
An FDA-Approved Antifungal, Ketoconazole, and Its Novel Derivative Suppress tGLI1-Mediated Breast Cancer Brain Metastasis by Inhibiting the DNA-Binding Activity of Brain Metastasis-Promoting Transcription Factor tGLI1
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell-Based Chemical Screens
2.3. Mammosphere Assay
2.4. Selective Knockdown of tGLI1 Using Antisense Oligonucleotides (AS-ON)
2.5. Quantitative RT-PCR
2.6. Animal Studies
2.7. Production of tGLI1 Recombinant Protein
2.8. Electrophoretic Mobility Shift Assay
2.9. Chromatin Immunoprecipitation
2.10. Western Blotting
2.11. Promoter Reporter Assay
2.12. Statistical Analysis
3. Results
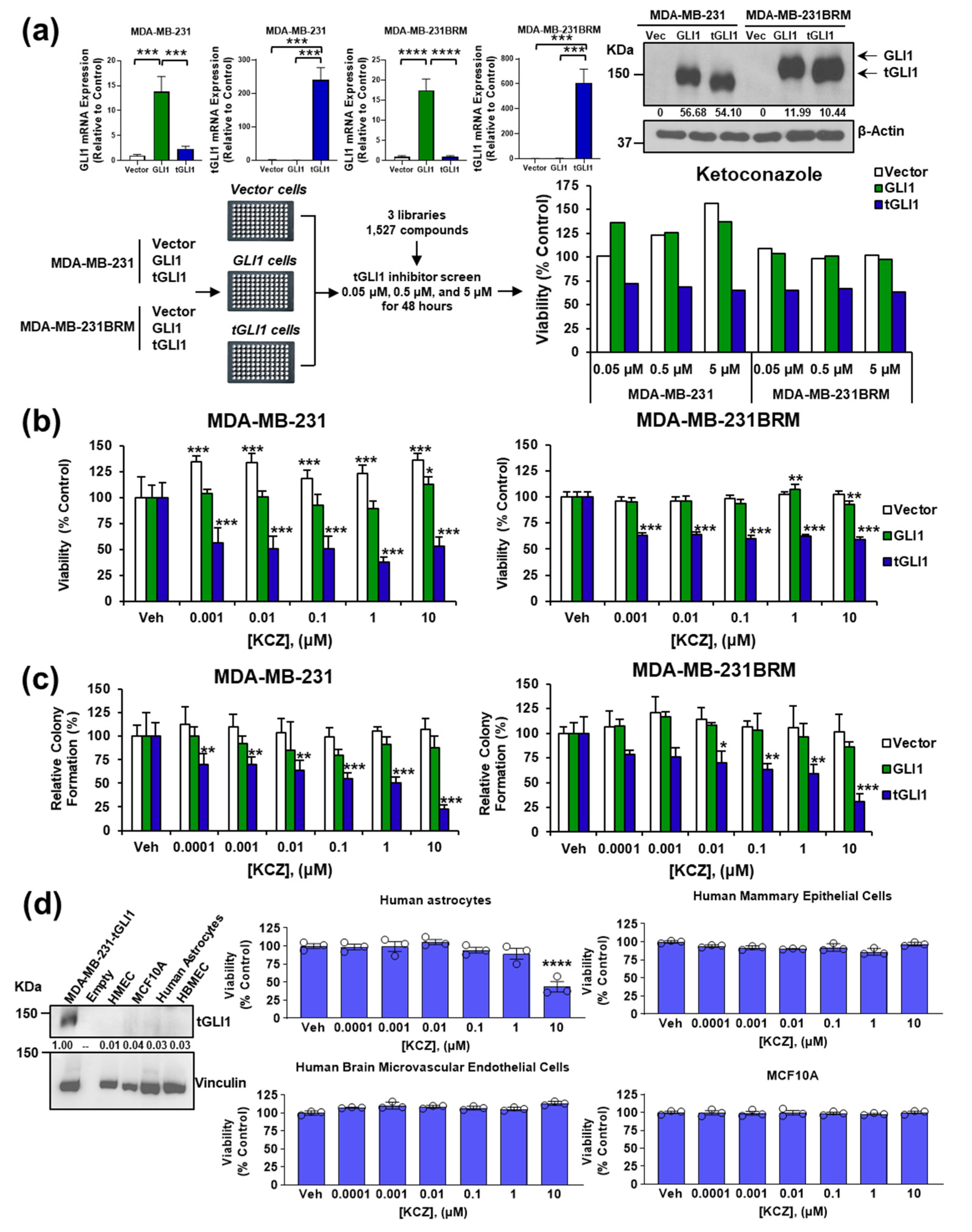
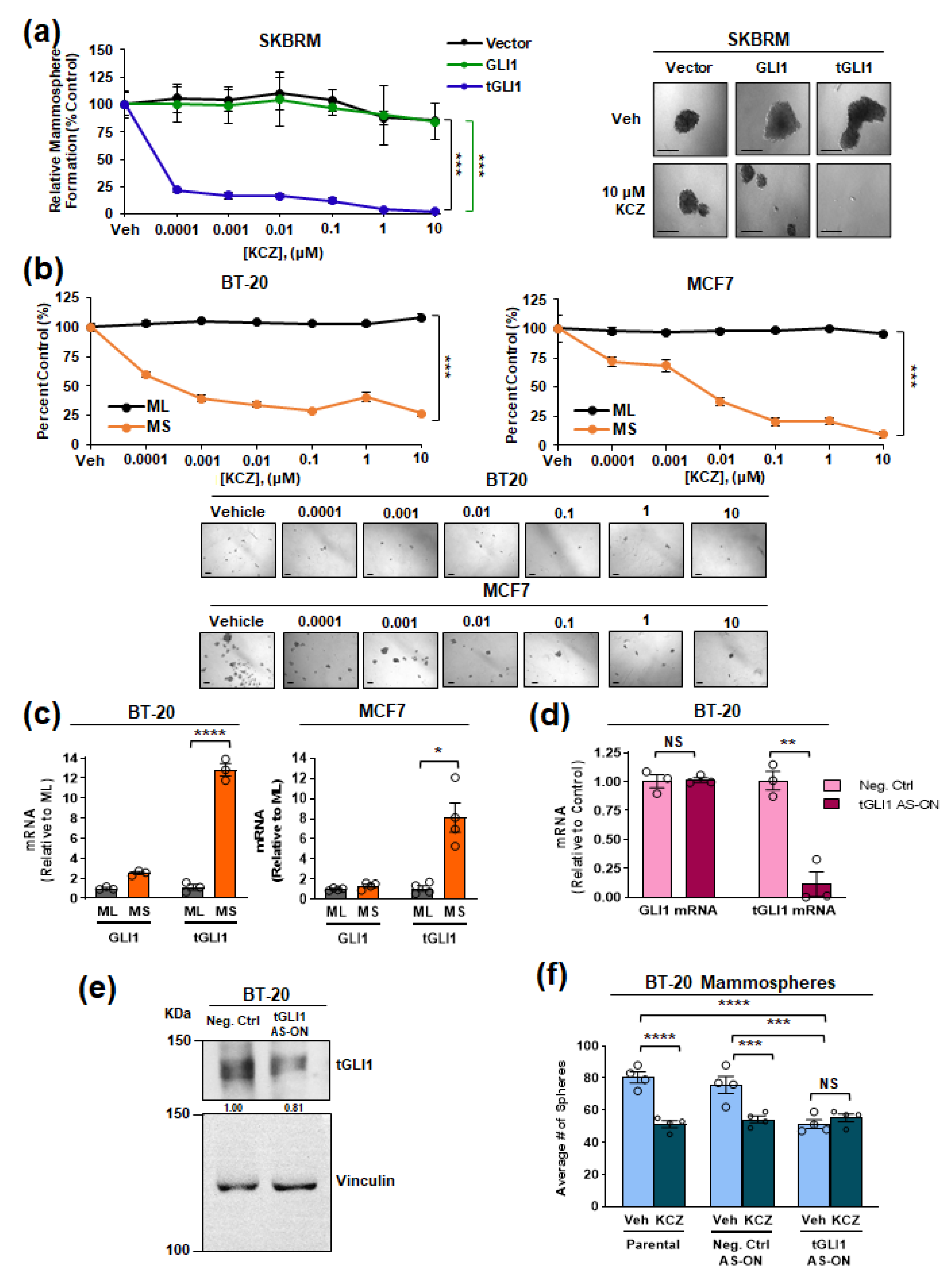
3.1. KCZ Selectively Inhibits Breast Cancer Cells Expressing tGLI1 and Displays Increased Potency against the CSC Population
3.2. tGLI1 Expression Is Required for the KCZ-Induced Suppression of Breast CSCs
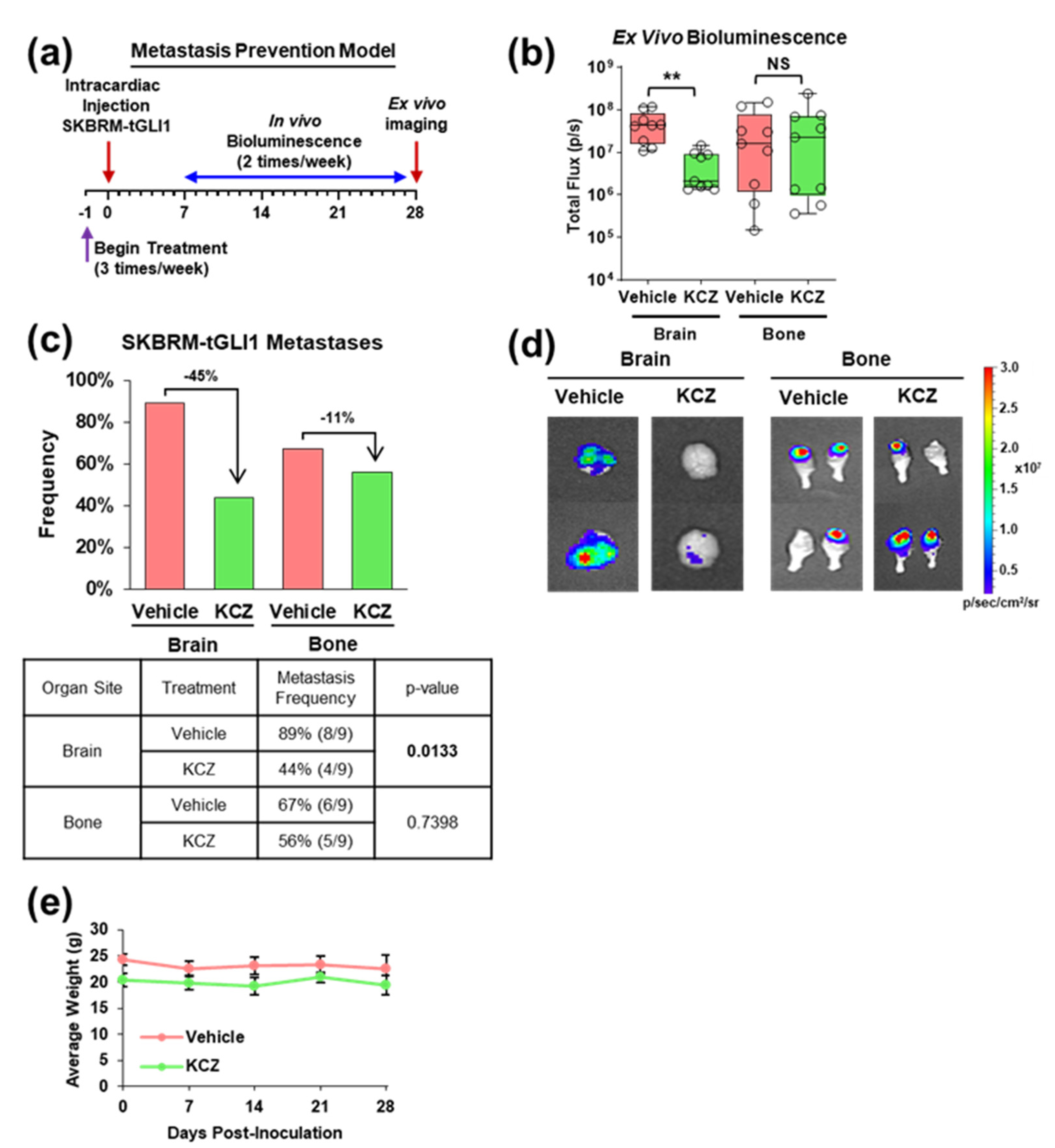
3.3. KCZ Reduces the Ability of tGLI1-Positive Circulating Breast Cancer Cells to Undergo Colonization and Form Brain Metastases In Vivo
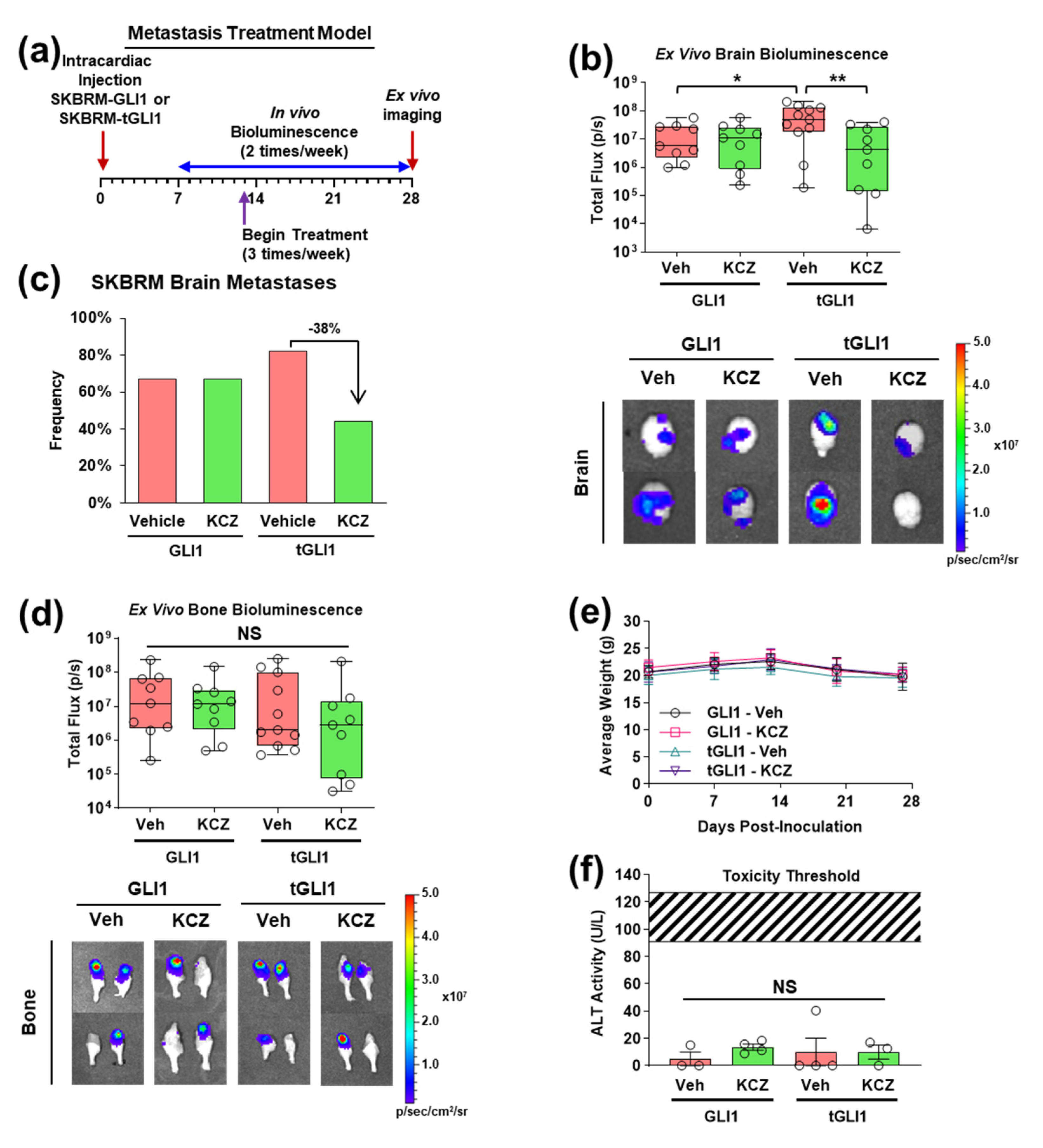
3.4. KCZ Selectively Inhibits the Progression of Established tGLI1-Positive Breast Cancer Brain Metastases In Vivo
3.5. Novel KCZ Derivative KCZ-7 Retains tGLI1 Selectivity and In Vivo Efficacy While Readily Permeating the BBB
