Simple Summary
In this review article, we highlight the process and importance of identifying neoantigens for immunotherapy for cancer. Although the process of identifying neoantigens may be time-consuming and costly, various efforts are being conducted at different stages of pre-clinical and clinical development to improve the process and identify neoantigens. This initiative will be imperative in developing the next generation of cost-efficient and potent immunotherapies against cancer.
Abstract
The era of personalized cancer therapy is here. Advances in the field of immunotherapy have paved the way for the development of individualized neoantigen-based therapies that can translate into favorable treatment outcomes and fewer side effects for patients. Addressing challenges related to the identification, access, and clinical application of neoantigens is critical to accelerating the development of individualized immunotherapy for cancer patients.
1. Introduction
Immunotherapy has rapidly become a primary mode of cancer treatment due to the remarkable success and clinical improvement seen in patients with advanced-stage or aggressive, recurrent cancers such as melanoma, ovarian cancer, breast cancer, and gastrointestinal cancers [1]. A significant number of therapies that have seen success to date involve the utilization of T cells. This is because of the tumor cell’s unique ability to suppress the immune system by modulating its surrounding microenvironment, also known as the tumor microenvironment (TME). By targeting the TME with antigen-specific immune cells, we are able to essentially activate the immune system to mount an anti-cancer attack.
There has been much interest in identifying, developing, and producing immune cells and proteins that are able to mount a potent immune response against the tumor cells and their TME. This vested interest has led to the production of various immunotherapies including monoclonal antibodies (mAbs), tumor-infiltrating lymphocytes (TIL), and chimeric antigen receptor T-cells (CAR-T). Despite these advancements, due to the metabolic reprogramming and adaptability of cancer cells, these immunotherapies have been met with both innate and acquired resistance [2,3]. This has led to the search for new immunotherapy targets and has opened a new hallmark of cancer immunotherapy.
Neoantigens are tumor-specific proteins synthesized by tumor cells as protein by-products [4,5]. The rapid division and proliferation of tumor cells lead to various mutations in coding and non-coding loci [4,5]. The changes in the amino acid sequence that occur due to mutations in coding regions leads to the production of proteins that are not found in normal cells and are unique to tumor cells [4,5,6]. These tumor-specific by-products termed neoantigens are unique in that multiple patients can share the same neoantigens (shared neoantigens), but they can also be specific to an individual (personalized neoantigens) [4,5,6]. In addition, neoantigens are highly immunogenic, making them a favorable immunotherapy target [5,7]. Thus, the identification of neoantigens and the development of therapies targeting them highlights a promising field that may yield the next generation of cancer treatments and advance the field of personalized medicine. This review will focus on the current available immunotherapies as well as the identification, manufacturing, and clinical applications of neoantigens.
2. Cancer and Immunotherapy
The relationship between cancer and the immune system is very dynamic. Cancer cells modulate the immune system to survive, proliferate, and metastasize. For instance, tumor cells express immuno-suppressive ligands on the cell surface such as programmed death ligand-1 (PDL-1) to evade immune cell recognition and inhibit the mounting of an immune response. In addition, tumor cells can also hijack and employ crosstalk to mediate immune cells and inflammation to aid with metastasis [8,9]. The basis of immunotherapy is to target these specific areas of crosstalk, intervene, and modulate the immune response to target the cancer cells. The ultimate goal of immunotherapy is to enhance recognition, target, and mount a toxic response against the cancer cells [4].
Monoclonal antibodies (mAbs) are a type of targeted immunotherapy that has shown promising therapeutic results that is currently being used to treat various cancers including breast cancer, colorectal cancer, and leukemias [5,7,10]. For instance, mAbs can mount a potent immune response and can be employed to trigger cytotoxicity and inhibit further tumorigenesis (Figure 1A) [1,11,12]. They are able to recognize both cell surface antigens and secreted antigens with high specificity, indicating their versality [10].
