Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma

 ,
, 
Abstract
1. Introduction
2. Results
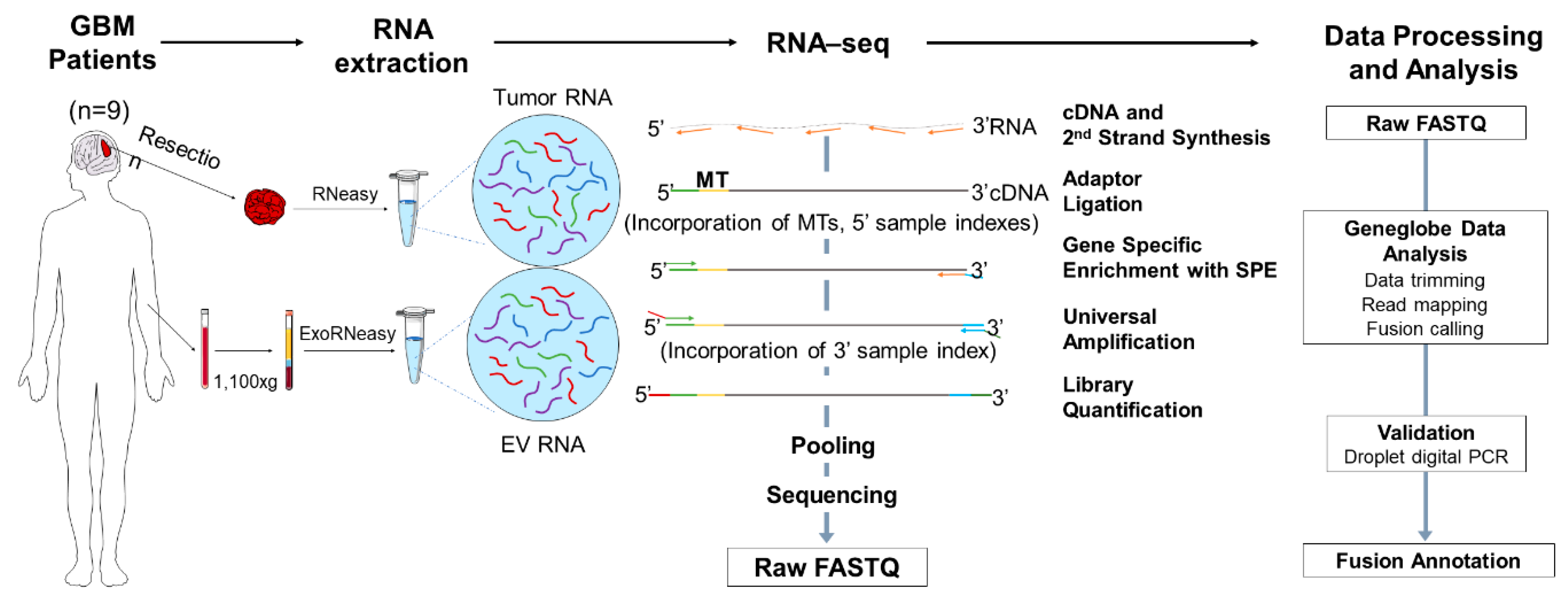
2.1. Experimental Design and Overview of the Library Preparation Methodologies
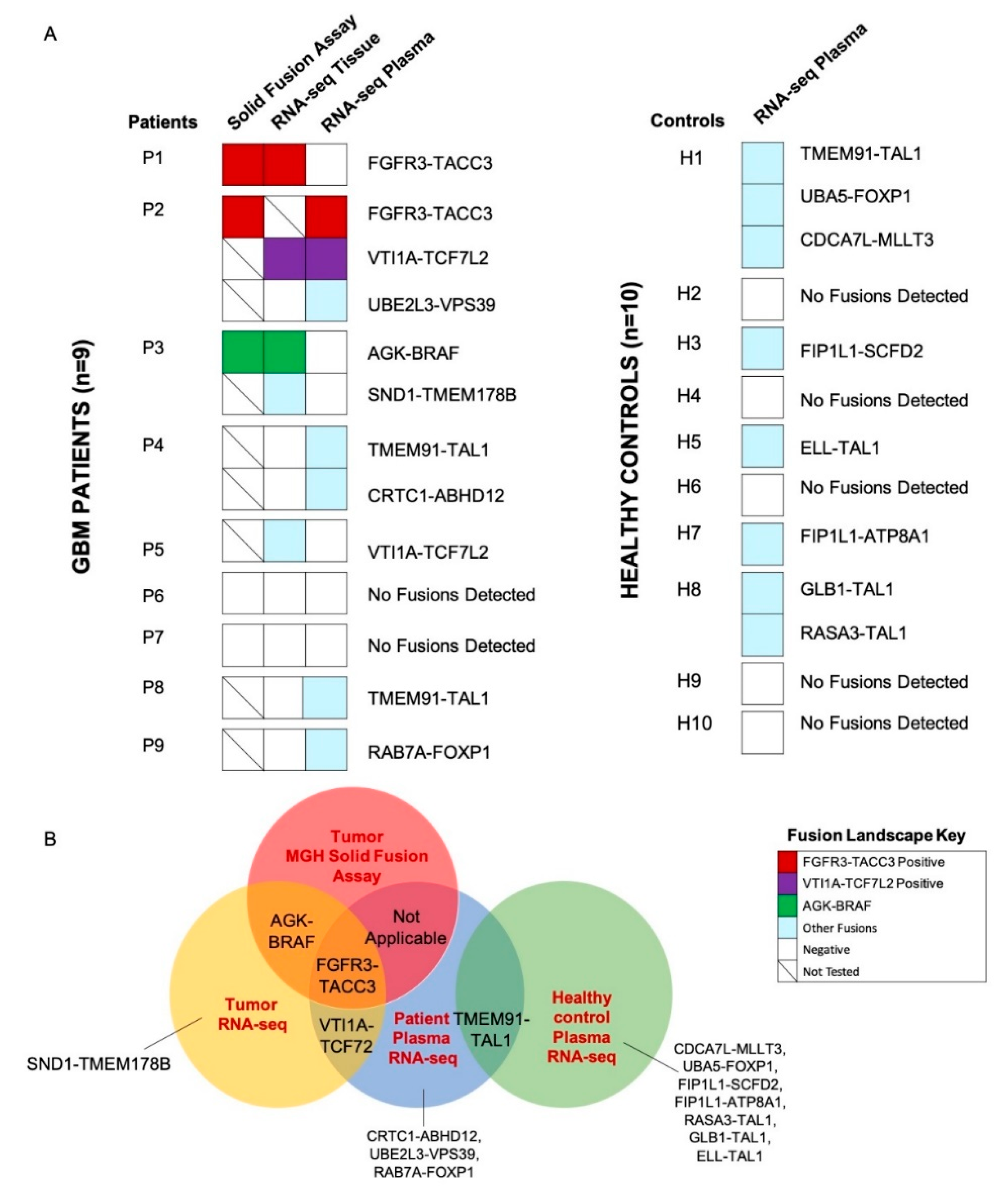
2.2. Detection of Fusions in Tumor Tissue and Matched Plasma of Patients with GBM and Plasma of Healthy Controls
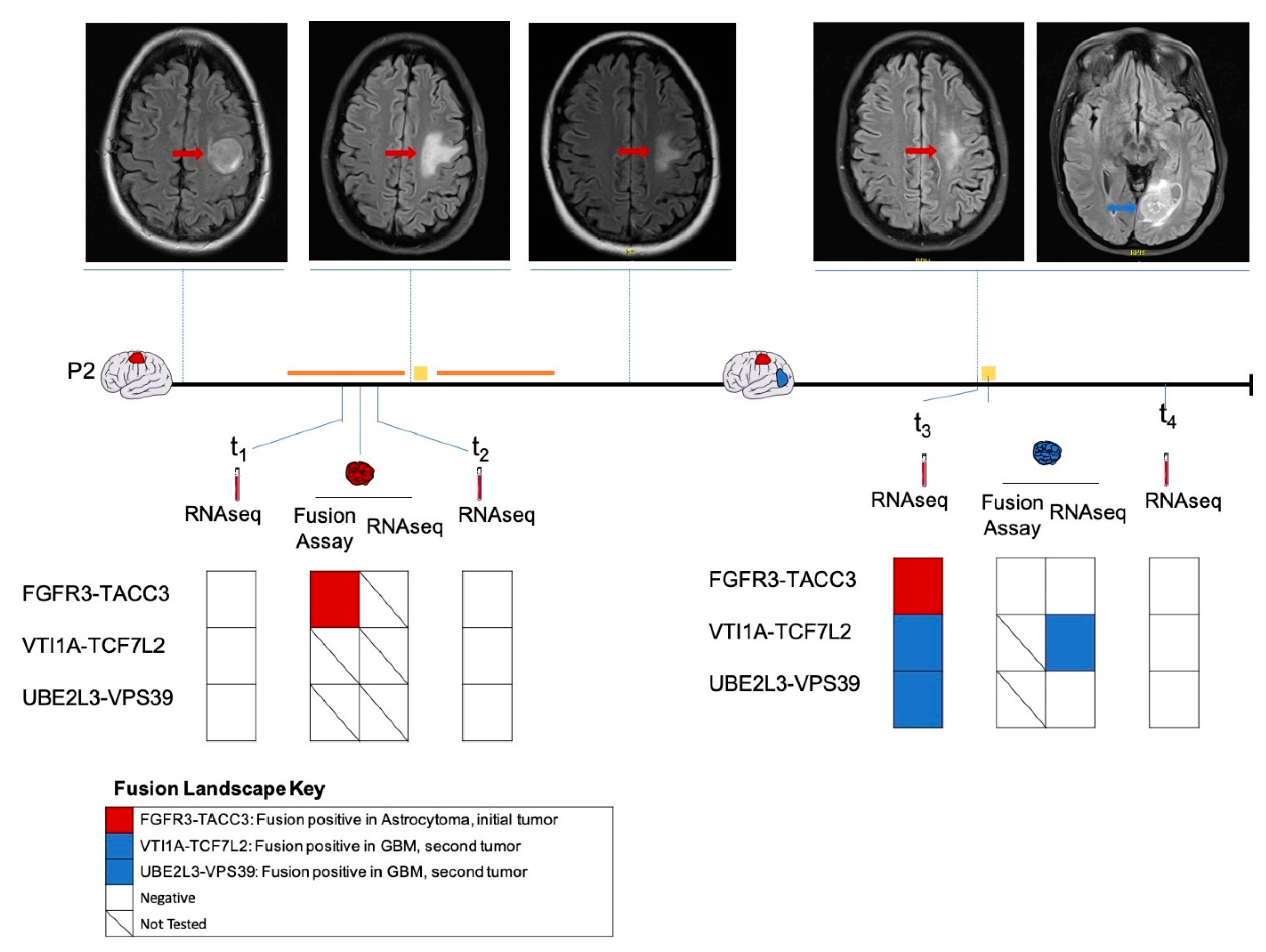
2.3. Longitudinal Follow-Up of Patient 2
3. Discussion
4. Materials and Methods
4.1. Study Subject Characteristics
4.2. Tumor Tissue Processing and RNA Extraction
4.3. Plasma processing
4.4. Plasma EV RNA Extraction
4.5. RNA Quantification and Quality Control
4.6. Library Preparation and Sequencing
4.7. Sequencing Data Processing
4.8. Fusion Calling and Reportable Range
4.9. Validation of Fusions by Droplet Digital PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro-Oncology 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Lankerovich, M.; Lee, H.; Yoon, J.