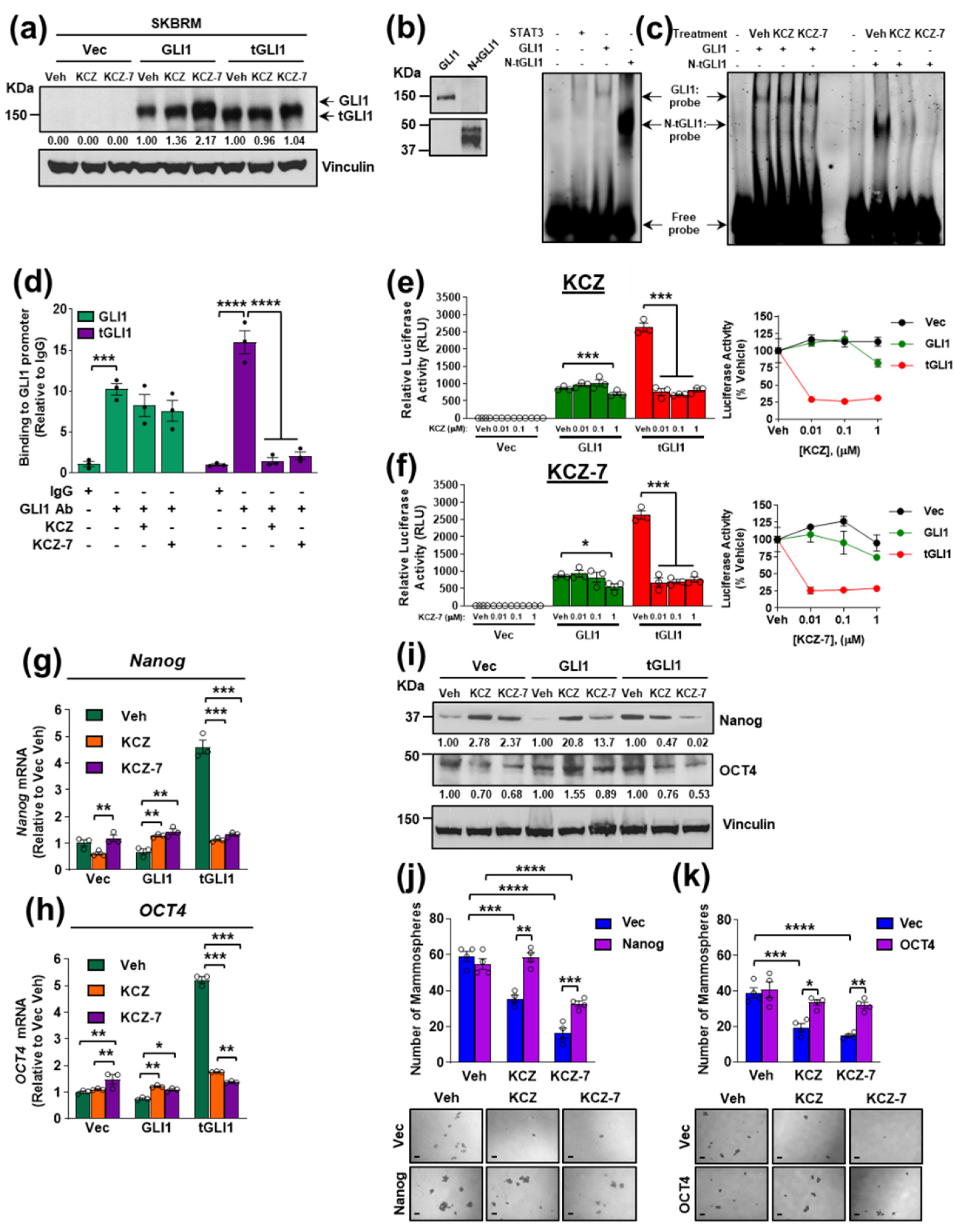
3.6. KCZ and the Novel Derivative KCZ-7 Inhibit tGLI1’s DNA-Binding Activity, Leading to Reduced Expression of tGLI1-Targeted Stemness Genes Nanog and OCT4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Arslan, C.; Dizdar, O.; Altundag, K. Systemic treatment in breast-cancer patients with brain metastasis. Expert Opin. Pharmacother. 2010, 11, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Amiri-Kordestani, L.; Palmieri, D.; Liewehr, D.J.; Steeg, P.S. CNS metastases in breast cancer: Old challenge, new frontiers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6404–6418. [Google Scholar] [CrossRef]
- Dawood, S.; Broglio, K.; Esteva, F.J.; Yang, W.; Kau, S.W.; Islam, R.; Albarracin, C.; Yu, T.K.; Green, M.; Hortobagyi, G.N.; et al. Survival among women with triple receptor-negative breast cancer and brain metastases. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Bellon, J.R.; Winer, E.P. CNS metastases in breast cancer. J. Clin. Oncol. 2004, 22, 3608–3617. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef]
- Lo, H.W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Paw, I.; Zhu, H.; Sirkisoon, S.; Xing, F.; Watabe, K.; Debinski, W.; Lo, H.W. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget 2015, 6, 22653–22665. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Sirkisoon, S.; Zhu, D.; Pasche, B.C.; Chan, M.D.; Lesser, G.J.; Tatter, S.B.; Watabe, K.; Debinski, W.; et al. Truncated Glioma-Associated Oncogene Homolog 1 (tGLI1) Mediates Mesenchymal Glioblastoma via Transcriptional Activation of CD44. Cancer Res. 2018, 78, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Carpenter, R.L.; Han, W.; Lo, H.W. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014, 343, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Doheny, D.; Zhu, D.; Aguayo, N.R.; Xing, F.; Chan, M.; Ruiz, J.; Metheny-Barlow, L.J.; et al. TGLI1 transcription factor mediates breast cancer brain metastasis via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene 2020, 39, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Carpenter, R.L.; Lo, H.-W. TGLI1 Upregulates Expression of VEGFR2 and VEGF-A, Leading to a Robust VEGF-VEGFR2 Autocrine Loop and Cancer Cell Growth. Cancer Hallm. 2013, 1, 28–37. [Google Scholar] [CrossRef]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef]
- Riaz, S.K.; Ke, Y.; Wang, F.; Kayani, M.A.; Malik, M.F.A. Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci. Rep. 2019, 9, 6620. [Google Scholar] [CrossRef]
- Gricius, D.; Kazlauskas, A. Analysis of oncogene GLI1 protein expression levels between differing grades of astrocytoma. BIOLOGIJA 2014, 60, 142–147. [Google Scholar] [CrossRef][Green Version]