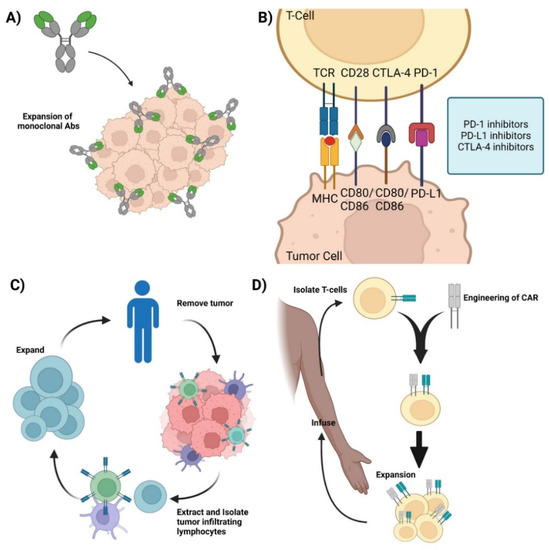
Figure 1.
Current immunotherapies. (A) Monoclonal antibodies—representation of mAbs and the mechanism involved in mounting an immune response against cancer cells. (B) Immune checkpoint inhibitors—Graphic showcasing the immune crosstalk that occurs between T cells and tumor cells and the respective ligands and receptors targeted using mAbs. (C) Adoptive Cell Therapy—Graphic showing how TILs are synthesized by removing the respective tumor from the patient, isolating, extracting, and expanding the appropriate TILs, and its re-infusion back into the patient for therapy. (D) Chimeric Antigen Receptor T-cell therapy—Representation of CAR-T synthesis and production. Isolate T cells from the patient and mount a synthetic CAR onto the host T cells. Expand the modified T cell population and re-infuse back into the patient. Credits: Images were created using BioRender.
One of the more successful therapeutic mAbs are immune checkpoint inhibitors (ICIs) [4] This is possible because of the T cells’ unique ability to moderate their own immune response. T cells keep the immune system from becoming overly active and destructive. This self-modulating mechanism is conducted by immune checkpoint proteins such as CTLA-1 and PD-1, where the binding of the respective receptors and ligands on T cells induces a regulatory response and curtails the immune response [13,14]. Cancer cells take advantage of this self-regulating function of T cells by synthesizing and presenting these markers to the cell surface, inhibiting the T cell from mounting a response, allowing the cancer cell to evade apoptosis and survive [1,11,12]. ICIs target these immunosuppressive ligands and receptors and renders them inactive, thereby turning on and activating the immune system (Figure 1B). By targeting these immune checkpoint markers, cancer cells are unable to regulate or evade the immune system, making them susceptible to apoptosis. Retrospective studies have shown that ICIs, such as the CTLA-4 mAb, ipilimumab, are effective for the treatment of metastatic melanoma [15,16,17,18]. Additionally, the combination of anti-PD-1/PD-L1 and anti-CTLA-4 mAbs has been shown to be therapeutically responsive in treating various cancers including hepatocellular carcinoma and ovarian cancer [19,20,21,22].
Adoptive cell therapy (ACT) is a passive immunotherapy that uses tumor-infiltrating lymphocytes (TIL) to induce tumor-suppressive or cytotoxic effects. ACT entails isolating and extracting immune cells from patients, inducing cell differentiation ex vivo, and expanding and re-infusing the cells back into the patients (Figure 1C). It is used to grow a large army of immune cells outside the body that when re-infused back into the patient lead a potent immune response against antigen-specific tumor cells [6,7]. TIL therapy has shown significant results in the treatment of several cancers, especially metastatic melanoma [15,16,17,18,22]. One of the major advantages of utilizing TILs for immunotherapy is the diverse repertoire of antigens that TILs can recognize.
Other ACTs include T-cell Receptor-engineered T cell (TCR-T) and Chimeric Antigen Receptor T-cell (CAR-T) therapies. TCR-T therapy uses genetically modified natural T cells to target and destroy tumors and relies on the interaction between the peptide-MHC complex to mount a potent cytotoxic response against the tumor. TCR-T has been used to target various tumors and hematologic malignancies and current studies have shown TCR-T to be more effective against solid tumors rather than hematologic malignancies [7,23]. In contrast, CAR-T therapy uses genetically engineered T cells that can mount a cytotoxic response without the peptide–MHC complex interaction (Figure 1D) and has shown clinical efficacy against hematologic malignancies, with efficacy against solid tumors currently being thoroughly investigated [24,25].
