COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma

 , and
, and
Abstract
1. Introduction
2. Results
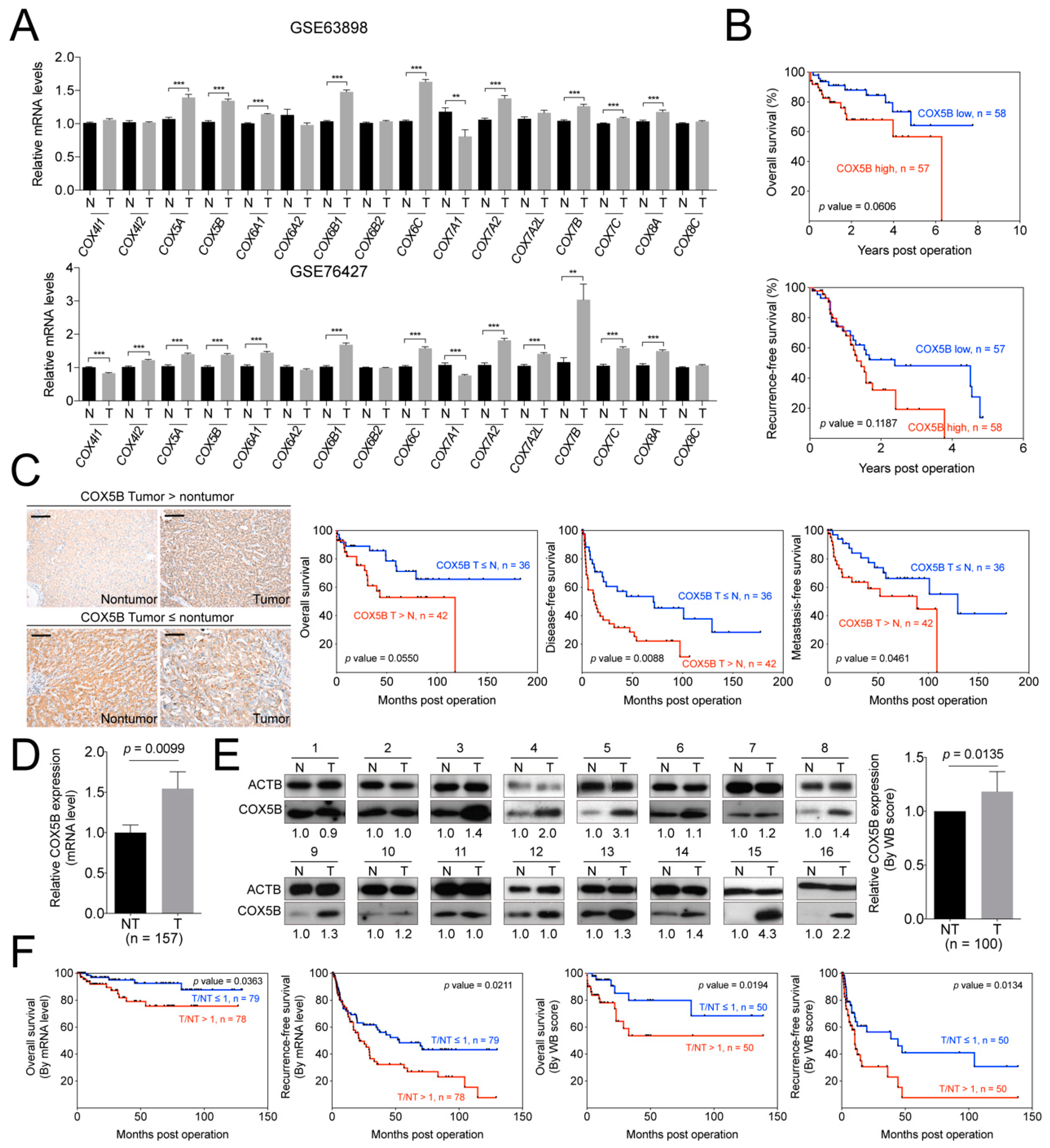
2.1. COX5B Level is Up-Regulated in Hepatoma and Associated with Unfavorable Postoperative Outcomes
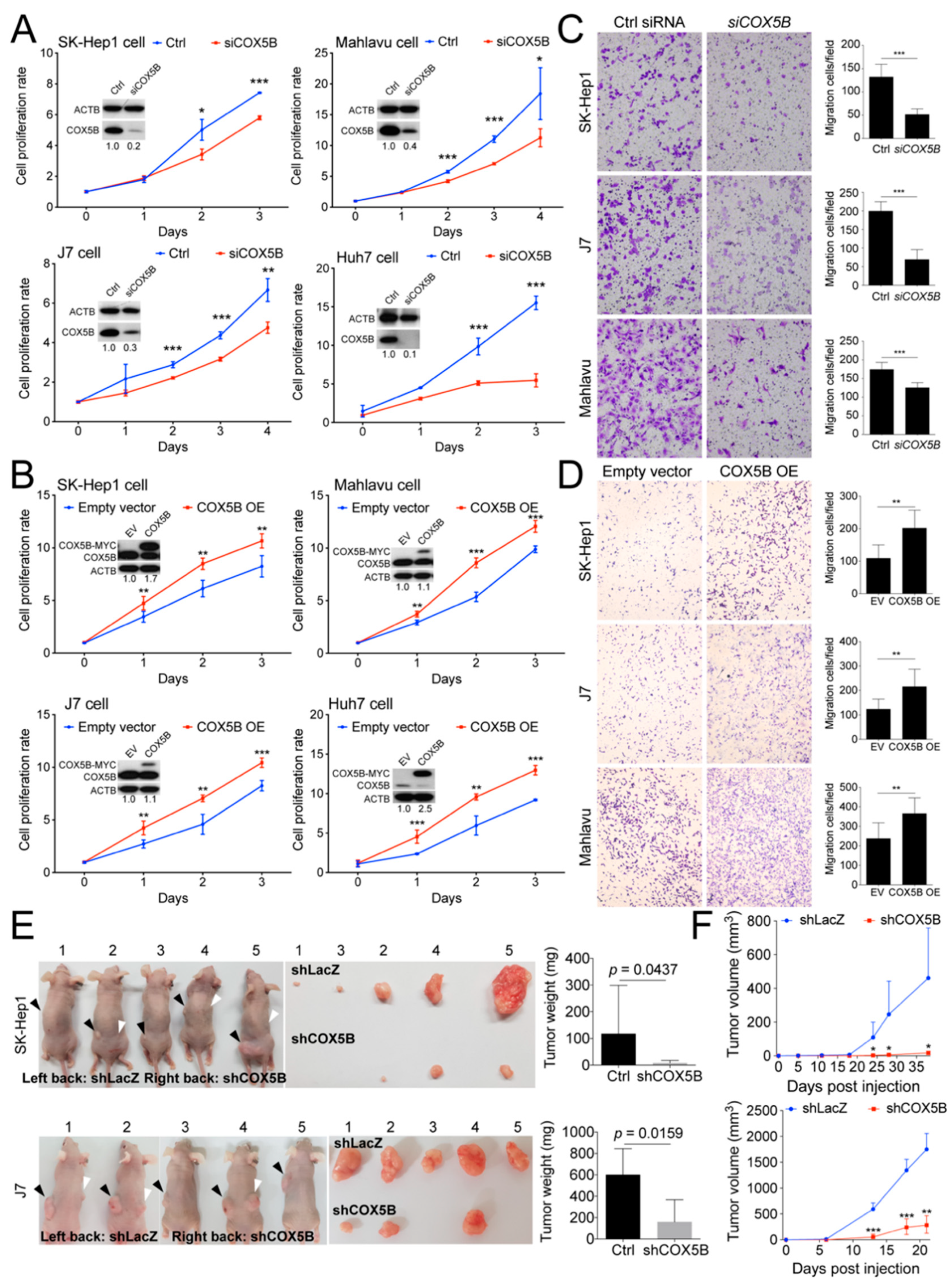
2.2. COX5B Promotes Proliferation and Migration in Hepatoma Cells
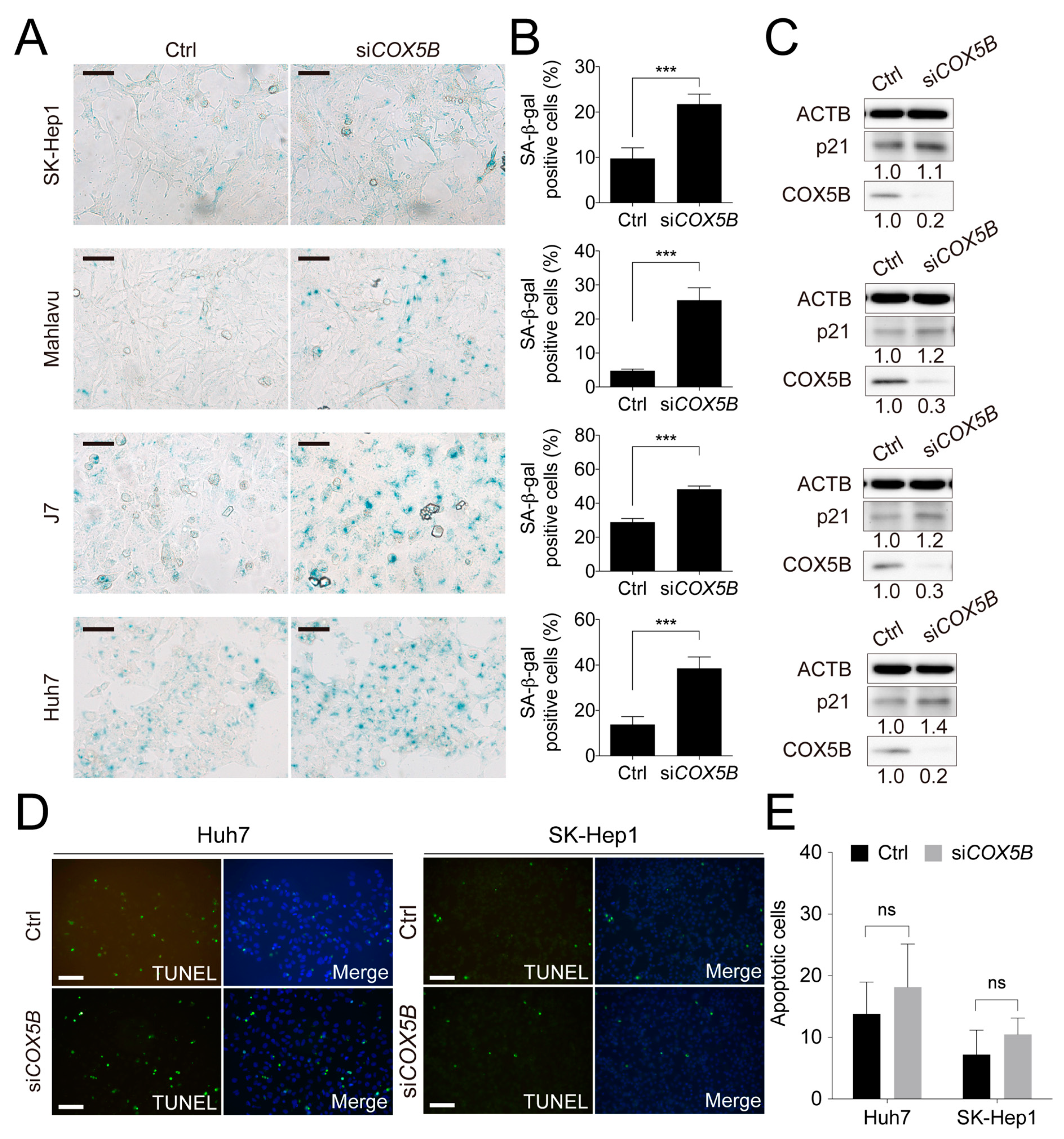
2.3. Loss of COX5B Represses Cell Proliferation Partly through Induction of Cell Senescence
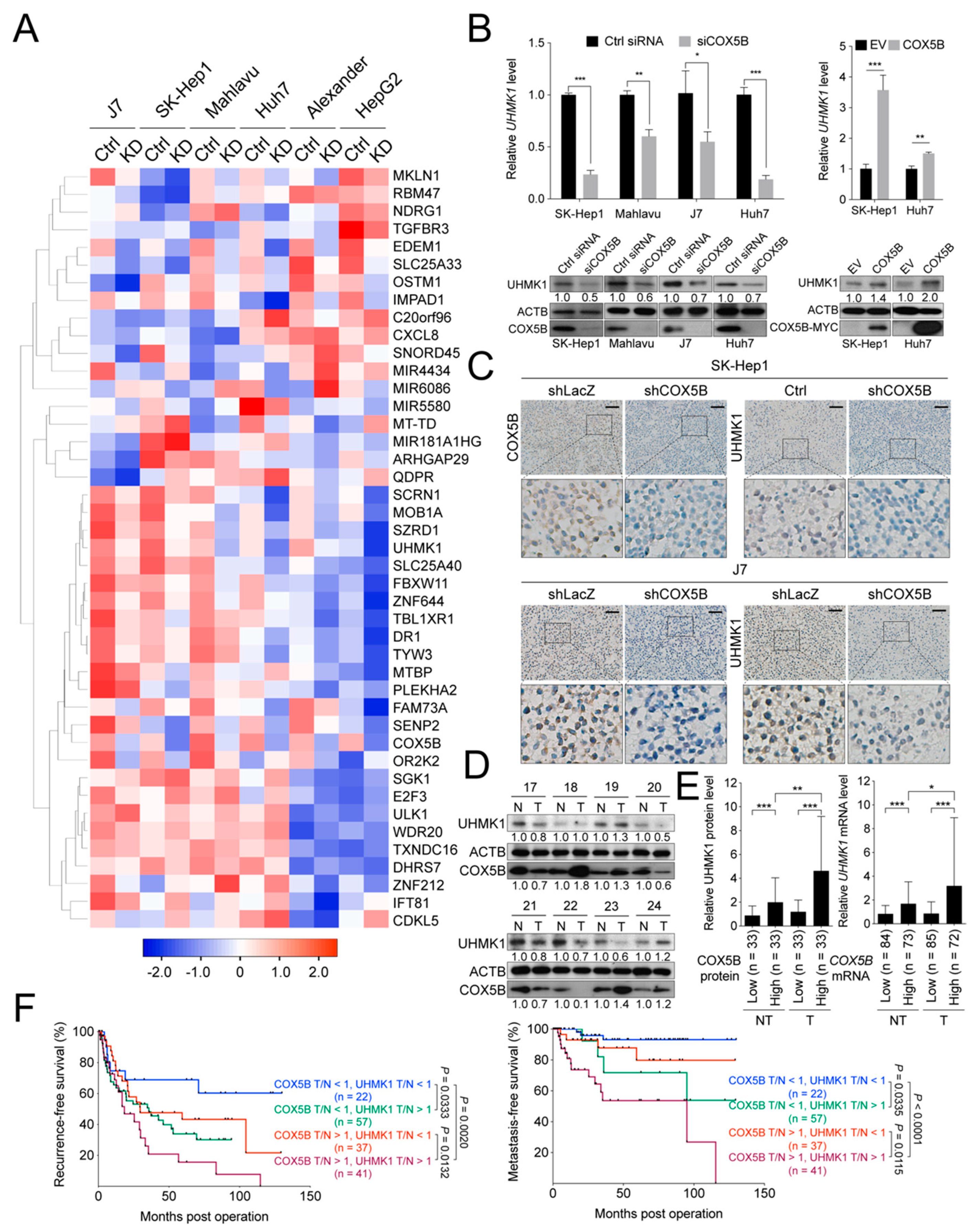
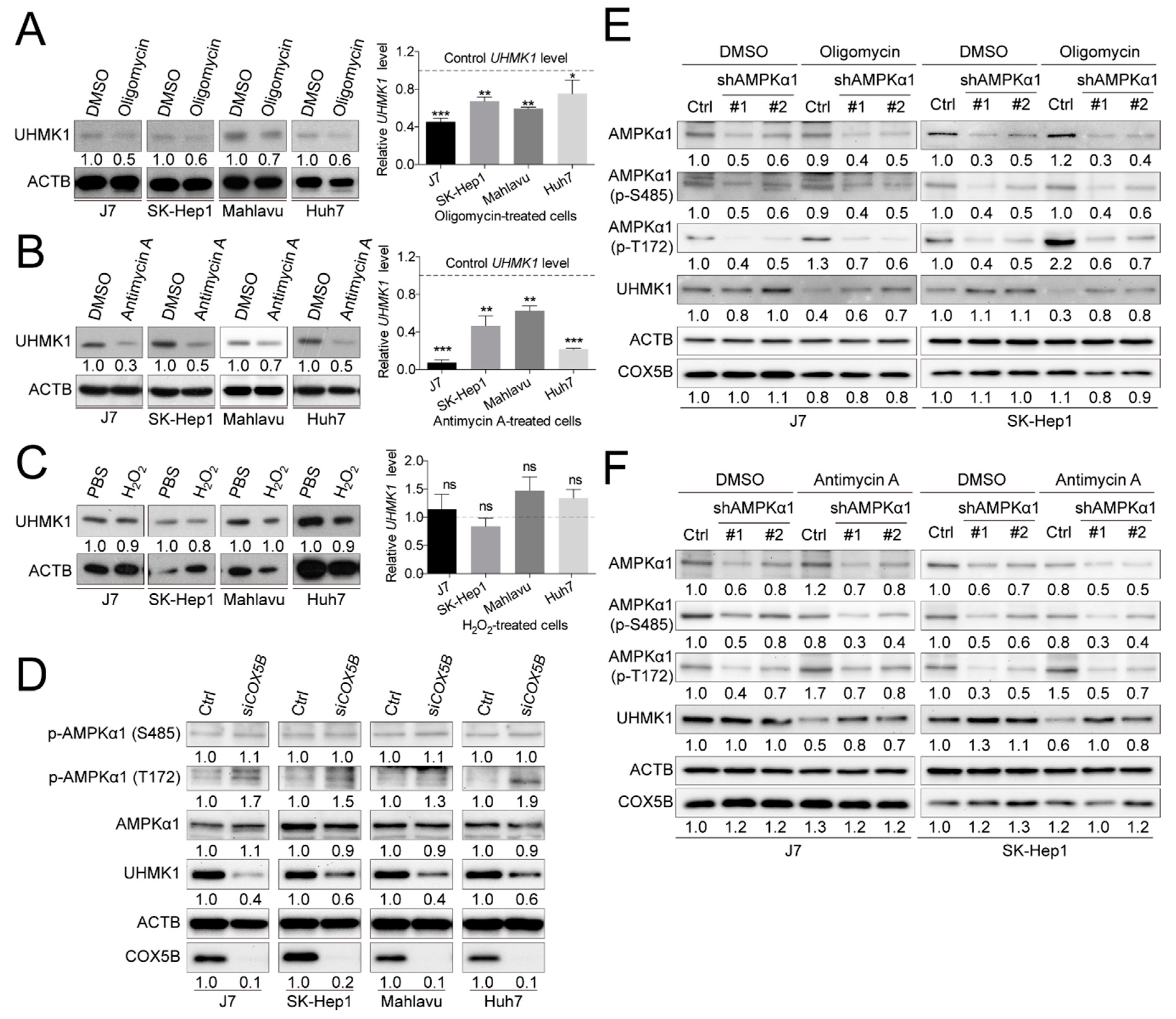
2.4. COX5B Modulates an Oncogene UHMK1 and a Potent Tumor Suppressor ULK1 Expressions in Hepatoma
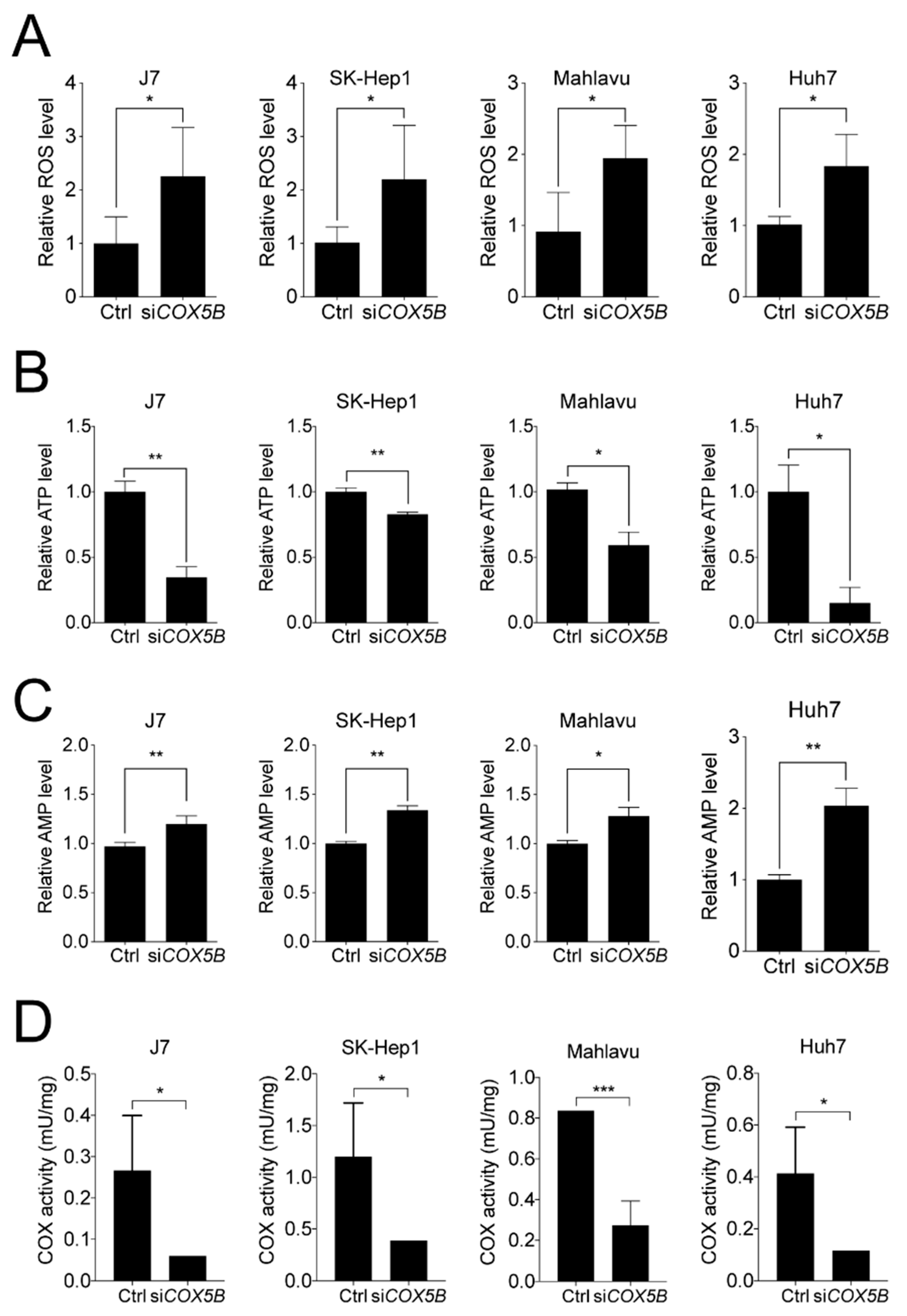
2.5. COX5B-Mediated Regulation of UHMK1 and ULK1 Expression May Be AMPK Activation-Dependent
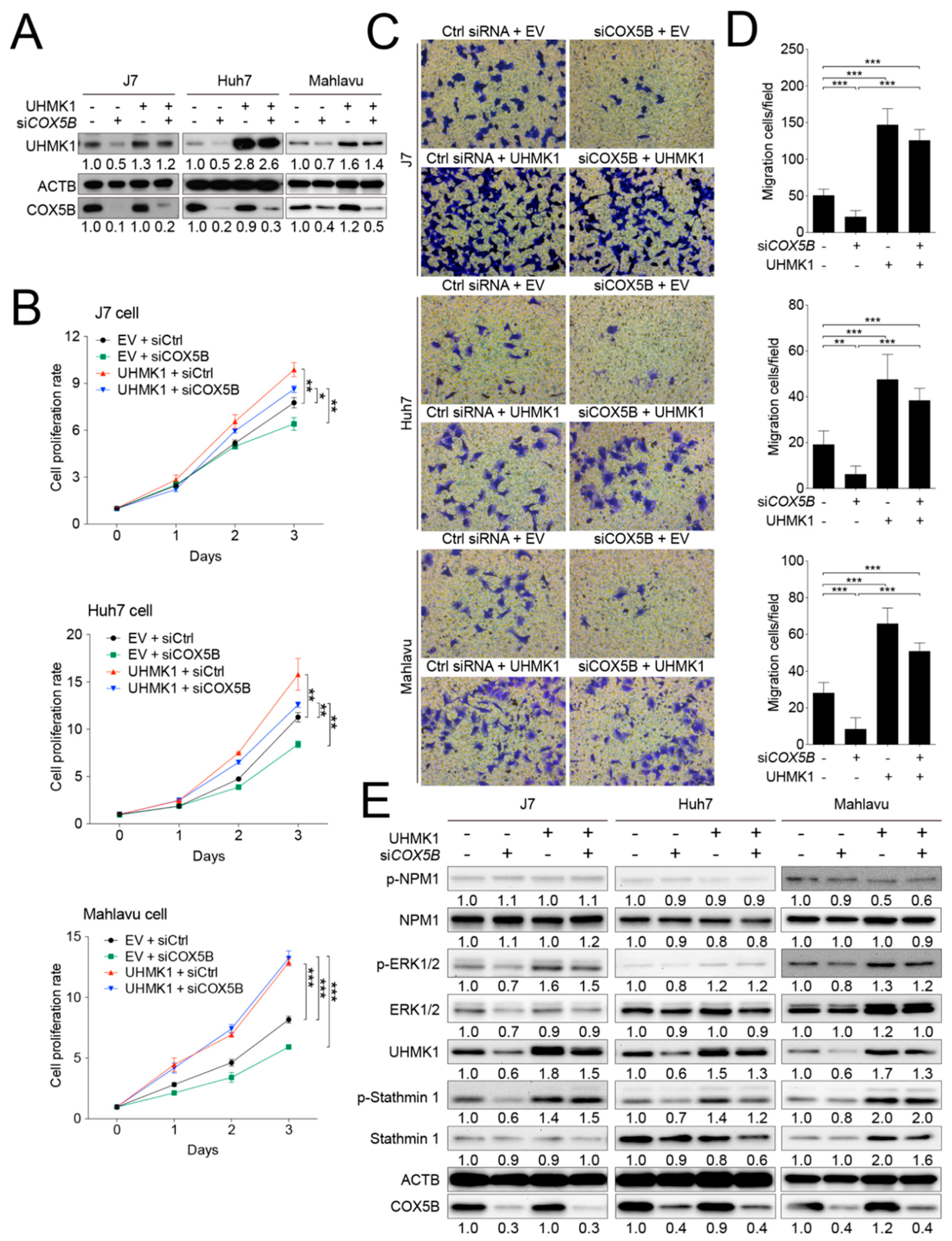
2.6. COX5B-UHMK1 Axis Modulates Cell Proliferation and Migration in Hepatoma
2.7. Elevation of UHMK1 Promotes Phosphorylation of ERK1/2 and Stathmin1 in Hepatoma Cells
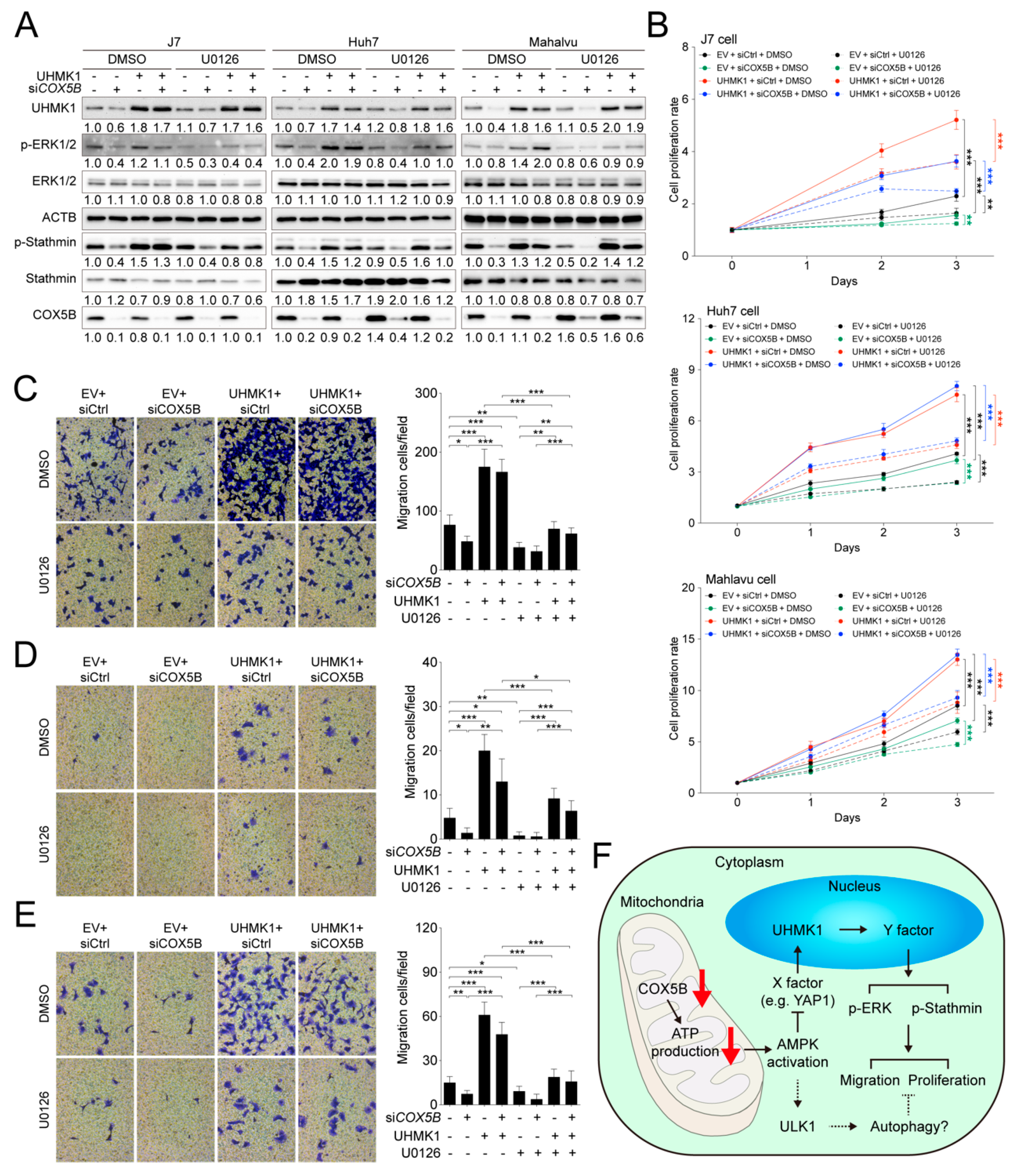
2.8. COX5B-UHMK1-ERK Signal Loop Regulates Cell Proliferation and Migration in Hepatoma Cells
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Immunohistochemistry (IHC) Staining
4.3. Lysate Preparation and Western Blot
4.4. Plasmid Construction and Preparation for Expression
4.5. Cell Culture and Transfection
4.6. Lentivirus-Mediated Knockdown of COX5B and AMPKα1
4.7. Senescence Beta-Galactosidase Cell Staining Assay
4.8. Apoptosis Detection Assay
4.9. Oligomycin, Antimycin A, H2O2 and U0126 Treatment
4.10. Cell Proliferation Assay
4.11. Cell Migration Assay
4.12. ROS, ATP and AMP Detection Assays
4.13. Cytochrome c Oxidase Activity Detection Assay
4.14. Xenograft Model
4.15. cDNA Microarray
4.16. RNA Isolation and Quantitative RT-PCR