-G.; Schroeder, B.; Foltz, G. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genom. 2013, 14, 818. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, H.; Huang, X.; Li, W.; Huang, Q.; Yan, Y.; Chen, J. Gene Fusion in Malignant Glioma: An Emerging Target for Next-Generation Personalized Treatment. Transl. Oncol. 2018, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Razzaq, S.K.; Vo, A.D.; Gautam, M.; Li, H. Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip Rev. RNA 2016, 7, 811–823. [Google Scholar] [CrossRef]
- Sabir, S.R.; Yeoh, S.; Jackson, G.; Bayliss, R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers 2017, 9, E118. [Google Scholar] [CrossRef]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA fusion genes in acute promyelocytic leukemia: A review. Expert Rev. Hematol. 2014, 7, 347–357. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Penson, R.T.; Sales, E.; Sullivan, L.; Borger, D.R.; Krasner, C.N.; Goodman, A.K.; del Carmen, M.G.; Growdon, W.B.; Schorge, J.O.; Boruta, D.M.; et al. A SNaPshot of potentially personalized care: Molecular diagnostics in gynecologic cancer. Gynecol. Oncol. 2016, 141, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-L.; Ye, H.-M.; Zheng, L.-M.; Ruan, R.-S.; Tzeng, C.-M. Circulating tumor cells (CTCs) as a liquid biopsy material and drug target. Curr. Drug Targets 2014, 15, 965–972. [Google Scholar] [PubMed]
- Roy, S.; Lin, H.-Y.; Chou, C.-Y.; Huang, C.-H.; Small, J.; Sadik, N.; Ayinon, C.M.; Lansbury, E.; Cruz, L.; Yekula, A.; et al. Navigating the Landscape of Tumor Extracellular Vesicle Heterogeneity. Int. J. Mol. Sci. 2019, 20, 1349. [Google Scholar] [CrossRef]
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro. Oncol. 2017, 19, 1494–1502. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC. Genomics 2013, 14, 319. [Google Scholar] [CrossRef]