Despite significant increases in overall survival and the therapeutic success found with these immunotherapies, they are not without adverse effects or complications. Immunotherapeutic agents have led to the development of serious (and sometimes fatal) immune-related adverse events (irAEs) in a subset of patients [26]. Therapeutic response rates have also varied among patients and different solid tumor types [14,27] In addition, resistance to these immunotherapies is an unfortunate complication that limits the duration and efficacy of treatment. Although there have been various methods to combat these barriers including the use of a combination of mAbs to induce a polyclonal response, there still exists room for optimization and further development [1,11,12,24,25]. With the advancements in high-throughput sequencing, prediction algorithms, and screening and characterization technology, the focus of immunotherapy has turned towards neoantigens.
3. Neoantigens
Tumor antigens can be divided into three different categories: Tumor Associated Antigens (TAAs), Cancer Testis Antigens (CTAs), and Tumor Specific Antigens (TSAs). TAAs are considered self-antigens that are expressed in both normal and cancer cells. These TAAs are abundantly expressed in cancer cells and can elicit an anti-tumor immune response. CTAs are another type of tumor-specific antigens that are only expressed in germline cells. Developing therapies against CTAs thus allows us to target germline antigens without damaging the somatic cells [28,29]. Recent progress in cancer therapy has focused on targeting TSAs. TSAs are foreign, typically arising from genomic mutations or viruses, and thus are not considered self-antigens [4,5,6,30]. They are unique to the tumor genome, making them excellent therapeutic targets. Targeting TSAs allows us to attack the cancer cells specifically without affecting normal cells.
Neoantigens are a type of TSA. They are unique protein by-products that arise from the genetic instability and aberrant mutations of tumor cells [5,6,7,8]. These neoantigens are specific to cancer cells and are not found in normal tissue. They vary from patient to patient and can also be found across multiple cancers [4,5,6,7]. Neoantigens that are unique to the individual cancer patient are referred to as personalized neoantigens. Targeting these personalized neoantigens leads to the development of personalized therapy. On the other hand, neoantigens can also be found across multiple cancer patients that are not found in the normal genome and are known as shared neoantigens (Figure 2). Shared neoantigens play a role in driving tumor growth and consist of most major oncogenes, including BRAF mutations in melanoma and KRAS mutations in pancreatic, colorectal, and endometrial cancers [5,6,7,13]. Additionally, these molecules are less prone to being lost as targets due to the essential nature of their mutations and imperative role in tumor growth and metastasis. The development of therapies against shared neoantigens is favorable as they hold the potential to treat a broad population of patients [4].
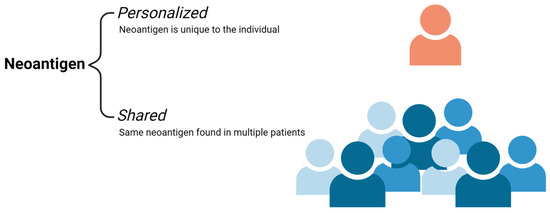
Figure 2.
Neoantigen Subtypes. Personalized neoantigens are antigens secreted from the tumor that is unique to the individual. Shared neoantigens are tumor unique antigens that are found across multiple individuals or a subset of a patient population. Credits: Images were created using BioRender.
4. Identification of Neoantigens
The current methodology of identifying neoantigens includes an exhaustive list of sequencing, quality control checks, realignments of tumor and normal sequencing data, and comparing the normal and tumor alignments to determine somatic mutations [31]. These somatic mutations must then be converted to their corresponding mutated peptide sequences and undergo human leukocyte antigens (HLA)-allele typing [31]. Once the HLA-allele and mutated epitope affinity is assessed using prediction algorithms such as netMHCpan, it is henceforth called a neoantigen. Afterwards, the ability of these neoantigens to be recognized by immune cells are determined by running various immunological assays [31,32].
There are multiple areas within these steps that require quality control including the maintenance and handling of the tumor samples and the sequencing and alignment data. The purity of the tumor samples must be maintained as it can easily decrease the sensitivity of the somatic mutation calling [31,32]. In addition, there are multiple methods and algorithms that are used to help optimize and lower the false positive rates with the sequencing and alignments [31]. The most important step in this multi-step process is the prediction of the HLA binding capacity to the mutated peptide. These prediction algorithms assess and rank the HLA-peptide by their binding affinity which corresponds to the neoantigen’s ability to be recognized by the immune system [31,32,33].