4.17. Phospho-Peptide Enrichment Analysis
4.18. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| COX | cytochrome c oxidase |
| COX5B | Cytochrome c oxidase subunit 5B |
| IHC | immunohistochemistry |
| MFS | metastasis-free survival |
| OS | overall survival |
| OXPHOS | oxidative phosphorylation |
| RFS | recurrence-free survival |
| ROS | reactive oxygen species |
| RT-qPCR | real-time quantitative PCR |
| UHMK1 | U2AF homology motif kinase1 |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Bolondi, L.; Sofia, S.; Siringo, S.; Gaiani, S.; Casali, A.; Zironi, G.; Piscaglia, F.; Gramantieri, L.; Zanetti, M.; Sherman, M. Surveillance programme of cirrhotic patients for early diagnosis and treatment of hepatocellular carcinoma: A cost effectiveness analysis. Gut 2001, 48, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.H.; Xiang, B.D.; Gong, W.F.; Ke, Y.; Mo, Q.G.; Ma, L.; Liu, X.; Li, L.Q. Comparison of long-term survival of patients with BCLC stage B hepatocellular carcinoma after liver resection or transarterial chemoembolization. PLoS ONE 2013, 8, e68193. [Google Scholar] [CrossRef]
- Fay, J.R.; Steele, V.; Crowell, J.A. Energy homeostasis and cancer prevention: The AMP-activated protein kinase. Cancer Prev. Res. 2009, 2, 301–309. [Google Scholar] [CrossRef]
- Iommarini, L.; Ghelli, A.; Gasparre, G.; Porcelli, A.M. Mitochondrial metabolism and energy sensing in tumor progression. Biochim Biophys Acta Bioenerg 2017, 1858, 582–590. [Google Scholar] [CrossRef]
- Bost, F.; Decoux-Poullot, A.G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef]
- Lin, W.R.; Chiang, J.M.; Lim, S.N.; Su, M.Y.; Chen, T.H.; Huang, S.W.; Chen, C.W.; Wu, R.C.; Tsai, C.L.; Lin, Y.H.; et al. Dynamic bioenergetic alterations in colorectal adenomatous polyps and adenocarcinomas. EBioMedicine 2019, 44, 334–345. [Google Scholar] [CrossRef]