- Fan, Y.H.; Ding, J.; Nguyen, S.; Liu, X.J.; Xu, G.; Zhou, H.Y.; Duan, N.N.; Yang, S.M.; Zern, M.A.; Wu, J. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene 2015, 35, 116–124. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Xing, F.; Liu, Y.; Sharma, S.; Wu, K.; Chan, M.D.; Lo, H.W.; Carpenter, R.L.; Metheny-Barlow, L.J.; Zhou, X.; Qasem, S.A.; et al. Activation of the c-Met Pathway Mobilizes an Inflammatory Network in the Brain Microenvironment to Promote Brain Metastasis of Breast Cancer. Cancer Res. 2016, 76, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hui, C.; Nakafuku, M.; Kondoh, H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 1997, 124, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Anichini, G.; Sala, C.; Manetti, F.; Almada, L.L.; Pepe, S.; Carr, R.M.; Paradise, B.D.; Sarkaria, J.N.; Davila, J.I.; et al. ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. Nat. Commun. 2020, 11, 5865. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef]
- Gandillet, A.; Vidal, I.; Alexandre, E.; Audet, M.; Chenard-Neu, M.P.; Stutzmann, J.; Heyd, B.; Jaeck, D.; Richert, L. Experimental models of acute and chronic liver failure in nude mice to study hepatocyte transplantation. Cell Transplant. 2005, 14, 277–290. [Google Scholar] [CrossRef]
- Greenblatt, H.K.; Greenblatt, D.J. Liver injury associated with ketoconazole: Review of the published evidence. J. Clin. Pharmacol. 2014, 54, 1321–1329. [Google Scholar] [CrossRef]