Though neoantigens are tumor specific, immunogenic, and have shown good clinical results, the proportion of neoantigens with immunological significance is surprisingly low [4,31]. In addition, the identification of these immunogenic neoantigens is laborious, costly, and time-consuming, highlighting one of the challenges in developing therapies using personalized neoantigens. This limitation can be addressed by targeting shared neoantigens which are known to be broadly recurrent and immunogenic amongst cancer patients [31].
5. Clinical Applications of Neoantigens
Once a neoantigen is identified and its immunological significance has been verified, it can be applied in various clinical scenarios including diagnostic and treatment screening and various immune-based therapies.
6. Neoantigen-Based Biomarkers
Multiple studies have investigated the relationship between neoantigens and disease progression and have shown that a higher neoantigen load is associated with better overall survival in cancer patients [6,34]. Furthermore, studies have also shown metastatic progression occurring with the loss of neoantigens [6]. These findings suggest the possible role of neoantigens as biomarkers of tumor burden and disease progression as well as patient survival, adding to the growing interest in identifying neoantigens. Currently, there are nine actively recruiting clinical trials dedicated to identifying neoantigens across various cancers and patient populations (Table 1).

Table 1.
Clinical Trials Identifying Neoantigens.
7. Neoantigen-Based Therapies
As mentioned, tumor neoantigens are specific to the tumors of individual patients and are highly immunogenic making them a favorable target in cancer therapy [29,35]. They also are highly specific, have numerous targets, and have broad efficacy [35]. By capitalizing on new sequencing technologies that help identify particularly relevant neoantigens, immunotherapy agents including neoantigen-specific immune cells and neoantigen-based vaccines can be further developed and optimized for clinical potency [7,36].
7.1. Neoantigen-Specific TCR-T Cell Therapy
An extension of neoantigen specific T cells is TCR-T cell therapy. It is an adaptation of the current TCR-T cell therapy, where a TCR is genetically engineered to be specific for the identified neoantigens. These engineered TCR T cells (TCR-T) targeting tumor-specific antigens are expanded and infused into the patient as a form of therapy. This therapy combines the genetic engineering of TCRs and the sequencing and mutation alignment algorithms in identifying neoantigens to create a synthetic TCR-T targeting individual or multiple tumor specific antigens.
Neoantigen-specific T cells can be seen as an adaptation to ACT. The idea is to essentially build a large army of T-cells that recognize and bind the patient or tumor-specific neoantigens to mount a cytotoxic response against the tumor. Once a neoantigen is identified, autologous T cells from the patient are obtained and undergo stimulation by neoantigen presentation by APCs. The stimulated T cells are then isolated, expanded, and infused back into the patient (Figure 3A). These neoantigen-specific T cells are expected to recognize the same neoantigens and launch a cytotoxic immune response against the tumor.
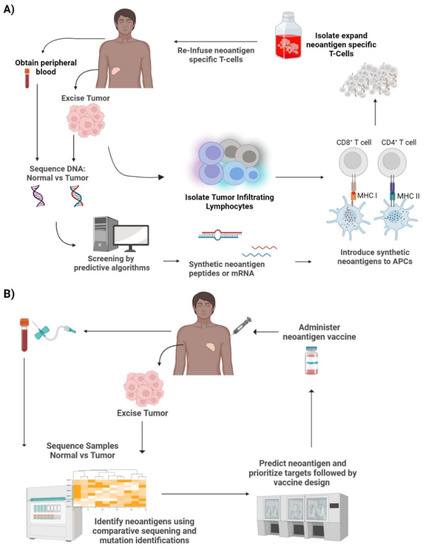
Figure 3.
Neoantigen therapies. (A) Neoantigen specific T-cell therapy—Tumor cells biopsied from the patient can be used for exome/transcriptome sequencing and computational screening to create synthetic neoantigen DNA, mRNA, or peptides. These antigens are processed by APCs and presented to T cells leading to T cell activation. The activated T cells with TCRs specific to the neoantigens are then isolated and expanded. This is then reinfused back into the patient. (B) Neoantigen vaccines—Tumor cells excised from the patient undergo comparative sequencing and mutation identification with the patient’s normal cells. This is then used to predict neoantigens and prioritize targets. This is followed by vaccine design, production, and finally administered to the patient. Credits: Images were created using BioRender.