- Herrera, A.S.; Esparza, M.a.d.C.A.; Ashraf, G.M.; Zamyatnin, A.A., Jr.; Aliev, G. Beyond Mitochondria, What Would be the Energy Source of the Cell? Cent Nerv. Syst. Agents Med. Chem. 2015, 15, 32–41. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Avadhani, N.G. Mitochondrial respiratory defects promote the Warburg effect and cancer progression. Mol. Cell Oncol. 2016, 3, e1085120. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- Kadenbach, B.; Hüttemann, M.H.; Arnold, S.; Lee, I.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free. Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Ludwig, B.; Bender, E.; Arnold, S.; Hüttemann, M.; Lee, I.; Kadenbach, B. Cytochrome c Oxidase and the Regulation of Oxidative Phosphorylation. Chembiochem 2001, 2, 392–403. [Google Scholar] [CrossRef]
- Kadenbach, B. Regulation of Mammalian 13-Subunit Cytochrome c Oxidase and Binding of other Proteins: Role of NDUFA4. Trends Endocrinol. Metab. 2017, 28, 761–770. [Google Scholar] [CrossRef]
- Deng, S.; Li, Y.; Yi, G.; Lei, B.; Guo, M.; Xiang, W.; Chen, Z.; Liu, Y.; Qi, S. Overexpression of COX7A2 is associated with a good prognosis in patients with glioma. J. Neurooncol. 2018, 136, 41–50. [Google Scholar] [CrossRef]
- Gao, S.-P.; Sun, H.-F.; Fu, W.-Y.; Li, L.-D.; Zhao, Y.; Chen, M.-T.; Jin, W. High expression of COX5B is associated with poor prognosis in breast cancer. Future Oncol. 2017, 13, fon–2017–0058–1719. [Google Scholar] [CrossRef]
- Hewedi, I.H.; Farid, R.M.; Sidhom, K.F.; Salman, M.I.; Mostafa, R.G. Differential Expression of Cytochrome C Oxidase Subunit I Along the Colorectal Adenoma: Carcinoma Progression. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 689–696. [Google Scholar] [CrossRef]
