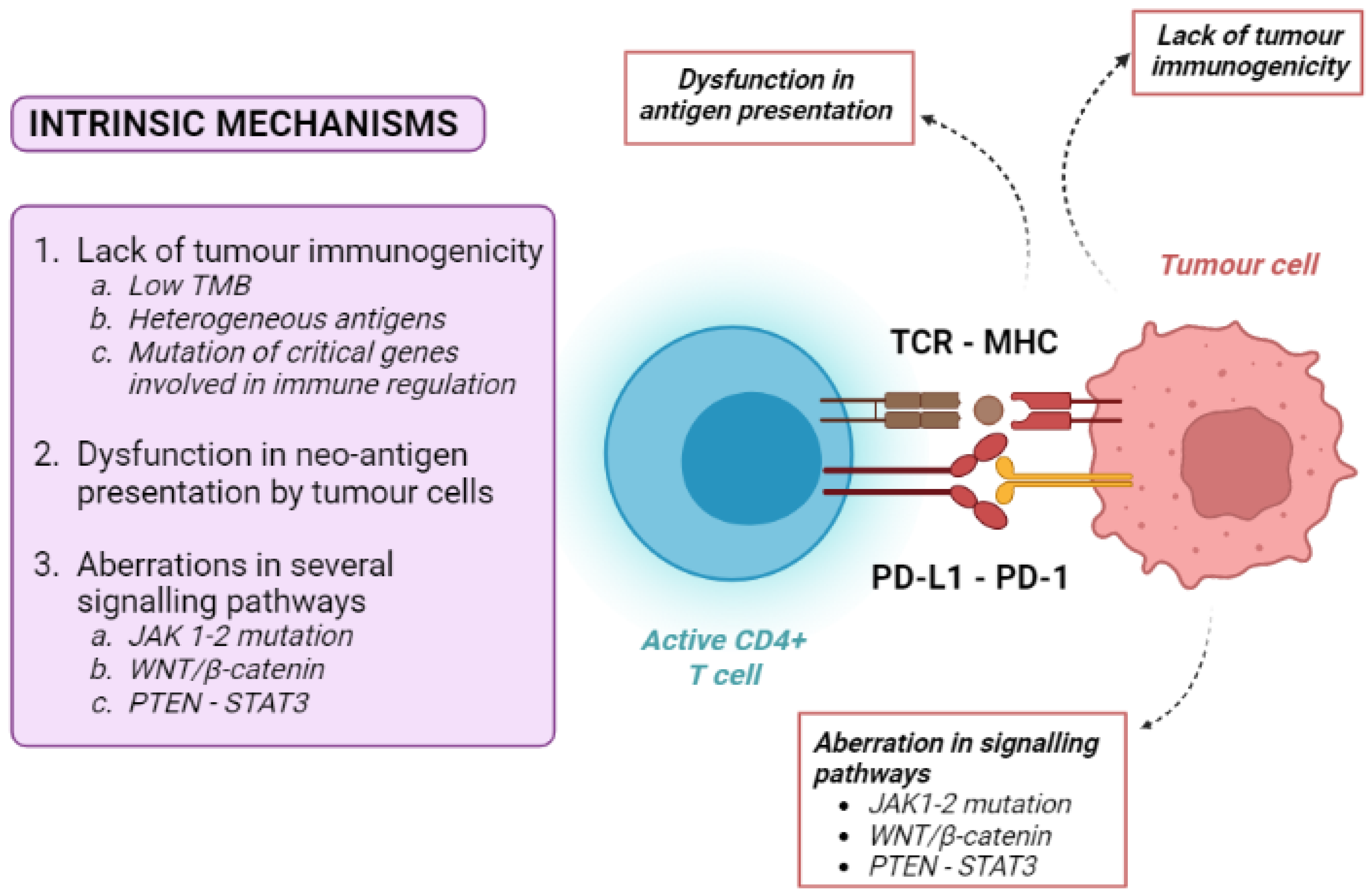
4.1. Tumour-Intrinsic Mechanisms
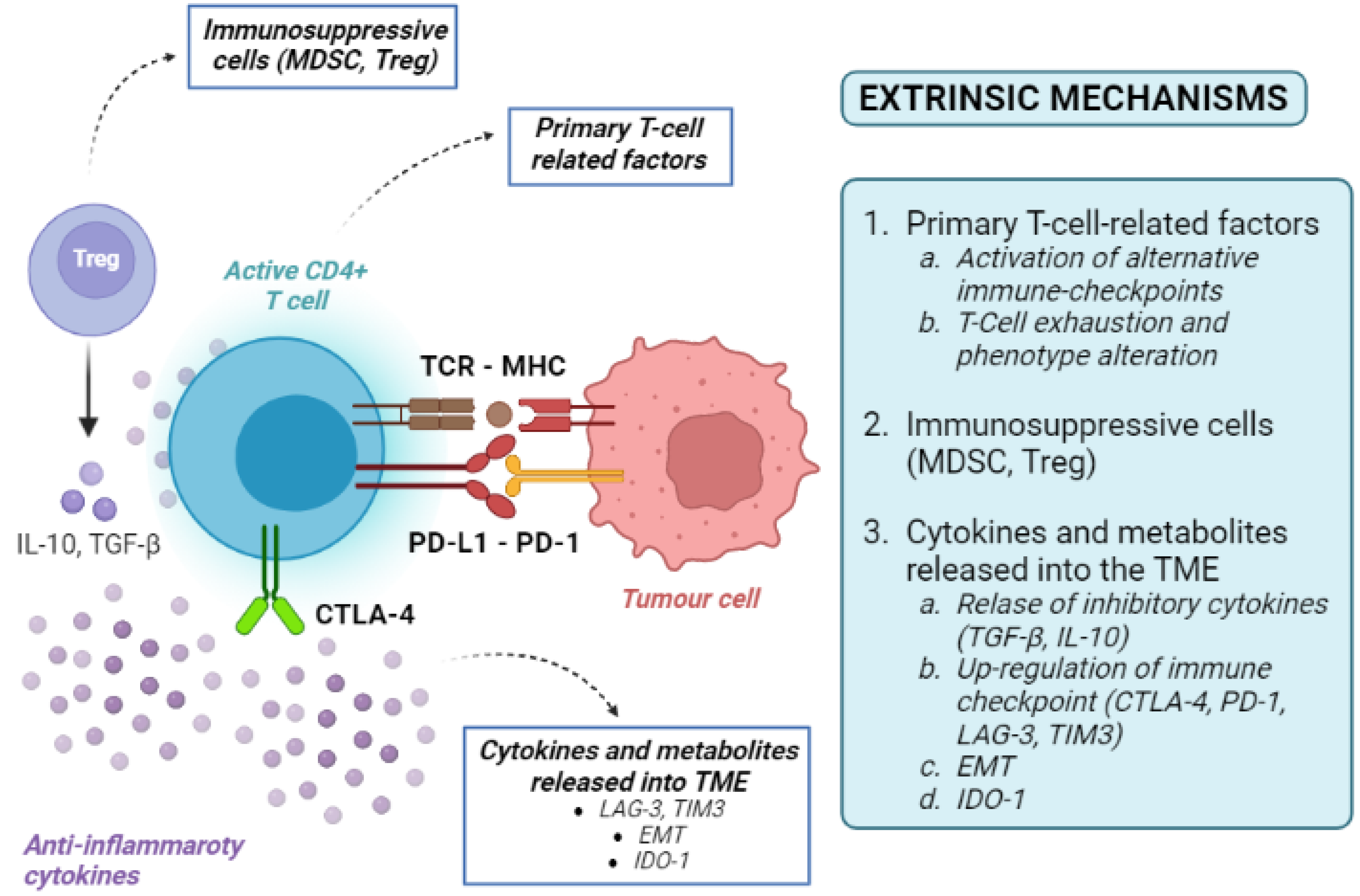
4.2. Tumour-Extrinsic Mechanisms
5. Mechanisms of Acquired ICI Resistance
6. Current Strategies in Immunotherapy for HCC
6.1. Established Strategies
6.1.1. Anti PD1/PD-L1 in Association with Intravenous Anti-VEGF Agents
6.1.2. Combination of PD-1 and CTLA-4 Inhibitors (Dual-Checkpoint Blockade)
6.1.3. Anti PD1/PD-L1 in Association with TKIs
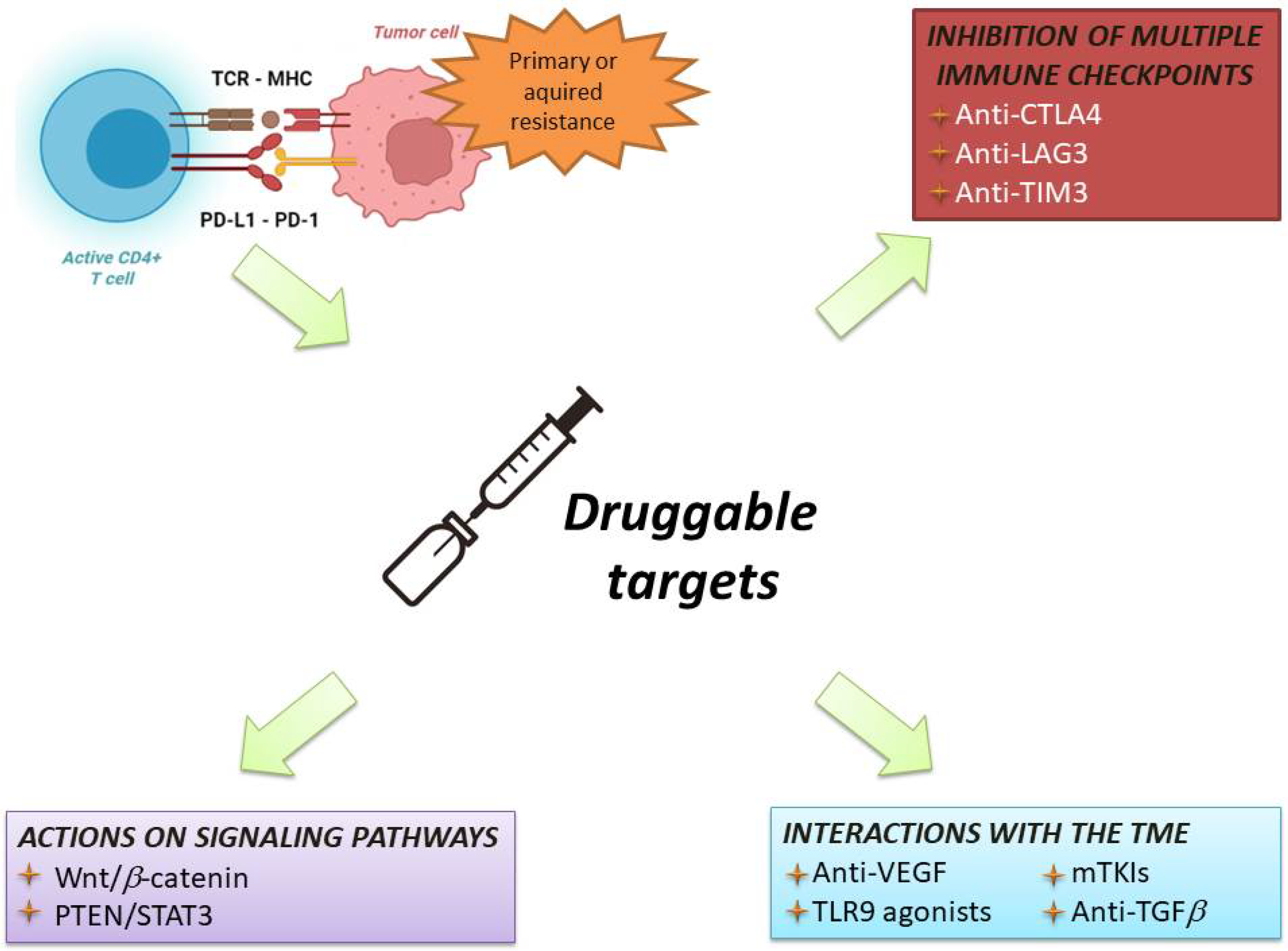
6.2. Novel Strategies
6.2.1. Combination with Drugs Acting on Tumour-Intrinsic Mechanisms of Resistance
6.2.2. Combination with Drugs Acting on Tumour-Extrinsic Mechanisms of Resistance
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Rumgay, H.; Ferlay, J.; De Martel, C.; Georges, D.; Ibrahim, A.S.; Zheng, R.; Wei, W.; Lemmens, V.E.P.P.; Soerjomataram, I. Global, Regional and National Burden of Primary Liver Cancer by Subtype. Eur. J. Cancer 2022, 161, 108–118. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Abou-Alfa Ghassan, K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive Resistance to Therapeutic PD-1 Blockade Is Associated with Upregulation of Alternative Immune Checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; Van der Bruggen, P.; Boon, T. Tumour Antigens Recognized by T Lymphocytes: At the Core of Cancer Immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, Q.; Zhang, J.; Zhu, B. Neoantigens in Precision Cancer Immunotherapy: From Identification to Clinical Applications. Chin. Med. J. 2022. [Google