- Patel, V.; Liaw, B.; Oh, W. The role of ketoconazole in current prostate cancer care. Nat. Rev. Urol. 2018, 15, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Goh, B.C.; Wang, L.Z.; Tan, S.H.; Chuah, B.Y.S.; Lim, S.E.; Iau, P.; Buhari, S.A.; Chan, C.W.; Sukri, N.B.; et al. Pharmacokinetics and pharmacodynamics of docetaxel with or without ketoconazole modulation in chemonaive breast cancer patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2010, 21, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Furtado, C.M.; Marcondes, M.C.; Carvalho, R.S.; Sola-Penna, M.; Zancan, P. Phosphatidylinositol-3-kinase as a putative target for anticancer action of clotrimazole. Int. J. Biochem. Cell Biol. 2015, 62, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Forgue-Lafitte, M.E.; Coudray, A.M.; Fagot, D.; Mester, J. Effects of ketoconazole on the proliferation and cell cycle of human cancer cell lines. Cancer Res. 1992, 52, 6827–6831. [Google Scholar] [PubMed]
- Bae, S.H.; Park, J.H.; Choi, H.G.; Kim, H.; Kim, S.H. Imidazole Antifungal Drugs Inhibit the Cell Proliferation and Invasion of Human Breast Cancer Cells. Biomol. Ther. 2018, 26, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-itraconazole as an anti-cancer agent. Ecancermedicalscience 2015, 9, 521. [Google Scholar] [CrossRef]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef]