- Lin, W.R.; Lim, S.N.; McDonald, S.A.; Graham, T.; Wright, V.L.; Peplow, C.L.; Humphries, A.; Kocher, H.M.; Wright, N.A.; Dhillon, A.P.; et al. The histogenesis of regenerative nodules in human liver cirrhosis. Hepatology 2010, 51, 1017–1026. [Google Scholar] [CrossRef]
- Le, P.H.; Huang, S.C.; Lim, S.N.; Chou, C.H.; Yeh, T.S.; Chen, T.C.; Yen, T.H.; Su, M.Y.; Chiu, C.T.; Yeh, C.T.; et al. Complex IV subunit 1 defect predicts postoperative survival in hepatocellular carcinoma. Oncol. Lett. 2014, 7, 1430–1438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, S.-P.; Sun, H.-F.; Jiang, H.-L.; Li, L.-D.; Hu, X.; Xu, X.-E.; Jin, W. Loss of COX5B inhibits proliferation and promotes senescence via mitochondrial dysfunction in breast cancer. Oncotarget 2015, 6, 43363–53374. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Dyk, D.E.V.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a Novel Inhibitor of Mitogen-activated Protein Kinase Kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef]
- Hu, T.; Xi, J. Identification of COX5B as a novel biomarker in high-grade glioma patients. Onco Targets 2017, 10, 5463–5470. [Google Scholar] [CrossRef]
- Sotgia, F.; Lisanti, M.P. Mitochondrial biomarkers predict tumor progression and poor overall survival in gastric cancers: Companion diagnostics for personalized medicine. Oncotarget 2017, 8, 67117–67128. [Google Scholar] [CrossRef]
- Krupar, R.; Hautmann, M.G.; Pathak, R.R.; Varier, I.; McLaren, C.; Gaag, D.; Hellerbrand, C.; Evert, M.; Laban, S.; Idel, C.; et al. Immunometabolic Determinants of Chemoradiotherapy Response and Survival in Head and Neck Squamous Cell Carcinoma. Am. J. Pathol. 2018, 188, 72–83. [Google Scholar] [CrossRef]
- Stein, J.; Tenbrock, J.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. Int. J. Urol. 2019, 26, 910–916. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef]