Scholar] [CrossRef]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Dufait, I.; Bricogne, C.; Lanna, A.; Pen, J.; Breckpot, K.; Escors, D. PD-L1/PD-1 Co-Stimulation, a Brake for T Cell Activation and a T Cell Differentiation Signal. J. Clin. Cell Immunol. 2012, S12, 006. [Google Scholar] [CrossRef]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 615317. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T Cell Exhaustion during Chronic Viral Infection by Multiple Inhibitory Receptors. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Abdel-Wahab, N.; Shah, M.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E. Use of Immune Checkpoint Inhibitors in the Treatment of Patients With Cancer and Preexisting Autoimmune Disease: A Systematic Review. Ann. Intern. Med. 2018, 168, 121–130. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef]
- Ichinokawa, K.; Nakanishi, Y.; Hida, Y.; Tsuchikawa, T.; Kato, T.; Itoh, T.; Kaji, M.; Kaga, K.; Hirano, S. Downregulated Expression of Human Leukocyte Antigen Class I Heavy Chain Is Associated with Poor Prognosis in Non-Small-Cell Lung Cancer. Oncol. Lett. 2019, 18, 117–126. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-Specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.-Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; De Achaval, S.; Alibhai, I.; Kaseb, A.O. First-in-Man Phase I Clinical Trial Evaluating TTI-101, an Orally Bioavailable, Small Molecule Inhibitor of STAT3, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2021, 39, TPS3158. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef]