The development of neoantigen-specific T cells is a multi-step process that requires the meticulous handling of samples and expertise in computational analysis (Figure 3A). First, samples from the tumor and normal tissue or blood are taken from the patient. This is then followed by genomic sequencing and somatic mutation calling followed by sequence comparison and alignment. Once the tumor-specific mutations are identified, exome and transcriptome sequencing and computational screening are performed to create synthetic neoantigen DNA, mRNA, or peptides. These synthetic neoantigens are introduced to APCs, where they are processed and mounted onto their respective HLAs. Following the identification of neoantigens, T cells are also isolated and enriched for antigen-specific T cells in lieu of bulk T cells. These cells undergo antigen stimulation by the processed neoantigen-HLA bound APCs. In order to identify the stimulated T cells, co-stimulatory molecules, such as CD137, are screened using flow cytometry to ensure antigen-specific T cell activation [13,17,18,22]. The expression of CD137 is transient and can be seen on CD4+ and CD8 + T cells. Therefore, it is imperative to investigate the role of antigen-specific T cells in both CD4+ and CD8+ subsets. Once antigen-specific T cell activation has been confirmed, the activated T cell populations are collected and expanded in vitro. This subset of T cells is neoantigen-specific TCR-T cells. These autologous T cells are infused back into the patient where they will launch an immune response upon recognition of the neoantigen.
Currently, there are nine active clinical trials involving neoantigen-specific TCR-T cell therapy and one study that has been terminated (Table 2). In addition, there are two actively recruiting neoantigen-specific TCR-T cell clinical trials (Table 2). These TCR-T therapies have been on the rise and have shown promising therapeutic results against cancers particularly in ovarian cancer and epithelial cancer [37,38,39].
Table 2.
Clinical Trials with Neoantigen specific TCR-T cell therapy.
7.2. Neoantigen Vaccines
Historically, cancer vaccines were developed to target TAAs. However, TAA vaccines met limited therapeutic success as they were often unable to elicit a potent immune response to avoid autoimmunity [2]. This can be attributed to the lack of tumor specificity as TAAs are present in both normal and tumor cells [28,29]. Neoantigen-based vaccines are more beneficial than TAA-based vaccines as neoantigens are expressed solely by tumor cells [40]. The unique expression pattern of neoantigens can be utilized to avoid autoimmunity and mount a potent immune response possibly leading to immunological memory development and protection against tumor recurrence [7,12,36].
Neoantigen-based vaccines are composed of immunogenic neoantigens that are designed to ultimately induce de novo cytotoxic T cell responses [7,29,35]. Once the vaccine is administered, the neoantigens are processed and presented to T cells by APCs. Once recognized, the T cells will become activated and subsequently mount a cytotoxic response and infiltrate into the tumor cells, turning “cold” tumors into “hot” tumors [27,41]. Cold tumors are tumors that generally do not respond to immunotherapy, while hot tumors usually respond to immunotherapy due to the infiltration of T cells and pro-inflammatory cytokines [27,41].
The development and manufacturing of neoantigen vaccines is a multi-step and laborious process (Figure 3B). There are various types of neoantigen cancer vaccines that have been developed including peptide vaccines, mRNA vaccines, DNA vaccines, viral vaccines, and dendritic cell (DC) vaccines [30,42]. The first step involves obtaining a biopsy of tumor tissue and normal tissue or peripheral blood samples. These samples are then screened and compared to identify and verify tumor-specific mutations by exome sequencing. This is then followed by sequence alignment and computational analysis to predict the affinity of potential neoantigens to respective HLAs. Once the prediction algorithms rank the neoantigen-HLA binding affinity and neoantigen candidates are selected, vaccine design and development ensues and finally administered to the patient. There is not a set schedule for vaccine administration; they seem to differ based on vaccine type, cancer type, response rate, and the patient.