- Angireddy, R.; Kazmi, H.R.; Srinivasan, S.; Sun, L.; Iqbal, J.; Fuchs, S.Y.; Guha, M.; Kijima, T.; Yuen, T.; Zaidi, M.; et al. Cytochrome c oxidase dysfunction enhances phagocytic function and osteoclast formation in macrophages. Faseb. J. 2019, 33, 9167–9181. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Yoshimoto, T.; Crook, M.F.; Nallamshetty, S.; True, A.; Nabel, G.J.; Nabel, E.G. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. Embo. J. 2002, 21, 3390–3401. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Okinaka, K.; Hirano, I.; Ono, T.; Sugimoto, Y.; Shigeno, K.; Fujisawa, S.; Shinjo, K.; Ohnishi, K. KIS induces proliferation and the cell cycle progression through the phosphorylation of p27Kip1 in leukemia cells. Leuk. Res. 2008, 32, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J. AMPK -> ULK1 -> autophagy. Mol Cell Biol 2011, 31, 3082–3084. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Wei, T.; Weiler, S.M.E.; Toth, M.; Sticht, C.; Lutz, T.; Thomann, S.; De La Torre, C.; Straub, B.; Merker, S.; Ruppert, T.; et al. YAP-dependent induction of UHMK1 supports nuclear enrichment of the oncogene MYBL2 and proliferation in liver cancer cells. Oncogene 2019, 38, 5541–5550. [Google Scholar] [CrossRef]
- Petrovic, V.; Costa, R.H.; Lau, L.F.; Raychaudhuri, P.; Tyner, A.L. FoxM1 regulates growth factor-induced expression of kinase-interacting stathmin (KIS) to promote cell cycle progression. J. Biol. Chem. 2008, 283, 453–460. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef]
- Maucuer, A.; Camonis, J.H.; Sobel, A. Stathmin interaction with a putative kinase and coiled-coil-forming protein domains. Proc. Natl. Acad. Sci. USA 1995, 92, 3100–3104. [Google Scholar] [CrossRef]