- Matsuda, Y.; Yamagiwa, Y.; Fukushima, K.; Ueno, Y.; Shimosegawa, T. Expression of Galectin-3 Involved in Prognosis of Patients with Hepatocellular Carcinoma. Hepatol. Res. 2008, 38, 1098–1111. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 Fosters HCC Development by Enhancing TGF-β-Mediated Alternative Activation of Macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Yarchoan, M.; Xing, D.; Luan, L.; Xu, H.; Sharma, R.B.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the Immune Microenvironment in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New Insights into the Role of EMT in Tumor Immune Escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef]
- Ye, L.-Y.; Chen, W.; Bai, X.-L.; Xu, X.-Y.; Zhang, Q.; Xia, X.-F.; Sun, X.; Li, G.-G.; Hu, Q.-D.; Fu, Q.-H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune Escape to PD-L1/PD-1 Blockade: Seven Steps to Success (or Failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-Inducible Antagonist of TGF-Beta Signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of Transforming Growth Factor Beta in Human Disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef]
- Eisenstein, E.M.; Williams, C.B. The T(Reg)/Th17 Cell Balance: A New Paradigm for Autoimmunity. Pediatr. Res. 2009, 65, 26R–31R. [Google Scholar] [CrossRef]
- Roes, J.; Choi, B.K.; Cazac, B.B. Redirection of B Cell Responsiveness by Transforming Growth Factor Beta Receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 7241–7246. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Santangelo, A.; Castioni, N.; Portararo, P.; Gulino, A.; Botti, L.; Parenza, M.; Cappetti, B.; Orlandi, R.; et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep. 2016, 17, 233–248. [Google Scholar] [CrossRef]
- Audrito, V.; Managò, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Martin, A.M.; Nirschl, T.R.; Nirschl, C.J.; Francica, B.J.; Kochel, C.M.; Van Bokhoven, A.; Meeker, A.K.; Lucia, M.S.; Anders, R.A.; DeMarzo, A.M.; et al. Paucity of PD-L1 Expression in Prostate Cancer: Innate and Adaptive Immune Resistance. Prostate Cancer Prostatic Dis. 2015, 18, 325–332. [Google Scholar] [CrossRef]
- Tovoli, F.; De Lorenzo, S.; Trevisani, F. Immunotherapy with Checkpoint Inhibitors for Hepatocellular Carcinoma: Where Are We Now? Vaccines 2020, 8, 578. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Kudo, M. Scientific Rationale for Combined Immunotherapy with PD-1/PD-L1 Antibodies and VEGF Inhibitors in Advanced Hepatocellular Carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated Efficacy and Safety Data from IMbrave150: Atezolizumab plus Bevacizumab vs. Sorafenib for Unresectable Hepatocellular Carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Krug, S.; Mattheis, L.; Haemmerle, M.; Rosendahl, J.; Kleeff, J.; Michl, P. The Impact of Atezolizumab and Bevacizumab in Hepatocellular Carcinoma with Activated SS-Catenin Signaling. Cancer Rep. 2022, 5, e1493. [Google Scholar] [CrossRef]