- Cheng, L.; Sun, X.; Scicluna, B.J.; Coleman, B.M.; Hill, A.F. Characterization and deep sequencing analysis of exosomal and non-exosomal miRNA in human urine. Kidney Int. 2014, 86, 433–444. [Google Scholar] [CrossRef]
- Miranda, K.C.; Bond, D.T.; Levin, J.Z.; Adiconis, X.; Sivachenko, A.; Russ, C.; Brown, D.; Nusbaum, C.; Russo, L.M. Massively parallel sequencing of human urinary exosome/microvesicle RNA reveals a predominance of non-coding RNA. PLoS ONE 2014, 9, e96094. [Google Scholar] [CrossRef]
- Ogawa, Y.; Taketomi, Y.; Murakami, M.; Tsujimoto, M.; Yanoshita, R. Small RNA transcriptomes of two types of exosomes in human whole saliva determined by next generation sequencing. Biol. Pharm. Bull. 2013, 36, 66–75. [Google Scholar] [CrossRef] [PubMed]
- RNA sequencing: advances, challenges and opportunities.: MGH OneSearch n.d. Available online: https://phstwlp2.partners.org:3699/eds/pdfviewer/pdfviewer?vid=1&sid=ea4e5203-5f5d-40c0-9f0e-60a37775633b%40sdc-v-sessmgr03 (accessed on 8 October 2019).