- Alam, M.R.; Caldwell, B.D.; Johnson, R.C.; Darlington, D.N.; Mains, R.E.; Eipper, B.A. Novel Proteins That Interact with the COOH-terminal Cytosolic Routing Determinants of an Integral Membrane Peptide-processing Enzyme. J. Biol. Chem. 1996, 271, 28636–28640. [Google Scholar] [CrossRef]
- Caldwell, B.D.; Darlington, D.N.; Penzes, P.; Johnson, R.C.; Eipper, B.A.; Mains, R.E. The Novel Kinase Peptidylglycine. J. Biol. Chem. 1999, 274, 34646–34656. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Steveson, T.C.; Johnson, R.C.; Bäck, N.; Abraham, B.; Mains, R.E.; Eipper, B.A. Signaling Mediated by the Cytosolic Domain of Peptidylglycine α-Amidating Monooxygenase. Mol. Biol. Cell 2001, 12, 629–644. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Archangelo, L.F.; Greif, P.A.; Maucuer, A.; Manceau, V.; Koneru, N.; Bigarella, C.L.; Niemann, F.; dos Santos, M.T.; Kobarg, J.; Bohlander, S.K.; et al. The CATS (FAM64A) protein is a substrate of the Kinase Interacting Stathmin (KIS). Biochim. Et Biophys. Acta 2013, 1833, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Chatrikhi, R.; Wang, W.; Gupta, A.; Loerch, S.; Maucuer, A.; Kielkopf, C.L. SF1 Phosphorylation Enhances Specific Binding to U2AF(65) and Reduces Binding to 3’-Splice-Site RNA. Biophys J. 2016, 111, 2570–2586. [Google Scholar] [CrossRef]
- Langenickel, T.H.; Olive, M.; Boehm, M.; San, H.; Crook, M.F.; Nabel, E.G. KIS protects against adverse vascular remodeling by opposing stathmin-mediated VSMC migration in mice. J. Clin. Invest. 2008, 118, 3848–3859. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio. Protoc. 2019, 9. [Google Scholar] [CrossRef]
- Chu, Y.D.; Wang, W.C.; Chen, S.A.; Hsu, Y.T.; Yeh, M.W.; Slack, F.J.; Chan, S.P. RACK-1 regulates let-7 microRNA expression and terminal cell differentiation in Caenorhabditis elegans. Cell Cycle 2014, 13, 1995–2009. [Google Scholar] [CrossRef][Green Version]