- Kuwano, A.; Yada, M.; Narutomi, F.; Nagasawa, S.; Tanaka, K.; Kurosaka, K.; Ohishi, Y.; Masumoto, A.; Motomura, K. Therapeutic Efficacy of Atezolizumab plus Bevacizumab for Hepatocellular Carcinoma with WNT/β-Catenin Signal Activation. Oncol. Lett. 2022, 24, 216. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Kudo, M. Combination Cancer Immunotherapy in Hepatocellular Carcinoma. Liver Cancer 2018, 7, 20–27. [Google Scholar] [CrossRef]
- Kelley, R.K.; Rimassa, L.; Cheng, A.-L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus Atezolizumab versus Sorafenib for Advanced Hepatocellular Carcinoma (COSMIC-312): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Merck and Eisai Provide Update on Phase 3 LEAP-002 Trial Evaluating KEYTRUDA® (Pembrolizumab) Plus LENVIMA® (Lenvatinib) versus LENVIMA Monotherapy in Patients with Unresectable Hepatocellular Carcinoma. Available online: https://www.merck.com/news/merck-and-eisai-provide-update-on-phase-3-leap-002-trial-evaluating-keytruda-pembrolizumab-plus-lenvima-lenvatinib-versus-lenvima-monotherapy-in-patients-with-unresectable-hepatocellul/ (accessed on 23 August 2022).
- Lipson: Initial Experience Administering BMS-986016. Available online: https://scholar-google-com.ezproxy.unibo.it/scholar_lookup?hl=en&volume=4&publication_year=2016&pages=232-232&author=E+Lipsonauthor=A+Gopalauthor=SS+Neelapu&title=Initial+experience+administering+BMS-986016%2C+a+monoclonal+antibody+that+targets+lymphocyte+activation+gene+%28LAG%29-3%2C+alone+and+in+combination+with+nivolumab+to+patients+with+hematologic+and+solid+malignancies (accessed on 23 August 2022).