- Pflueger, D.; Terry, S.; Sboner, A.; Habegger, L.; Esgueva, R.; Lin, P.-C.; Svensson, M.A.; Kitabayashi, N.; Moss, B.J.; MacDonald, T.Y.; et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011, 21, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar] [CrossRef]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef]
- Lasorella, A.; Sanson, M.; Iavarone, A. FGFR-TACC gene fusions in human glioma. Neuro-Oncology 2017, 19, 475–483. [Google Scholar] [CrossRef]
- Na, K.; Kim, H.-S.; Shim, H.S.; Chang, J.H.; Kang, S.-G.; Kim, S.H. Targeted next-generation sequencing panel (TruSight Tumor 170) in diffuse glioma: a single institutional experience of 135 cases. J. Neurooncol. 2019, 142, 445–454. [Google Scholar] [CrossRef]
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Q.; Gao, G.; Wang, Y.; Fang, Z.; Li, G.; Yu, M.; Kong, L.; Xing, Y.; Gao, X. The effects of ponatinib, a multi-targeted tyrosine kinase inhibitor, against human U87 malignant glioblastoma cells. OncoTargets and Therapy 2014, 2013. [Google Scholar] [CrossRef]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Hsiung, C.A.; Matsuo, K.; Hong, Y.-C.; Seow, A.; Wang, Z.; Hosgood, H.D., 3rd; Chen, K.; Wang, J.C.; Chatterjee, N.; et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 2012, 44, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Deng, Z.; Wang, M.; Li, R.; Xu, G.; Bao, G. Additional evidence supports association of common genetic variants in VTI1A and ETFA with increased risk of glioma susceptibility. J. Neurol. Sci. 2017, 375, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Flowerdew, S.E.; Burgoyne, R.D. A VAMP7/Vti1a SNARE complex distinguishes a non-conventional traffic route to the cell surface used by KChIP1 and Kv4 potassium channels. Biochemical Journal 2009, 418, 529–540. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Bass, A.J.; Lawrence, M.S.; Brace, L.E.; Ramos, A.H.; Drier, Y.; Cibulskis, K.; Sougnez, C.; Voet, D.; Saksena, G.; Sivachenko, A.; et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011, 43, 964–968. [Google Scholar] [CrossRef]
- Porcher, C.; Chagraoui, H.; Kristiansen, M.S. SCL/TAL1: a multifaceted regulator from blood development to disease. Blood 2017, 129, 2051–2060. [Google Scholar] [CrossRef]
- Tan, T.K.; Zhang, C.; Sanda, T. Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia. Int. J. Hematol 2019, 109, 5–17. [Google Scholar] [CrossRef]
- Prange, K.H.M.; Mandoli, A.; Kuznetsova, T.; Wang, S.-Y.; Sotoca, A.M.; Marneth, A.E.; van der Reijden, B.A.; Stunnenberg, H.G.; Martens, J.H.A. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene 2017, 36, 3346–3356. [Google Scholar] [CrossRef]
- Chen, X.; Burkhardt, D.B.; Hartman, A.A.; Hu, X.; Eastman, A.E.; Sun, C.; Wang, X.; Zhong, M.; Krishnaswamy, S.; Guo, S. MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat. Commun. 2019, 10, 5767. [Google Scholar] [CrossRef]
- Calvanese, V.; Nguyen, A.T.; Bolan, T.J.; Vavilina, A.; Su, T.; Lee, L.K.; Wang, Y.; Lay, F.D.; Magnusson, M.; Crooks, G.M.; et al. MLLT3 governs human haematopoietic stem-cell self-renewal and engraftment. Nature 2019, 576, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Holstege, H.; Pfeiffer, W.; Sie, D.; Hulsman, M.; Nicholas, T.J.; Lee, C.C.; Ross, T.; Lin, J.; Miller, M.A.; Ylstra, B.; et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 2014, 24, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Wang, J.; Zhou, S.; Wang, C.L.; Ye, M.Z.; Wang, X.Y.; Song, Y.; Wang, Y.Q.; Zhang, L.T.; et al. Biological background of the genomic variations of cf-DNA in healthy individuals. Ann. Oncol. 2019, 30, 464–470. [Google Scholar] [CrossRef]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef]
- Parra, G. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2005, 16, 37–44. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B.; Parmigiani, G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad Sci. USA. 2013, 110, 1999–2004. [Google Scholar] [CrossRef]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef]