There have been multiple clinical studies that developed and administered neoantigen vaccines to treat cancers, including gastrointestinal cancers, melanoma, and nasopharyngeal cancers, with promising results [38,43,44]. Previous studies found that neoantigen vaccines are best used in cancers with high rates of somatic mutations and vaccines with a larger load of specific neoantigen peptides increased the potential of a favorable clinical response [4,5,6,30]. In addition, a prior study used genomic and bioinformatics to identify shared neoantigens in patients with refractory solid tumors and demonstrated a durable response and stabilization of the diseases [45]. Currently, there are 71 clinical trials that have utilized or are utilizing neoantigen-based vaccines to treat multiple neoplasms (Table 3). Personalized neoantigen vaccines and/or neoantigen vaccines in conjunction with other immunotherapies are being studied in various cancers including small cell lung cancer, pancreatic cancer, kidney tumors, and more broadly solid tumors to determine its efficacy and therapeutic potential (Table 3).
Table 3.
Clinical Trials with Neoantigen Vaccines.
Though prior studies have shown promising results, these current ongoing trials will help further our understanding of neoantigen and its vaccines including its safety, duration of efficacy, and more. These studies also highlight a quickly advancing niche in cancer immunotherapy that can lead to the production of numerous immunotherapies against a wide array of cancers and personalized medicine.
8. Limitations and Challenges
There are several limitations to the identification of neoantigens and the implementation of neoantigen-based therapies in the clinical setting. First, tumor mutational burden may affect the effectiveness of such therapies, particularly neoantigen-targeted vaccines. For example, in a study looking into the identification and the use of mutations for neoantigen-targeted vaccines, a total of 962 mutations were reported [23]. Given that most human tumors contain an average of two to six mutations, the utility of neoantigen-targeted vaccination is questionable for tumors with low mutational burden [37,38].
The identification of neoantigens and biomarkers has been made possible by the emergence of techniques such as next-generation sequencing (NGS) and other high-throughput molecular technologies [45]. However, performing the large-scale identification of mutations can present a health care barrier for subsets of the population. For example, studies have found that compared with non-Hispanic whites, Black and Hispanic individuals were less likely to have had NGS testing, despite the Medicare national coverage determination [46,47,48]. Although more research is needed on this subject, these findings suggest that the identification of neoantigens is not equitable, paving the way for health care disparities in the field of immune-targeted therapy.
While TIL therapy is highly effective, the isolation and expansion of mature T cells from the TME increases the risk of terminal differentiation, which may limit the therapeutic effect when reintroducing the cells to the patient [16,49]. The optimal differentiation stage of the T cells, as well as the identification of the T cell receptors that recognize the neoantigens is currently being explored in both CD4+ and CD8+ subsets [7,13,28,50]. In addition, the majority of neoantigens that have been identified are restricted to MHC-I, whereas the role of MHC-II and CD4+ T cells is still being explored [6].
Another technical obstacle related to the creation of neoantigen-targeted therapies, is the identification and prioritization of neoantigens that are present on the cell surface of HLAs. Thus, optimizing the identification of immunogenic neoantigens is of great priority. Additionally, the accuracy in predicting the immunogenicity of tumor neoantigens is another limiting factor. Scientists are attempting to address this challenge by coming up with unique ways of computationally predicting which neoantigens are of interest [39].
The cost of neoantigen research as well as the time for development also presents as a limiting factor. As neoantigens are specific to each person, the treatment is highly personalized. This translates into a costly and time-consuming endeavor. For example, the timing of the development of neoantigen vaccines has been a deterrent, particularly with the pressure on laboratories for production [7,36]. Similarly, from the identification of gene mutations and neoantigens to the validation and production of the neoantigen-based treatment, the cost of personalized neoantigen treatment can be very expensive [7,36]. One strategy for cost reduction may include the development of vaccines targeted at shared neoantigens; this would also potentially significantly increase access of neoantigen vaccines to an appropriately applicable population [29,37,51]. Another strategy to reduce cost is the development of combination therapies of traditional immunotherapy drugs with neoantigen based therapies [7,36]. The development of a more improved and streamlined process for neoantigen-based therapy production to reduce labor and cost as well as the continued analysis of large cancer databases to identify shared neoantigens are some challenges and advances this field will have to address moving forward.
Last but not least, although the developments in immunotherapeutic agents have been promising and have advanced the field of cancer immunotherapy, the long-term safety and efficacy of these agents have yet to be thoroughly investigated. The irAEs, unknown clinical adverse events, rate of recurrence to remission, and rate of morbidity and mortality are possible limitations to the widespread utilization of neoantigen-based immunotherapies.