- Arnold, K.M.; Pohlig, R.T.; Sims-Mourtada, J. Co-activation of Hedgehog and Wnt signaling pathways is associated with poor outcomes in triple negative breast cancer. Oncol. Lett. 2017, 14, 5285–5292. [Google Scholar] [CrossRef]
- Maubant, S.; Tesson, B.; Maire, V.; Ye, M.; Rigaill, G.; Gentien, D.; Cruzalegui, F.; Tucker, G.C.; Roman-Roman, S.; Dubois, T. Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS ONE 2015, 10, e0122333. [Google Scholar] [CrossRef]
- Maeda, O.; Kondo, M.; Fujita, T.; Usami, N.; Fukui, T.; Shimokata, K.; Ando, T.; Goto, H.; Sekido, Y. Enhancement of GLI1-transcriptional activity by beta-catenin in human cancer cells. Oncol. Rep. 2006, 16, 91–96. [Google Scholar] [PubMed]
- Takenaka, K.; Kise, Y.; Miki, H. GSK3beta positively regulates Hedgehog signaling through Sufu in mammalian cells. Biochem. Biophys. Res. Commun. 2007, 353, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Tonge, P.J. Drug–Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9, 29–39. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doheny, D.; Manore, S.; Sirkisoon, S.R.; Zhu, D.; Aguayo, N.R.; Harrison, A.; Najjar, M.; Anguelov, M.; Cox, A.O.; Furdui, C.M.; et al. An FDA-Approved Antifungal, Ketoconazole, and Its Novel Derivative Suppress tGLI1-Mediated Breast Cancer Brain Metastasis by Inhibiting the DNA-Binding Activity of Brain Metastasis-Promoting Transcription Factor tGLI1. Cancers 2022, 14, 4256. https://doi.org/10.3390/cancers14174256