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Klempner, S.J.; Chao, J.; Uronis, H.E.; Sirard, C.A.; Kagey, M.; Baum, J.; Song, J.; Wang, J.; Kim, I.-H.; Lee, K.W.; et al. DKN-01 and Tislelizumab ± Chemotherapy as a First-Line (1L) and Second-Line (2L) Investigational Therapy in Advanced Gastroesophageal Adenocarcinoma (GEA): DisTinGuish Trial. J. Clin. Oncol. 2022, 40, 292. [Google Scholar] [CrossRef]
- FDA Grants ODD Status to TTI-101 for Hepatocellular Carcinoma. Available online: https://www.targetedonc.com/view/fda-grants-odd-status-to-tti-101-for-hepatocellular-carcinoma (accessed on 23 August 2022).
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Brun, S.; Bestion, E.; Raymond, E.; Bassissi, F.; Jilkova, Z.M.; Mezouar, S.; Rachid, M.; Novello, M.; Tracz, J.; Hamaï, A.; et al. GNS561, a Clinical-Stage PPT1 Inhibitor, Is Efficient against Hepatocellular Carcinoma via Modulation of Lysosomal Functions. Autophagy 2022, 18, 678–694. [Google Scholar] [CrossRef]
- Harding, J.J.; Awada, A.; Roth, G.; Decaens, T.; Merle, P.; Kotecki, N.; Dreyer, C.; Ansaldi, C.; Rachid, M.; Mezouar, S.; et al. First-In-Human Effects of PPT1 Inhibition Using the Oral Treatment with GNS561/Ezurpimtrostat in Patients with Primary and Secondary Liver Cancers. Liver Cancer 2022, 11, 268–277. [Google Scholar] [CrossRef]
- Fierce Biotech. Eli Lilly Cuts 3 Cancer Drugs Amid Q4 Clear-Out. Available online: https://www.fiercebiotech.com/biotech/eli-lilly-cuts-three-cancer-drugs-amid-q4-clear-out (accessed on 24 August 2022).
- Hsu, C.; Chang, Y.-F.; Yen, C.-J.; Lu, L.-C.; Zhu, X.; Xu, Y.; Zhou, Q.; Dong, X.; Tong, Y. Safety and Efficacy of Combination of GT90001, an Anti-Activin Receptor-like Kinase-1 (ALK-1) Antibody, and Nivolumab in Patients with Metastatic Hepatocellular Carcinoma (HCC). J. Clin. Oncol. 2021, 39, 326. [Google Scholar] [CrossRef]
- Melisi, D.; Frizziero, M.; Tamburrino, A.; Zanotto, M.; Carbone, C.; Piro, G.; Tortora, G. Toll-Like Receptor 9 Agonists for Cancer Therapy. Biomedicines 2014, 2, 211–228. [Google Scholar] [CrossRef]
- Humbert, M.; Guery, L.; Brighouse, D.; Lemeille, S.; Hugues, S. Intratumoral CpG-B Promotes Antitumoral Neutrophil, CDC, and T-Cell Cooperation without Reprograming Tolerogenic PDC. Cancer Res. 2018, 78, 3280–3292. [Google Scholar] [CrossRef]
- Writer, G.S. Incyte, Merck & Co. Halt Phase III Trial after Epacadostat/Keytruda Combination Fails in Melanoma; GEN Genetic Engineering and Biotechnology News: New Rochelle, NY, USA, 2018. [Google Scholar]
- Albillos, A.; Lario, M.; ÁLvarez-Mon, M. Cirrhosis-Associated Immune Dysfunction: Distinctive Features and Clinical Relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef]
- Nguyen, P.H.D.; Wasser, M.; Tan, C.T.; Lim, C.J.; Lai, H.L.H.; Seow, J.J.W.; DasGupta, R.; Phua, C.Z.J.; Ma, S.; Yang, J.; et al. Trajectory of Immune Evasion and Cancer Progression in Hepatocellular Carcinoma. Nat. Commun. 2022, 13, 1441. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 Inhibition Enables NASH-HCC Immunotherapy. Gut 2022, 71, 2093–2106. [Google Scholar] [CrossRef]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed Death Ligand 1 Expression in Hepatocellular Carcinoma: Relationship With Clinical and Pathological Features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- The 2019 WHO Classification of Tumours of the Digestive System—PMC. Available online: https://www-ncbi-nlm-nih-gov.ezproxy.unibo.it/pmc/articles/PMC7003895/ (accessed on 19 September 2022).