9. Conclusions
The field of immunotherapy has revolutionized the treatment of cancer. In recent years, neoantigen-based therapies have provided patients with alternative treatment options to whom standard immunotherapies were unable to be used or rendered ineffective. Although vaccine development against neoantigens is time-consuming, complex, and expensive, the potential to offset resistance, target specific cancers, and most importantly, personalize therapy holds immense clinical value and merit. The continued optimizations in engineering neoantigen-based therapies and continued investigations of its therapeutic efficacy and safety in clinical trials will be imperative in developing the next generation of cost-efficient and potent therapies against the ever-evolving battle against cancer.
Author Contributions
Conceptualization, Y.K., L.N., S.G., R.Y., K.T., J.C., F.R. and S.D.; figures and tables, Y.K., S.G., H.R., L.F., M.S. and N.H.; software, Y.K. and H.R.; writing—original draft preparation, Y.K., L.N., S.G., L.F., F.R. and J.C.; writing—review and editing, Y.K., F.R., J.C. and M.S.; visualization, H.R., L.F., M.S. and Y.K.; supervision, S.G. and J.C.; project administration, S.G. and J.C. All authors have read and agreed to the published version of the manuscript.
Funding
No funding was received for this review article.
Acknowledgments
S.G. is a CPRIT scholar in cancer research and is supported by a First-time Faculty Recruitment Award from the Cancer Prevention and Research Institute of Texas (CPRIT; RR170020). S.S.G. is also supported by the Lizanell and Colbert Coldwell Foundation. J.A.C is a CPRIT sub awardee for a pilot grant funded through the University of Texas Medical Branch (UTMB) Data Management and Analysis Core (DMAC).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACT | Adoptive Cell Therapy |
| APC | Antigen Presenting Cell |
| CAR-T | Chimeric Antigen Receptor-T cell |
| CTA | Cancer Testis Antigens |
| DC | Dendritic Cell |
| HLA | Human Leukocyte Antigen |
| IFNγ | Interferon-gamma |
| ICI | Immune Checkpoint Inhibitors |
| irAEs | Immune-Related Adverse Events |
| KRAS | Kirsten rat sarcoma virus |
| mAb | monoclonal antibodies |
| MHC | Major Histocompatibility Complex |
| NK cell | Natural Killer cell |
| NGS | Next Generation Sequencing |
| NSCLC | Non-Small Cell Lung Carcinoma |
| TAA | Tumor Associated Antigens |
| TSA | Tumor Specific Antigen |
| TCR | T-Cell Receptor |
| TCRm | T-Cell Receptor Mimic Antibodies |
| TIL | Tumor Infiltrating Lymphocyte |
| TME | Tumor Microenvironment |
References
- Marrocco, I.; Romaniello, D.; Yarden, Y. Cancer Immunotherapy: The Dawn of Antibody Cocktails. In Human Monoclonal Antibodies; Steinitz, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1904, pp. 11–51. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2019, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Zhang, Q.; Jia, Q.; Zhang, J.; Zhu, B. Neoantigens in precision cancer immunotherapy: From identification to clinical applications. Chin. Med. J. 2022; Publish Ah. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Itoh, Y.; Nagase, H. Matrix metalloproteinases in cancer. Essays Biochem. 2002, 38, 21–36. [Google Scholar] [CrossRef]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef]
- Pelletier, J.P.R.; Mukhtar, F. Passive Monoclonal and Polyclonal Antibody Therapies. In Immunologic Concepts in Transfusion Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 251–348. [Google Scholar] [CrossRef]
- Zahavi, D.J.; Weiner, L.M. Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy. Int. J. Mol. Sci. 2019, 20, 158. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Pasetto, A.; Jia, L.; Deniger, D.C.; Stevanović, S.; Robbins, P.F.; Rosenberg, S.A. Tumor-infiltrating human CD4 + regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol. 2019, 4, eaao4310. [Google Scholar] [CrossRef]