- Chu, Y.D.; Lai, H.Y.; Pai, L.M.; Huang, Y.H.; Lin, Y.H.; Liang, K.H.; Yeh, C.T. The methionine salvage pathway-involving ADI1 inhibits hepatoma growth by epigenetically altering genes expression via elevating S-adenosylmethionine. Cell Death Dis. 2019, 10, 240. [Google Scholar] [CrossRef]
- Lin, Y.H.; Wu, M.H.; Huang, Y.H.; Yeh, C.T.; Cheng, M.L.; Chi, H.C.; Tsai, C.Y.; Chung, I.H.; Chen, C.Y.; Lin, K.H. Taurine up-regulated gene 1 functions as a master regulator to coordinate glycolysis and metastasis in hepatocellular carcinoma. Hepatology 2018, 67, 188–203. [Google Scholar] [CrossRef]
- Lai, M.W.; Liang, K.H.; Lin, W.R.; Huang, Y.H.; Huang, S.F.; Chen, T.C.; Yeh, C.T. Hepatocarcinogenesis in transgenic mice carrying hepatitis B virus pre-S/S gene with the sW172* mutation. Oncogenesis 2016, 5, e273. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chu, Y.D.; Chen, H.K.; Huang, T.; Chan, S.P. A novel function for the DEAD-box RNA helicase DDX-23 in primary microRNA processing in Caenorhabditis elegans. Dev. Biol. 2016, 409, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, M.; Glassman, J.R. Categorizing a prognostic variable: Review of methods, code for easy implementation and applications to decision-making about cancer treatments. Stat. Med. 2000, 19, 113–132. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Localization | Sequence | Position | Site | Ratio 1 a | Ratio 2 b | Ratio 3 c |
|---|---|---|---|---|---|---|---|
| NPM1 | Nuclear | DELHIVEAEAMNYEGSPIK | 55–73 | S70 | 0.8 | 1.3 | 1.9 |
| MYH10 | Cytoplasmic | QLHLEGASLELSDDDTESK | 1976–1994 | S1987 | 0.8 | 1.2 | 1.3 |
| C11orf58 | Cytoplasmic | RSASPDDDLGSSNWEAADLGNEER | 14–37 | S17 | 0.8 | 1.3 | 1.7 |
| ERK2 | Cytoplasmic | VADPDHDHTGFLTEYVATR | 190–208 | Y204 | 0.5 | 1.8 | 3.5 |