- Omori, G.; Osuga, T.; Miyanishi, K.; Hamaguchi, K.; Tanaka, S.; Ohnuma, H.; Murase, K.; Takada, K.; Nagayama, M.; Kimura, Y.; et al. Programmed Cell Death Ligand 1 Expression in a Case of Poorly Differentiated Lymphocyte-Rich Hepatocellular Carcinoma. Clin. Case Rep. 2021, 9, e04764. [Google Scholar] [CrossRef]
- Reig, M.; Rimola, J.; Torres, F.; Darnell, A.; Rodriguez-Lope, C.; Forner, A.; Llarch, N.; Ríos, J.; Ayuso, C.; Bruix, J. Postprogression Survival of Patients with Advanced Hepatocellular Carcinoma: Rationale for Second-Line Trial Design. Hepatology 2013, 58, 2023–2031. [Google Scholar] [CrossRef]
- Terashima, T.; Yamashita, T.; Takata, N.; Nakagawa, H.; Toyama, T.; Arai, K.; Kitamura, K.; Yamashita, T.; Sakai, Y.; Mizukoshi, E.; et al. Post-Progression Survival and Progression-Free Survival in Patients with Advanced Hepatocellular Carcinoma Treated by Sorafenib. Hepatol. Res. 2016, 46, 650–656. [Google Scholar] [CrossRef]
- Bruix, J. Endpoints in Clinical Trials for Liver Cancer and Their Value in Evidence-Based Clinical Decision Making: An Unresolved Gordian Knot. J. Hepatol. 2021, 74, 1483–1488. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Villanueva, A. Randomized Trials and Endpoints in Advanced HCC: Role of PFS as a Surrogate of Survival. J. Hepatol. 2019, 70, 1262–1277. [Google Scholar] [CrossRef]
- Stefanini, B.; Bucci, L.; Santi, V.; Reggidori, N.; Rampoldi, D.; Lani, L.; Granito, A.; Sangiovanni, A.; Cabibbo, G.; Farinati, F.; et al. Potential Feasibility of Atezolizumab-Bevacizumab Therapy in Patients with Hepatocellular Carcinoma Treated with Tyrosine-Kinase Inhibitors. Dig. Liver Dis. 2022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTLA-4 | PD-1 | PD-L1 | LAG-3 |
|---|---|---|---|
| Ipilimumab | Nivolumab | Durvalumab | Relatlimab |
| Tremelimumab | Pembrolizumab | Avelumab | |
| Camrelizumab | Atezolizumab | ||
| Dostarlimab | |||
| Toripalimab | |||
| Spartalizumab | |||
| Cempilimab | |||
| Sintilimab | |||
| Serpulimab | |||
| Nofazinlimab | |||
| Penpulimab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lorenzo, S.; Tovoli, F.; Trevisani, F. Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers 2022, 14, 4616. https://doi.org/10.3390/cancers14194616
De Lorenzo S, Tovoli F, Trevisani F. Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers. 2022; 14(19):4616. https://doi.org/10.3390/cancers14194616
Chicago/Turabian StyleDe Lorenzo, Stefania, Francesco Tovoli, and Franco Trevisani. 2022. "Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma" Cancers 14, no. 19: 4616. https://doi.org/10.3390/cancers14194616
APA StyleDe Lorenzo, S., Tovoli, F., & Trevisani, F. (2022). Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers, 14(19), 4616. https://doi.org/10.3390/cancers14194616