- Evans, S.M.; Putt, M.; Yang, X.-Y.; Lustig, R.A.; Martinez-Lage, M.; Williams, D.; Desai, A.; Wolf, R.; Brem, S.; Koch, C.J. Initial evidence that blood-borne microvesicles are biomarkers for recurrence and survival in newly diagnosed glioblastoma patients. J. Neurooncol. 2016, 127, 391–400. [Google Scholar] [CrossRef]
- Osti, D.; Del Bene, M.; Rappa, G.; Santos, M.; Matafora, V.; Richichi, C.; Faletti, S.; Beznoussenko, G.V.; Mironov, A.; Bachi, A.; et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin. Cancer Res. 2019, 25, 266–276. [Google Scholar] [CrossRef] [PubMed]
- André-Grégoire, G.; Bidère, N.; Gavard, J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie 2018, 155, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, Y.; Liu, H.; Liang, Z.; Zhu, Q.; Wang, L.; Tang, B.; Miao, S.; Ma, N.; Cen, X.; et al. Establishment and characterization of a DOT1L inhibitor-sensitive human acute monocytic leukemia cell line YBT-5 with a novel KMT2A-MLLT3 fusion. Hematol. Oncol. 2019, 37, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, R.M.; Aplan, P.D.; Humphries, R.K. NUP98-topoisomerase I acute myeloid leukemia-associated fusion gene has potent leukemogenic activities independent of an engineered catalytic site mutation. Blood 2004, 104, 1127–1136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jang, J.S.; Lee, A.; Li, J.; Liyanage, H.; Yang, Y.; Guo, L.; Asmann, Y.W.; Li, P.W.; Erickson-Johnson, M.; Sakai, Y.; et al. Common Oncogene Mutations and Novel SND1-BRAF Transcript Fusion in Lung Adenocarcinoma from Never Smokers. Sci. Rep. 2015, 5, 9755. [Google Scholar] [CrossRef]
- Yekula, A.; Muralidharan, K.; Kang, K.M.; Wang, L.; Balaj, L.; Carter, B.S. From laboratory to clinic: Translation of extracellular vesicle based cancer biomarkers. Methods 2020. [Google Scholar] [CrossRef]
- Li, J.; Ding, Y.; Li, A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J. Surg. Oncol. 2016, 14, 297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IDs | Age | Sex | Diagnosis | IDH | MGMT | EGFR | MET | TERT | CDKN2A |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 72 | F | GBM | WT | Methylated | Unamplified | Unamplified | Mutant | Loss |
| P2 | 41 | F | GBM | WT | Methylated | NK | NK | WT | Maintained |
| P3 | 52 | F | GBM | WT | Methylated | NK | NK | Mutant | Maintained |
| P4 | 73 | F | GBM | WT | Unmethylated | Unamplified | Unamplified | Mutant | Loss |
| P5 | 71 | M | GBM | WT | Unmethylated | Amplified | Amplified | WT | Maintained |
| P6 | 61 | F | GBM | WT | Unmethylated | Unamplified | Unamplified | WT | Maintained |
| P7 | 68 | F | GBM | WT | Unmethylated | Amplified | NK | Mutant | Maintained |
| P8 | 65 | F | GBM | WT | Methylated | Unamplified | Unamplified | WT | Loss |
| P9 | 74 | F | GBM | WT | Methylated | Unamplified | Amplified | WT | Maintained |
| H1 | NK | NK | - | - | - | - | - | - | - |
| H2 | 24 | F | - | - | - | - | - | - | - |
| H3 | NK | NK | - | - | - | - | - | - | - |
| H4 | NK | NK | - | - | - | - | - | - | - |
| H5 | 25 | M | - | - | - | - | - | - | - |
| H6 | 24 | F | - | - | - | - | - | - | - |
| H7 | 24 | F | - | - | - | - | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Yekula, A.; Muralidharan, K.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Carter, B.S.; Balaj, L. Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma. Cancers 2020, 12, 1219. https://doi.org/10.3390/cancers12051219
Wang L, Yekula A, Muralidharan K, Small JL, Rosh ZS, Kang KM, Carter BS, Balaj L. Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma. Cancers. 2020; 12(5):1219. https://doi.org/10.3390/cancers12051219
Chicago/Turabian StyleWang, Lan, Anudeep Yekula, Koushik Muralidharan, Julia L. Small, Zachary S. Rosh, Keiko M. Kang, Bob S. Carter, and Leonora Balaj. 2020. "Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma" Cancers 12, no. 5: 1219. https://doi.org/10.3390/cancers12051219
APA StyleWang, L., Yekula, A., Muralidharan, K., Small, J. L., Rosh, Z. S., Kang, K. M., Carter, B. S., & Balaj, L. (2020). Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma. Cancers, 12(5), 1219. https://doi.org/10.3390/cancers12051219