| Stathmin1 | Cytoplasmic | SKESVPEFPLSPPK | 28–41 | S38 | 0.7 | 1.3 | 1.4 |
| ZC3H13 | Nuclear | GNIETTSEDGQVFSPK | 980–995 | S993 | 0.8 | 1.3 | 1.3 |
| DYNC1LI2 | Cytoplasmic | DFQDYMEPEEGCQGSPQRR | 180–198 | S194 | 0.9 | 1.4 | 1.1 |
| STIP1 | Cytoplasmic | HDSPEDVK | 526–533 | S528 | 0.8 | 1.1 | 1.1 |
| FIP1L1 | Nuclear | ERDHSPTPSVFNSDEER | 488–504 | S492 | 0.8 | 1.1 | 1.2 |
| HIRIP3 | Nuclear | TLDSDEERPRPAPPDWSHMR | 527–546 | S530 | 0.9 | 1.1 | 1.1 |
| TCEA1 | Nuclear and cytoplasmic | EPAITSQNSPEAR | 92–104 | S100 | 0.7 | 1.2 | 1.3 |
| MYBBP1A | Nuclear | DPAQPMSPGEATQSGARPADR | 5–25 | S11 | 0.8 | 1.2 | 1.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Y.-D.; Lin, W.-R.; Lin, Y.-H.; Kuo, W.-H.; Tseng, C.-J.; Lim, S.-N.; Huang, Y.-L.; Huang, S.-C.; Wu, T.-J.; Lin, K.-H.; et al. COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma. Cancers 2020, 12, 1646. https://doi.org/10.3390/cancers12061646
Chu Y-D, Lin W-R, Lin Y-H, Kuo W-H, Tseng C-J, Lim S-N, Huang Y-L, Huang S-C, Wu T-J, Lin K-H, et al. COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma. Cancers. 2020; 12(6):1646. https://doi.org/10.3390/cancers12061646
Chicago/Turabian StyleChu, Yu-De, Wey-Ran Lin, Yang-Hsiang Lin, Wen-Hsin Kuo, Chin-Ju Tseng, Siew-Na Lim, Yen-Lin Huang, Shih-Chiang Huang, Ting-Jung Wu, Kwang-Huei Lin, and et al. 2020. "COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma" Cancers 12, no. 6: 1646. https://doi.org/10.3390/cancers12061646
APA StyleChu, Y.-D., Lin, W.-R., Lin, Y.-H., Kuo, W.-H., Tseng, C.-J., Lim, S.-N., Huang, Y.-L., Huang, S.-C., Wu, T.-J., Lin, K.-H., & Yeh, C.-T. (2020). COX5B-Mediated Bioenergetic Alteration Regulates Tumor Growth and Migration by Modulating AMPK-UHMK1-ERK Cascade in Hepatoma. Cancers, 12(6), 1646. https://doi.org/10.3390/cancers12061646