Doheny D, Manore S, Sirkisoon SR, Zhu D, Aguayo NR, Harrison A, Najjar M, Anguelov M, Cox AO, Furdui CM, et al. An FDA-Approved Antifungal, Ketoconazole, and Its Novel Derivative Suppress tGLI1-Mediated Breast Cancer Brain Metastasis by Inhibiting the DNA-Binding Activity of Brain Metastasis-Promoting Transcription Factor tGLI1. Cancers. 2022; 14(17):4256. https://doi.org/10.3390/cancers14174256
Chicago/Turabian StyleDoheny, Daniel, Sara Manore, Sherona R. Sirkisoon, Dongqin Zhu, Noah R. Aguayo, Alexandria Harrison, Mariana Najjar, Marlyn Anguelov, Anderson O’Brien Cox, Cristina M. Furdui, and et al. 2022. "An FDA-Approved Antifungal, Ketoconazole, and Its Novel Derivative Suppress tGLI1-Mediated Breast Cancer Brain Metastasis by Inhibiting the DNA-Binding Activity of Brain Metastasis-Promoting Transcription Factor tGLI1" Cancers 14, no. 17: 4256. https://doi.org/10.3390/cancers14174256
APA StyleDoheny, D., Manore, S., Sirkisoon, S. R., Zhu, D., Aguayo, N. R., Harrison, A., Najjar, M., Anguelov, M., Cox, A. O., Furdui, C. M., Watabe, K., Hollis, T., Thomas, A., Strowd, R., & Lo, H.-W. (2022). An FDA-Approved Antifungal, Ketoconazole, and Its Novel Derivative Suppress tGLI1-Mediated Breast Cancer Brain Metastasis by Inhibiting the DNA-Binding Activity of Brain Metastasis-Promoting Transcription Factor tGLI1. Cancers, 14(17), 4256. https://doi.org/10.3390/cancers14174256