- Pearlman, A.H.; Hwang, M.S.; Konig, M.F.; Hsiue, E.H.-C.; Douglass, J.; DiNapoli, S.R.; Mog, B.J.; Bettegowda, C.; Pardoll, D.M.; Gabelli, S.B.; et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer 2021, 2, 487–497. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ghisoni, E.; Wicky, A.; Bouchaab, H.; Imbimbo, M.; Delyon, J.; Moura, B.G.; Gérard, C.; Latifyan, S.; Özdemir, B.; Caikovski, M.; et al. Late-onset and long-lasting immune-related adverse events from immune checkpoint-inhibitors: An overlooked aspect in immunotherapy. Eur. J. Cancer 2021, 149, 153–164. [Google Scholar] [CrossRef]
- Uhara, H.; Kiyohara, Y.; Uehara, J.; Fujisawa, Y.; Takenouchi, T.; Otsuka, M.; Uchi, H.; Fukushima, S.; Minami, H.; Hatsumichi, M.; et al. Five-year survival with nivolumab in previously untreated Japanese patients with advanced or recurrent malignant melanoma. J. Dermatol. 2021, 48, 592–599. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- El-Osta, H.; Jafri, S. Predictors for clinical benefit of immune checkpoint inhibitors in advanced non-small-cell lung cancer: A meta-analysis. Immunotherapy 2019, 11, 189–199. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.-F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 1–22. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Kunte, S.; Abraham, J.; Montero, A.J. Novel HER2–targeted therapies for HER2–positive metastatic breast cancer. Cancer 2020, 126, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tagliamonte, M. Selecting Target Antigens for Cancer Vaccine Development. Vaccines 2020, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27. [Google Scholar] [CrossRef]
- Sun, C.; Xu, S. Advances in personalized neoantigen vaccines for cancer immunotherapy. Biosci. Trends 2020, 14, 349–353. [Google Scholar] [CrossRef]
- Gopanenko, A.V.; Kosobokova, E.N.; Kosorukov, V.S. Main Strategies for the Identification of Neoantigens. Cancers 2020, 12, 2879. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Luo, H.; Kong, Y.; Lai, W.-F.; Cui, L.; Zhu, X. Cancer neoantigen: Boosting immunotherapy. Biomed. Pharmacother. 2020, 131, 110640. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Salazar, G.T.; Zhang, N.; An, Z. T-cell receptor mimic (TCRm) antibody therapeutics against intracellular proteins. Antib. Ther. 2019, 2, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.R.; Kim, S.P.; et al. Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef]
- Paria, B.C.; Levin, N.; Lowery, F.J.; Pasetto, A.; Deniger, D.C.; Parkhurst, M.R.; Yossef, R.; Kim, S.P.; Florentin, M.; Ngo, L.T.; et al. Rapid Identification and Evaluation of Neoantigen-reactive T-Cell Receptors From Single Cells. J. Immunother. 2020, 44, 1–8. [Google Scholar] [CrossRef]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Zhou, L.; Zou, F.; Sun, W. Prioritizing candidate peptides for cancer vaccines through predicting peptide presentation by HLA-I proteins. Biometrics 2022. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, X.-L.; You, R.; Yang, Q.; Zou, X.; Yu, K.; Liu, Y.-P.; Zou, R.-H.; Hua, Y.-J.; Huang, P.-Y.; et al. Neoantigen landscape in metastatic nasopharyngeal carcinoma. Theranostics 2021, 11, 6427–6444. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Siniard, R.C.; Harada, S. Immunogenomics: Using genomics to personalize cancer immunotherapy. Virchows Arch. 2017, 471, 209–219. [Google Scholar] [CrossRef]
- Sheinson, D.M.; Wong, W.B.; Meyer, C.S.; Stergiopoulos, S.; Lofgren, K.T.; Flores, C.; Adams, D.V.; Fleury, M.E. Trends in Use of Next-Generation Sequencing in Patients With Solid Tumors by Race and Ethnicity After Implementation of the Medicare National Coverage Determination. JAMA Netw. Open 2021, 4, e2138219. [Google Scholar] [CrossRef]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Christian, L.S.; Wang, L.; Lim, B.; Deng, D.; Wu, H.; Wang, X.-F.; Li, Q.-J. Resident memory T cells in tumor-distant tissues fortify against metastasis formation. Cell Rep. 2021, 35, 109118. [Google Scholar] [CrossRef]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorganic Med. Chem. 2017, 26, 2842–2849. [Google Scholar] [CrossRef]
- Lozano-Rabella, M.; Gros, A. TCR Repertoire Changes during TIL Expansion: Clonal Selection or Drifting? Clin. Cancer Res. 2020, 26, 4177–4179. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).